

Abstract
B-RafV600E inhibitors have been suggested to promote tumor regression with the help of host immunity, but this hypothesis has not been examined directly in detail. In this study, we profiled immunological changes in the tumor microenvironment and tumor infiltrating lymphocytes (TILs) in a B-RafV600E/Pten-driven murine model of melanoma, after administration of the BRafV600E small molecule inhibitor PLX4720. In this model, we found that as tumors developed they gradually acquired immunosuppressive features, including accumulation of regulatory T cells (Tregs) and CD11b+/Gr-1+ myeloid cells and loss of Th1 effector functions on CD4+ TILs, such as CD40L and IFN-γ expression. PLX4720 administration promoted development of a more immune stimulatory microenvironment associated with a relative increase in CD40L and IFNγ expression on intratumoral CD4+ TILs and a reduced accumulation of Tregs and CD11b+/Gr-1+ myeloid cells. Strikingly, CD40L or IFNγ blockade compromised the ability of PLX4720 to inhibit melanoma growth. Supporting this result, agonistic CD40 antibody was sufficient to evoke anti-tumor immunity and suppress tumor growth in tumor-bearing mice. Taken together, our results establish the critical role of immune-related changes with key contributions for CD40L and IFNγ signaling in the anti-tumor responses triggered in vivo by BRafV600E inhibitors.
Keywords: Braf kinase, melanoma, PLX4720, CD40L, anti-tumor
INTRODUCTION
Melanoma is considered a highly aggressive form of cancer and its resistance to chemotherapy results in high mortality rates (1, 2). Over 50% of melanomas contain the V600E mutation in the B-Raf kinase gene (BrafV600E), which causes constitutive activation of B-Raf and the downstream mitogen-activated protein kinase (MAPK) pathway that contributes to malignancy by increasing cellular glycolysis, anabolic metabolism, proliferation and suppressing apoptosis (1, 3, 4). Selective inhibitors targeting the oncogenic BrafV600E, but not wild type B-Raf kinase, have been shown to trigger apoptosis and reduce proliferation in melanoma cell lines in in vitro cell culture and in vivo animal models. Importantly, treatment of metastatic melanoma patients harboring BrafV600E mutation with the oral Braf inhibitors, vemurafenib and dabrafenib, prevents tumor progression in a high frequency of patients and in some cases induces tumor regression, and vemurafenib improves overall survival compared to chemotherapy (1, 4, 5). Despite the initial therapeutic benefits of vemurafenib, resistance to treatment inevitably occurs within a few months and this progressive form of melanoma appears intractable to current therapies (4). Multiple mechanisms, including additional mutations in RAS kinase and other proteins in MAPK pathway, are involved in developing resistance to BrafV600E inhibitors (4).
Tumors are infiltrated by several types of immune cells, including T lymphocytes, natural killer cells, and macrophages that have the potential to kill tumor cells or curb their growth via production of cytotoxic molecules, chemokines and inflammatory cytokines (6, 7). However, their anti-tumor functions are commonly suppressed by other immunoregulatory immune cells, such as regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs: categorized by CD11b+/Gr-1+surface staining), alternative activated macrophages (often referred to as M2-like macrophages), and immature or tolerogenic dendritic cells that also accumulate within the tumor microenvironment (8). In addition to blocking anti-tumor immune responses, these immunomodulatory cells can also promote tumor growth and metastasis through secretion of angiogenic factors (e.g., VEGF). Intratumoral T cells also upregulate inhibitory receptors, such as PD-1, TIM3, CTLA-4 and LAG3, which further repress anti-tumor effector functions upon ligand binding in the tumor microenvironment (9, 10). Importantly, recent clinical trials have found that blocking CTLA-4 and PD-1 signaling with monoclonal antibodies can evoke preexisting anti-tumor immunity and cause partial or, in some cases, complete tumor regression in a fraction of patients (11, 12). In contrast to the negative signaling pathways controlled by PD-1 and CTLA-4, CD40:CD40L signaling and costimulatory ligands, including CD70 and CD86, have been suggested to provide positive signals that boost anti-tumor immunity (13-16). These studies demonstrate the potential power of immunotherapy in treating cancer, but the issue remains that a large number of patients are unresponsive to these immunotherapies for reasons that are not entirely clear.
The BrafV600E mutation has a direct role in driving cellular transformation, but multiple studies suggest that it also indirectly modulates the tumor microenvironment. For instance, tumors treated with BrafV600E inhibitors displayed increased T lymphocyte infiltration and expression of melanoma antigens, MHCI, and PDL1 expression (17-21). Similarly, mice engrafted with a melanoma cell line and treated with the vemurafenib analogue PLX4720 also demonstrated increased T cell infiltration in tumors and responsiveness to antigens (18, 22). The anti-tumor effects of PLX4720 in this engraftment model was particularly dependent on CD8 T cells and could be enhanced by CD137 agonistic mAb treatment, suggesting that BrafV600E inhibitors can sensitize tumors to certain immunotherapies (22). In contrast, another study concluded that PLX4720 decreased T cell infiltration in the tumors and were unable to enhance anti-tumor responses in conjunction with CTLA4 blockade (23). Thus, more investigation is needed to better characterize the nature of the tumor microenvironment in melanoma and how BrafV600E inhibitors affect the function of infiltrating immune cells. Elucidating signals that create a more immunostimulatory tumor microenvironment by BrafV600E inhibitors could offer mechanistic insight into drug action and potentially identify new drug targets for improving anti-cancer therapies.
This study focused on the effects of PLX4720 on anti-tumor immunity in an inducible murine melanoma model that provides a highly physiologically relevant setting for studying the complex interplay between tumor, stromal, and immune cells in the tumor microenvironment. We found that as tumors grew the tumor microenvironment gradually acquired immunosuppressive features, including accumulation of Tregs and CD11b+/Gr-1+ myeloid cells and reduced effector functions of tumor infiltrating CD4 T cells. Importantly, PLX4720-stimulated host anti-tumor immune responses that inhibited tumor progression through CD40L-and IFNγ-dependent mechanisms. Interestingly, blockade of CD40:CD40L or IFNγ signaling differentially affected the immunoregulatory cells in the tumor microenvironment, suggesting these signaling pathways operate in a non-overlapping manner. We also demonstrated that agonistic anti-CD40 antibody treatment alone was effective to inhibit tumor growth in this model. These findings reveal that host immunity is a critical component of B-RafV600E inhibitor-mediated tumor suppression and provide new immunotherapeutic targets, such as CD40 and IFNγ, for enhancing anti-melanoma immunity that could be used in conjunction with B-RafV600E inhibitors or other immunotherapies.
MATERIALS and METHODS
Mouse breeding and tumor induction
The BRafCA; Tyr::CreER; Ptenlox4-5 (Braf/Pten) mice were previously described (3) and Pmel TCR transgenic mice were purchased from Jackson laboratory (24). For local tumor induction, 3-week-old Braf/Pten mice were administrated with 1μl 4-hydroxytamoxifen (4-HT) (8mg/ml in ethanol) topically on the back skin and examined every week for melanoma growth. All animal experiments were performed according to the approved protocols of Institutional Animal Care and Use Committee.
Tumor digestion
Tumors were minced in digestion media (HBSS with 0.5mg/ml collagenase IV and 200μg/ml DNase), incubated at 37°C for 1h and filtered to remove undigested tumor pieces. Single cell suspensions were then incubated with 2ml ACK lysis buffer (Invitrogen), washed and resuspended in complete RPMI.
Flow cytometry and cell sorting
Single cell suspensions from tumors or splenocytes were incubated with anti-Fc receptor antibody (2.4G2) on ice for 15 mins in FACS buffer (PBS with 1% FBS and sodium azide). The cells were then stained with the appropriate antibodies in 2.4G2-containing FACS buffer on ice for 30 mins. For intracellular cytokine staining, T cells were stimulated with PMA/ionomycin (Sigma Aldrich) and Brefeldin A for 5h, stained with surface antibodies and then fixed in Cytofix/Cytoperm buffer (BD Biosciences) or transcription factor staining buffer (ebioscience) and stained with antibodies to detect intracellular cytokines or FoxP3, respectively. All samples were acquired on LSRII flow cytometer and analyzed Flowjo. Cell sorting was performed on BD FACS Aria in Yale Cell Sorter Core Facility. TIDCs were sorted based on the following markers: CD45+/ CD3−/CD11c+/Gr-1−. TAMs were sorted based on the following markers: CD45+/ CD3−/CD11b+/Gr-1−/F4/80+. Antibodies against CD45 (A20), CD8 (53-6.7), CD3 (145-2C11), CD44 (IM7), CD11c (N418), CD11b (M1/70), Gr-1 (RB6-8C5), IFNγ (XMG1.2), IL-2 (JES6-5H4), TNFα (TN3-19), FoxP3 (FJK-16s), MHCII (M5/114.15.2) and CD70 (FR70) were purchased from eBioscience. Antibodies against CD4 (RM4-5), F4/80 (BM8), CD40L (MR1), MHCI (KH95), CD80 (16-10A1) and CD40 (3/23) were from Biolegend and anti-CD86 (GL1) was from BD Biosciences.
In vivo treatment of PLX4720 and agonistic anti-CD40 antibody
Five weeks after tumor induction, tumor-bearing BrafV600E/Pten mice were fed with normal or PLX4720-containing chow (417mg PLX4720/kg; generously provided by Plexxikon) for another 21 days. Tumor weight and tumor-infiltrating immune cells were assessed following treatments. In some cases the Braf/Pten mice (either on normal or PLX4720-containing chow) were administered anti-CD40L (MR1) or -IFNγ (XMG1.2) (250μg/mouse i.p.) or control vehicle (PBS) every three days from weeks 5-7. Alternatively, Braf/Pten mice were treated with control IgG or agonistic anti-CD40 antibody (FGK45) (250μg/mouse) every three days from weeks 6-8. Tumor growth was determined by measuring the tumor height with a caliper every three days and at the end of treatment, tumors were resected and tumor weight and immune response were determined.
T cell proliferation assay
Splenic cells of Pmel TCR transgenic mice were collected and CD8+ Pmel T cells were enriched by negative CD8 T cell enrichment kit (Stemcell technologies). Naïve CD8+ Pmel T cells were further purified by removing CD44+ cells with biotin selection kit (Stemcell technologies). Purified naïve Pmel cells were then loaded with 2 M CFSE at room temperature for 15 mins. CFSE-labeled naïve CD8+ Pmel T cells were mixed with tumor-infiltrating DCs in a 5:1 ratio in the presence of 1μg/ml gp100. For agonistic anti-CD40 antibody, the cell mixtures were incubated with 50μg/ml of IgG (Bioxcell) or agonistic anti-CD40 antibody (FGK45) (Bioxcell) during this period. Proliferation of CD8+ Pmel T cells was determined by FACS analysis to measure CFSE dilution.
RNA purification and quantitative real-time RT-PCR
For RNA purification, Trizol (Invitrogen) reagent was used to extract total RNA according to manufacturer’s instruction and cDNAs were synthesized using SuperScript II Reverse Transcriptase (Invitrogen). Quantitative real-time PCR were performed on Mx3000P qPCR system (Aglient technologies) with iTaq Universal SYBR Green Supermix (Bio-Rad). Primer sequences to indicated mRNAs were listed in supplementary table. Ct values were measured and relative gene expression was calculated using Δ ΔCt method. Statistics Data were presented in means ± SD. Statistical analysis was performed by unpaired Student’s t test and P value < 0.05 was considered as statistically significant.
RESULTS
BrafV600E/Pten melanomas acquire an immunosuppressive microenvironment during tumorigenesis
We first examined the immune cell infiltrate in melanomas in the TyrcreERT2/BrafV600E/Ptenlox mice. In this system, application of 4-hydroxytamoxifen (4-HT) to the skin initiates melanoma formation by introducing constitutively active form of B-Raf kinase (BrafV600E) and Pten deletion in melanocytes (3, 25). It takes approximately 8 weeks for these tumors to grow to a size over 200 mm2 and mice usually die within 10 weeks post tumor induction (3). To examine if there were any changes in the tumor microenvironment associated with tumor progression, we collected tumor samples from mice at early stages (5 weeks) and advanced stages (8 weeks) post 4-HT induction and examined multiple properties of the intratumoral immune cells using flow cytometry (Figure 1A). Several notable observations were made during this longitudinal study. First, the frequency of tumor infiltrating CD4+ and CD8+ T lymphocytes increased as tumors progressed and CD4+ T cells were the dominant T cell population in this model (Figure 1B). In addition, we found that the frequency of CD11b+/Gr-1+ cells (commonly referred to as myeloid derived suppressor cells) increased, but the populations of tumor-infiltrating dendritic cells (TIDCs) (CD45+/CD11c+/Gr-1−) and tumor-associated macrophages (TAMs) (CD45+/CD11b+/F4/80+/Gr-1−) decreased as the tumor grew (Figure 1B). Gating criteria for above myeloid cell populations are shown in Supplementary Figure 1; note ~40% of TIDC expressed F4/80 as reported previously (26)). We also determined the frequency of FoxP3+ Tregs among tumor infiltrating CD4 T cells and found that FoxP3+ Tregs accumulated in the tumors over time (Figure 1C). Importantly, the frequency of FoxP3+ Tregs in the spleen of tumor-bearing animals did not change over this time period, indicating that the increase in intratumoral Tregs was unique to the tumor microenvironment and not due to systemic increases in Tregs in the animals.
Figure 1. Melanoma progression is associated with changes in T cell effector and helper functions.

(A) Experimental outline of tumor induction using 4-HT: 4-hydroxytamoxifen and analysis of murine BrafV600E/Pten melanomas. (B) Tumor infiltrating CD45+ immune cells from early (5 weeks) or advanced (8 weeks) staged melanomas were analyzed by flow cytometry using anti-CD4, CD8, CD3, CD11b, CD11c, Gr-1, and F4/80 mAbs to identify CD4 and CD8 T cells, tumor-infiltrating dendritic cells (TIDCs), tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs) (gating strategies shown in Supp. Fig. 1). Data are derived from 3 independent experiments (n=5-6 animals/group). (C) FACS plots (upper) and bar graphs (lower) show the percentage of FoxP3+ CD4+ Tregs in tumors and spleens at 5 and 8 weeks post tumor inductions as measured by flow cytometry (n=5 mice per group). (D) The expression of indicated effector cytokines of tumor infiltrating CD4 T cells isolated from tumors of indicated stages was measured by flow cytometry after PMA/ionomycin stimulation and intracellular cytokine staining (ICCS). (E) The expression of CD40L on FoxP3− tumor infiltrating CD4 T cells isolated directly ex vivo from tumors at indicated tumor stages (n=5 mice per group). *, P<0.05 in unpaired student t test.
The increase in two immunosuppressive cell types, CD11b+/Gr-1+ cells and Tregs, in BrafV600E/Pten melanomas prompted us to assess the effector functions of tumor-infiltrating T lymphocytes. We did not detect statistically significant changes in effector cytokine production in tumor infiltrating CD8 T cells (Supplementary Figure 2). However, the percentage of IFNγ, TNFα and IL-2-producing CD4 T cells decreased as tumors grew (Figure 1D). Intriguingly, the loss of cytokine production between early and advanced tumor stages also correlated with a decrease in expression of the costimulatory ligand CD40L on the FoxP3− (non-Treg) CD4 T cells (Figure 1E). Taken together, these results showed that as BrafV600E/Pten melanomas grew, the tumor microenvironment acquired multiple immunosuppressive features wherein Tregs and MDSCs accumulated, and critical CD4 T cell helper and effector functions decayed. These changes might lead to ineffective anti-tumor immune responses and the establishment of a protumorigenic microenvironment.
Accumulation of immature antigen-presenting cells and M2-like macrophages in BrafV600E/Pten melanomas
CD40:CD40L signaling is a critical licensing mechanism that enhances maturation of DCs and macrophages and helps boost CD8 T cell activation and expansion under less-inflammatory conditions (27, 28). In macrophages, CD40:CD40L signaling promotes the development of classically activated (M1)-like macrophages that produce IL-12 to help TH1 differentiation and reactive oxygen species (ROS) and nitric oxide (NO) to eliminate pathogens or cancer cells (13, 28-30). In contrast, tumors typically contain alternatively activated (M2)-like macrophages that produce IL-10, VEGF and matrix metalloproteinases (MMPs) that assist Type-2 immune responses and/or dampen immune responses to promote wound healing (31). Although a variety of mechanisms can regulate macrophage skewing from M1-→ M2-like states in tumors, it has not been well explored if this involves changes in CD40:CD40L signaling within the tumor microenvironment (8, 32, 33). Because the expression of CD40L declined on CD4 T cells as BrafV600E/Pten melanomas progressed, we postulated that the maturation or activation status of TIDCs and TAMs might also be affected. Interestingly, in early-stage tumors (5 weeks) we observed relatively high amounts of MHC Class I and II and costimulatory ligands (CD70, CD80 and CD86) expression on both TIDCs and TAMs. However, the expression of these proteins declined substantially in the advanced stage tumors present at 8 weeks (Figure 2A).
Figure 2. The accumulation of tolerogenic antigen-presenting cells during melanoma progression.
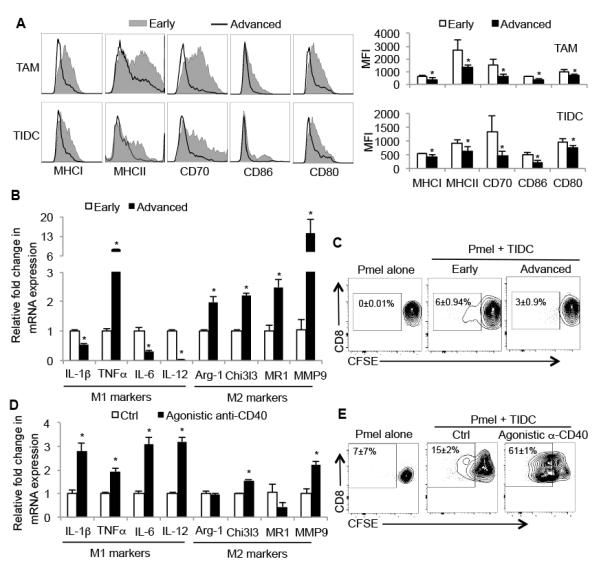
(A) FACS plots (left) and bar graphs (right) show the expression of indicated maturation markers on TAMs and TIDCs isolated from early (5 weeks) or advanced (8 weeks) staged melanomas as analyzed by flow cytometry. Bar graph represents cummulative data from one of three independent experiments (n=4-5 mice per group). MFI= mean fluorescence intensity. (B) The mRNA expression of conventional “M1” and “M2” genes in TAMs isolated directly from resected melanomas at indicated stages (n=8-12 mice per group). ‘M1’ refers to classic activation of macrophages and ‘M2’ refers to alternative activation. (C) Naïve Pmel CD8 T cells were labeled with CFSE and activated in vitro by gp100-pulsed TIDCs isolated from early (5 weeks) or advanced (8 weeks) staged melanomas. (D) Same as in (B), except the TAMs were treated in vitro with vehicle (Ctrl) or agonistic anti-CD40 mAb for 16 hrs prior to the analysis of gene expression (n=8 mice per group). Three days later the frequency of divided cells was assessed based on dilution of CFSE. (E) Same as in (C), except the TIDC and T cell cultures were treated with vehicle (Ctrl) or agonistic anti-CD40 mAb and analyzed 5 days later. Data are representative of 3-4 experiments. *, P<0.05 in unpaired student t test.
We next probed the macrophages for M1- and M2-like markers during tumor progression, and found that the expression of M1-like cytokines Il1β, Il6 and Il12 declined while the expression of Tnfα and M2-like genes including Arg1, Chi3l3, Mr1 and Mmp9 increased (Figure 2B). This result indicated more M2-like macrophages resided in late-stage tumors. Moreover, TIDCs isolated from advanced BrafV600E/Pten melanomas lost their capacity to stimulate gp100-specific (Pmel) CD8 T cell proliferation as compare to TIDCs isolated from early stage of tumors (Figure 2C). Taken together, these results showed a gradual loss of mature APCs, but an accumulation of M2-like macrophages as BrafV600E/Pten melanomas grew. To test whether APCs in advanced BrafV600E/Pten melanomas were responsive or non-responsive to CD40:CD40L signaling, we incubated the TAMs and TIDCs purified from advanced BrafV600E/Pten melanomas with control IgG or agonistic anti-CD40 antibody (FGK45) overnight. Indeed, TAMs were responsive to CD40:CD40L signaling and M1-like gene expression was upregulated more profoundly than M2-like gene expression (Figure 2D). In addition, FGK45 mAb enhanced the ability of the advanced staged TIDCs to stimulate naïve Pmel CD8 T cell activation and proliferation (Figure 2E). These data suggest that TIDCs and TAMs in advanced BrafV600E/Pten melanomas had developed tolerogenic features commonly associated with tumors such as reduced expression of MHC and costimulatory ligands and that the TAMs had undergone an M1→M2-like phenotypic switch. However, both TIDCs and TAMs of advanced staged melanoma were sensitive to CD40:CD40L signaling, suggesting a model whereby as the tumors grow, the loss of CD4 T cell CD40L expression may contribute to the decline in APC maturation and thus, T cell activity.
PLX4720 treatment inhibits accumulation of immunoregulatory cells and restores effector functions of tumor infiltrating CD4 T cells
Although host immunity has been implicated in the anti-tumor effects of BrafV600E inhibitors in melanoma, it is not clear which immune signaling pathways are most affected by BrafV600E inhibitors and how these pathways alter the tumor microenvironment and anti-tumor immune responses. Therefore, we next sought to determine whether PLX4720 therapy could alter the phenotype or function of tumor infiltrating immune cells. We induced BrafV600E/Pten melanomas and after 5 weeks the mice were treated with normal chow or that which contains PLX4720 (PLX diet) for another 3 weeks (Figure 3A). As expected, PLX4720 treatment significantly decreased tumor weight (Figure 3B). In agreement with recent studies, we also observed a fivefold increase in the number of CD4 and CD8 T lymphocytes (Figure 3C) infiltrating PLX4720-treated melanomas, but not the spleen, and this was confirmed by anti-CD3 immunohistochemical staining of tumor sections (Figure 3D and Supplementary Figure 3) (18, 19). In addition, the frequency of intratumoral FoxP3+ Treg CD4 T cells was reduced 3-4 fold after PLX4720 treatment, while no effect was observed in the spleen (Figure 3E). These results suggested that PLX4720 affected the recruitment of Tregs into tumors or directly modulated inducible-Treg (iTreg) formation in the tumor microenvironment. Moreover, these data imply that PLX4720 preferentially promoted the accumulation of FoxP3− CD4 T cells (non-Tregs) within the tumors, which may contain “helper” cells to boost anti-tumor responses.
Figure 3. BrafV600E inhibitor, PLX4720, elevates immune activity of intratumoral CD4 T cells and myeloid cells.

(A) Experimental outline of tumor induction and treatment. Melanomas were induced using 4-HT and at 5 weeks the mice were either maintained on normal chow diet or fed chow containing PLX4720 (PLX) for another three weeks. Infiltrating immune cells were then analyzed by flow cytometry. (B) Tumor weight of melanomas from BrafV600E/Pten mice fed with the indicated diets. (C) Bar graphs show the cell number of tumor infiltrating T cells and (D) representative images of CD3 immunohistochemical staining of melanomas at week 8. (E) FACS plots (upper) and bar graphs (bottom) show the percentage of FoxP3+ CD4+ Tregs in tumors and spleens at week 8 (n=5-7 mice per group). (F) The expression of indicated effector cytokines by tumor infiltrating CD4 T cells isolated from control or PLX-treated tumors was analyzed at week 8 by ICCS. (G) The expression of CD40L on tumor infiltrating CD4 T cells isolated directly ex vivo from control or PLX-treated tumors at week 8. (H) Quantitation of tumor infiltrating TIDCs, TAMs, and MDSCs at week 8 in tumor-bearing mice fed normal or PLX-containing chow based on flow cytometry as outlined in Supp. Fig. 1. Data are derived from 3 independent experiments (n=5-6 animals/group). (I) FACS plots (left) and bar graphs (right) show the expression of indicated maturation markers on TAMs and TIDCs isolated from control or PLX-treated melanomas. Data shown are pooled from 3 independent experiments that contain in total 10-12 mice per group.*, P<0.05 in unpaired student t test.
Since we found that expression of effector cytokines from intratumoral CD4 T cells declined during tumor progression, we next tested whether PLX4720 treatment affected CD4 T cell cytokine production. After PLX4720 therapy, a larger fraction of tumor infiltrating CD4 T cells produced IFNγ (i.e., TH1 cells) (Figure 3F). However, PLX4702 treatment did not affect the CD4 T cell production of IL-2 and TNFα. More notably, PLX4720 treatment also augmented the expression of CD40L on intratumoral CD4 T cells (Figure 3G). Intriguingly, PLX4720 treatment robustly promoted PD-1 expression on intratumoral CD4 T cells and the frequency of PD-1+ CD4 T cells (Supplementary Figure 4), supporting a similar observation in human melanoma following vemurafenib treatment (21). These results demonstrate that BrafV600E inhibitors have a potent immunostimulatory activity in the BrafV600E/Pten murine melanoma mouse and can greatly boost the effector T cell:Treg ratio and TH1 effector and helper functions within tumors.
Next, we investigated whether PLX4720 treatment also modulated the frequency or phenotype of tumor infiltrating myeloid cells. First, we noted that PLX4720 therapy reduced the frequency of intratumoral CD11b+/Gr-1+ cells by 4-5 fold and slightly decreased the frequency of TAMs, but no in the TIDCs (Figure 3H). PLX4720 therapy also enhanced the maturation status of both TAMs and TIDCs (Figure 3I), suggesting that intratumoral APCs might possess a higher capability to activate and expand T cells and promote TH1-polarized differentiation. Altogether, these findings demonstrated that PLX4720 treatment had potent immunostimulatory effects on the phenotype and function of innate and adaptive immune cells in the tumor microenvironment. PLX4720 treatment promoted the expression of IFNγ and CD40L on CD4 T cells, MHC and costimulatory ligands on APCs and repressed accumulation of Tregs and CD11b+/Gr-1+ population, which collectively, may lead to enhanced anti-tumor immune responses.
PLX4720 suppresses tumor growth is partially dependent on CD4 T cells
Next, we investigated whether T cells contribute to PLX4720 anti-tumor effect. Tumors were induced in BrafV600E/Pten mice and five weeks later the mice were treated with PLX4720 with or without TIB210 or GK1.5 mAbs for 15 days to deplete CD8 or CD4 T cells, respectively (Figure 4A). The depleting antibodies reduced both splenic and intratumoral CD8 or CD4 T cells after 15 days of treatment (Supplementary Figure 5). Surprisingly, the reduction in tumor growth by PLX4720 treatment did not depend on CD8 T cells, but was partially dependent on CD4 T cells (Figure 4B-E). These results suggest that CD4 T cells play a more dominant role in the PLX4720-mediated anti-tumor response than CD8 T cells. Note, we also examined whether the elevated CD70 expression on APCs after PLX4720 therapy contributed to its ability to reverse tolerant CTLs and retard tumor growth (14, 15, 34), but found that blocking CD70 did not perturb the ability of PLX4720 to inhibit tumor progression in any detectable manner (Supplementary Figure 6).
Figure 4. CD4 T cells contribute to PLX4720-mediated anti-tumor response.
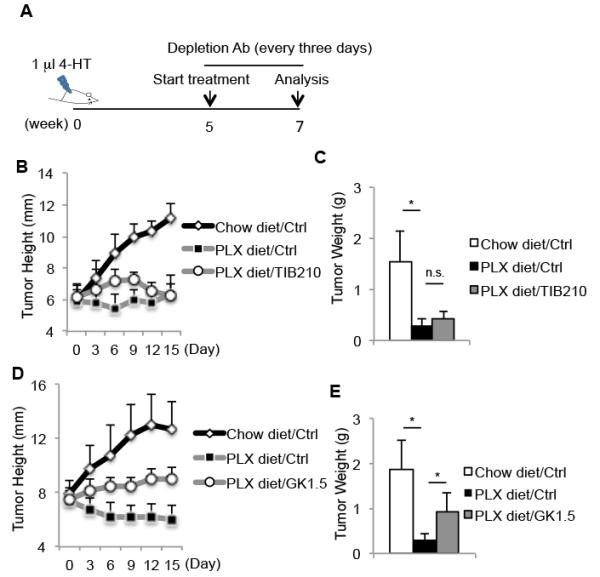
(A) Experimental outline of tumor induction and treatment. Melanomas were induced using 4-HT and at 5 weeks the mice were either maintained on a regular diet or fed chow containing PLX4720 (PLX). Several mice from each group were also treated with T cell-depleting mAbs (TIB210 or GK1.5 diluted in PBS) or vehicle control (PBS) every three days for 15-days. The growth rate (B, D) and weight (C, E) of the tumors in mice receiving the indicated treatments are shown. Data shown are pooled from 2 independent experiments that contain in total 5-7 mice (B,C) or 8-10 mice (D, E) per group. *, P<0.05 in unpaired student t test.
PLX4720 therapy enhances maturation of antigen-presenting cells and decreases accumulation of Tregs in BrafV600E/Pten melanomas in a CD40L-dependent manner
Since the anti-tumor response of PLX4720 treatment was compromised when CD4 T cells were depleted, it is possible that augmented effector and helper CD4 T cell functions contribute to PLX4720-mediated immunostimulation of tumor microenvironment. CD40:CD40L signaling can augment immune surveillance of tumors by enhancing APC maturation, boosting macrophages’ cytotoxicity, and elevating CTL proliferation and effector functions (13, 27-29, 35). To determine if increased CD40L expression on intratumoral CD4 T cells during PLX4720 treatment was functionally relevant, we blocked CD40L signaling during PLX4720 treatment using the anti-CD40L antibody (MR1) (36). Tumors were induced in BrafV600E/Pten mice and five weeks later the mice were treated with PLX4720 with or without anti-CD40L mAb (MR1) for two weeks. We found that blocking CD40:CD40L signaling strongly interfered with the anti-tumor effects of PLX4720 treatment because the BrafV600E/Pten melanomas grew in the PLX diet+MR1 treated animals at rates similar to, but slightly less than untreated vehicle-injected control animals (Figure 5A-C). MR1 treatment by itself did not have a significant effect on tumor growth in animals on normal chow diet. These results demonstrate that CD40:CD40L signaling was required for the suppression of tumor growth by PLX4720 therapy and suggest that host immunity contributes to the therapeutic effects of BrafV600E inhibition on melanoma growth.
Figure 5. PLX4720 suppresses regulatory T cell accumulation and boosts maturation of antigen-presenting cells in melanomas through a CD40:CD40L signaling-dependent mechanism.

Five weeks after tumor induction, BrafV600E/Pten melanoma-bearing mice were fed normal or PLX-containing chow for another 15 days. Control vehicle or CD40L-blocking mAb (MR1) was injected intraperitoneally (i.p.) every three days during this 15-day period and the growth rate (A), weight (B), and appearance (C) of the tumors were assessed. Data shown are pooled from 3 independent experiments that contain in total 9-12 mice per group. (D) FACS plots (left) and bar graphs (right) show the percentage of FoxP3+ CD4+ Tregs in tumors and spleens post treatment as measured by flow cytometry. (E-F) Bar graphs show the number of tumor infiltrating T cells (E) or percentage of MDSCs (F) post treatment as measured by flow cytometry (n=5-8 mice per group). (G) Histograms show the expression of indicated maturation markers on the TAMs and TIDCs isolated from the tumors following treatment. *, P<0.05 in unpaired student t test. n.s.= not significant in unpaired student t test.
Further comparison revealed that the reduction in infiltrating Tregs observed during PLX4720 treatment was also dependent on CD40L signaling because greater percentages of Tregs were found in PLX+MR1-treated tumors as compared to those given PLX-alone (Figure 5D). Likewise, CD40L signaling was important for the significant increase in total CD4 and CD8 T cells in the melanomas caused by PLX4720 treatment (Figure 5E). In contrast, CD40L blockade did not affect the ability of PLX4720 treatment to reduce CD11b+/Gr-1+ population frequencies (Figure 5F). Next, we sought to test if the enhanced maturation of intratumoral APCs observed during PLX4720 treatment was dependent on CD40:CD40L signaling. Indeed, CD40L blockade prevented PLX4720 therapy-induced elevation of MHC Class I and CD70 on both TAMs and TIDCs (Figure 5G). These experiments show that several of the notable immunostimulatory effects of PLX4720 on the microenvironment of melanoma depend on CD40L signaling, including the increased effector T cell:Treg ratio and maturation status of APCs. These findings provide novel mechanistic insight into therapeutic effects of Braf inhibitors in melanoma and implicate host immunity as a critical part of the anti-tumor effects of these types of drugs.
IFNγ contributes to reduction of intratumoral CD11b+/Gr-1+ myeloid cells upon PLX4720 treatment
In addition to CD40L, IFNγ is another anti-tumor immunological factor that can promote MHC expression on both APCs and tumor cells, trigger tumor cell senescence and polarize M1-like macrophages (37, 38). Since the frequency of IFNγ-producing CD4 T cells was increased in the tumors after PLX4720 therapy, we next investigated if IFNγ itself was also involved in PLX4720-mediated anti-tumor responses by blocking IFNγ with the mAb XMG1.2 at the time of PLX4720 treatment. This showed that the therapeutic effects of PLX4720 were compromised in mice receiving IFNγ-neutralizing mAb because, as observed with blocking CD40L, the tumors in these mice grew at rates comparable to those left untreated (Figure 6A-6C). IFNγ-neutralizing antibody did not escalate tumor growth in mice receiving chow diet. We then examined which changes in the tumor microenvironment induced by PLX4720 therapy were IFNγ-dependent. First, in contrast to anti-CD40L blockade, IFNγ blockade had minimal effect on altering intratumoral Treg frequency in either the normal- or PLX-diet fed mice (Figure 6D). However, we found that blocking IFNγ suppressed the PLX4720-induced increase in infiltrating CD4 and CD8 T cells into the tumors, albeit the effects on CD4 T cells were insignificant (Figure 6E). Although no notable changes in percentage of TAMs or TIDCs were observed when IFNγ was blocked during PLX4720 treatment (Supplementary Figure 7), the frequency of CD11b+/Gr-1+ myeloid cells increased considerably in the tumors (Figure 6F). This suggested that IFNγ–blockade affected CD11b+/Gr-1+ myeloid cells accumulation in tumors. We also examined the expression of MHCI and CD70 on TAMs and TIDCs and found that IFNγ blockade prevented PLX4720-induced upregulation of these proteins on APCs (Figure 6G). Overall, these data showed that IFNγ was necessary for the PLX4720-mediated maturation of APCs and reduction of intratumotal CD11b+/Gr-1+ myeloid cells, which likely contributed to anti-tumor effects of PLX4720 in BrafV600E/Pten melanomas.
Figure 6. PLX4720 inhibits intratumoral accumulation of myeloid suppressor cells in melanomas in an IFNγ-dependent mechanism.

Five weeks after tumor induction, BrafV600E/Pten melanoma-bearing mice were fed normal or PLX-containing chow for another 15 days. Control vehicle or IFNγ-neutralizing mAb (XMG1.2) was injected i.p. every three days during this 15-day period and the growth rate (A), weight (B), and appearance (C) of the tumors were assessed. (D) FACS plots (left) and bar graphs (right) show the percentage of FoxP3+ CD4+ Tregs in tumors and spleens post treatment as measured by flow cytometry. Data shown are pooled from 2 independent experiments that contain in total 7-12 mice per group. (E-F) Bar graphs show the cell number of tumor infiltrating T cells (n=5-8 mice per group) (E) or percentage of MDSCs (F) post treatment as measured by flow cytometry (n=7-11 mice per group). (G) Histograms show the expression of indicated maturation markers on the TAMs and TIDCs isolated from the tumors following treatment. *, P<0.05 in unpaired student t test. n.s.= not significant in unpaired student t test.
Agonistic anti-CD40 antibody elicits anti-tumor response and promotes maturation of intratumoral antigen-presenting cells in BrafV600E/Pten mice
The above findings reveal that CD40:CD40L and IFNγ signaling pathways have potent anti-tumor activities during PLX4720 treatment, identifying new therapeutic opportunities to treat melanoma. Because agonistic anti-CD40 mAb treatment can evoke anti-tumor immune responses, we sought to evaluate the anti-tumor responses of agonistic anti-CD40 mAb in the BrafV600E/Pten murine melanoma model. To this end, BrafV600E/Pten melanomas were allowed to progress for six weeks, and then the mice were injected with control IgG or agonistic anti-CD40 antibody (FGK45) (250μg/mouse) every three days for another 15 days. This experiment revealed that agonistic anti-CD40 mAb treatment could restrict tumor growth considerably (Figure 7A-7C). Importantly, mice treated with FGK45 mAb exhibited splenomegaly, yet no significant increases of intratumoral CD4 and CD8 T cells or CD11b+/Gr-1+ myeloid cells were observed (Supplementary Figure 8A-D). However, the number of splenic and intratumoral Tregs was increased by FGK45 treatment (Supplementary Figure 8E). Notably, agonistic anti-CD40 mAb treatment augmented the maturation status of both TAMs and TIDCs in BrafV600E/Pten melanomas (Figure 7D). We observed that neither CD45− cells (including both tumor and stromal cells) directly isolated from BrafV600E/Pten melanomas nor a Braf/Pten-derived melanoma cell line expressed CD40 by flow cytometry (Supplementary Figure 9), lessening the possibility that FGK45 triggers antibody-dependent cell-mediated cytotoxicity (ADCC) of tumor cells. We next examined whether T cells are required for FGK45-dependent restriction of tumor growth, and surprisingly, T cell depletion did not compromise the anti-tumor effects of FGK45 mAb treatment (Figure 7E-F). Altogether, these results suggest that the apparent decline in CD40:CD40L signaling that occurs during BrafV600E/Pten melanoma progression contributes to the formation of an immunosuppressive microenvironment, and augmentation of CD40 signaling via agonistic anti-CD40 mAb is a significant form of immunotherapy, that can operate independently of T cells, to reduce BrafV600E/Pten melanoma growth.
Figure 7. Agonistic anti-CD40 antibody impedes Braf/Pten melanoma growth in vivo.

Six weeks after tumor induction, BrafV600E/Pten melanoma-bearing mice were injected i.p. with control vehicle (ctrl) or agonistic anti-CD40 mAb (FGK45) every three days for 15 days. The rate of tumor growth (A), tumor weight (B) and appearance (C) were assessed 15 days post treatment. (D) Overlapping histograms show the expression of indicated maturation markers on the TAMs and TIDCs isolated from the tumors following treatment. As in (A), but mice were also injected with or without T cell-depleting mAbs (TIB210+GK1.5) and 15 days later the growth rate (E) and weight (F) of tumors were assessed. Data shown are pooled from 2 independent experiments with 6~9 mice per group. *, P<0.05 in unpaired student t test. n.s.= not significant in unpaired student t test.
DISCUSSION
BrafV600E is a common driver-oncogene in melanoma and other cancers. Clinically available BrafV600E inhibitors suppress tumor growth and it has been suggested that anti-tumor host immunity can be enhanced by these drugs (18-21). This study aimed to investigate the immune responses during tumor progression in an inducible BrafV600E/Ptenlox murine melanoma model and identify how the BrafV600E inhibitor PLX4720 altered the tumor microenvironment and infiltrating immune cells. We wanted to determine whether particular aspects of immune functions, cell types or signaling pathways contributed to the therapeutic effects of BrafV600E inhibition, as previously intimated by some studies in melanoma patients (18-23, 39, 40). We found that the tumor microenvironment in BrafV600E/Pten melanomas acquired multiple immunosuppressive features, including accumulation of Tregs, CD11b+/Gr-1+ myeloid cells, and immature APCs. Furthermore, intratumoral CD4 T cells lost their ability to exert effector (IFNγ) and helper (CD40L) functions. Surprisingly, PLX4720 therapy augmented the expression of CD40L and IFNγ on tumor infiltrating CD4 T cells and impeded intratumoral accumulation of Tregs and CD11b+/Gr-1+ myeloid cells. To our knowledge, this is the first report that these particular CD4 T cell functions can be modulated by BrafV600E inhibitor treatment. Moreover, we also found that treatment with an agonistic anti-CD40 mAb provided significant therapeutic benefits in suppressing melanoma growth and enhancing maturation of APCs in the BrafV600E/Pten melanoma model. Altogether, these findings support the model that CD40L is critical in mounting protective anti-melanoma immunity and have important implications in clinical trials involving immunotherapy. Possibly, combinational therapy with anti-CD40 agonists plus vemurafenib, anti-PD-1 or anti-CTLA-4 will enhance patient outcomes.
Vemurafenib and other BrafV600E inhibitors were originally believed to affect cell proliferation and trigger apoptosis and senescence on tumor cells harboring BrafV600E mutation. However, a number of studies revealed that BrafV600E inhibitors might boost immunosurveillance of tumors in both patients and mice. Using melanoma cell line engraftment models in mice, several effects of Braf inhibition have been reported, such as increased tumor-infiltrating T lymphocytes, reduced intratumoral Tregs and CD11b+/Gr-1+ myeloid cells, and decreased CCL2, IL-1α and IL-1β expression of tumor cells (18-23, 39, 40). While these studies suggested that host immunity is critical to maximize the therapeutic efficacy of BrafV600E inhibitors, tumors arising from engrafted cell lines are likely more immunogenic and most certainly do not fully recapitulate the microenvironment of tumors that arise endogenously in vivo. Therefore, elucidating which elements of the immune system are involved in the anti-tumor effects of BrafV600E inhibitors and how BrafV600E inhibitors relieve the immunosuppressive restraints imposed by the tumor microenvironment in a more pathophysiologically relevant murine model may enhance the development of anti-melanoma immunotherapies. For this reason, we took the advantage of the inducible BrafV600E/Pten melanoma model to investigate these important questions in a more physiologic manner. Our results showed that PLX4720 treatment significantly promoted tumor infiltration of T cells that depended on CD40:CD40L and IFNγ signaling. However, PLX4720 treatment has also been shown to decrease intratumoral lymphocyte frequencies in BrafV600E/Pten melanoma model (23, 40) and the discrepancy between these studies might be due to different methods of tumor infiltrating immune cell isolation and/or calculation of cell numbers (41). Our analysis went further and identified several previously unrecognized immunostimulatory effects of PLX4720 treatment on the immune microenvironment of tumors. Although adoptive CD4 T cell transfer can help to establish anti-tumor immune responses (42, 43), the precise mechanisms are poorly understood and a recent report showed that CD4 T lymphocytes are required to re-establish anti-tumor immunity upon oncogene inactivation (44). In agreement with these findings, our results reveal that CD4 T cells contribute to PLX4720-induced anti-tumor responses and delineate underlying immune signaling pathways (e.g., IFNγ and CD40L) that break an immunosuppressive tumor microenvironment.
Although we found that PLX4720 therapy augmented the effector and helper functions of tumor infiltrating CD4 T cells and diminished the accumulation of intratumoral Tregs in BrafV600E/Pten melanomas, the underlying mechanism(s) by which PLX4720 induces these immunostimulatory properties remain unclear. In addition to inhibiting B-Raf kinase, some BrafV600E inhibitors also paradoxically enhance C-Raf kinase activity by promoting heterodimerization between C-Raf and wild type B-Raf (4). Although recent studies showed that PLX4720 and vemurafenib do not affect T lymphocyte functions and viability in a broad range of concentrations (4), this does not necessarily rule out that the effects we observed are independent C-Raf activity alteration. Moreover, the BrafV600E mutation remodels the metabolic activities of cancer cells and BrafV600E inhibitors can decrease glycolysis and glutaminolysis and elevate oxidative phosphorylation in melanomas (45-47). Given that nutrients, such as glutamine, arginine, and glucose, are critical for effector T cell function and differentiation (48), it is possible that PLX4720-induced changes in tumor metabolism may indirectly lead to TH1 or M1-like polarization in T cells and macrophages, respectively. Additional studies are needed to further elucidate the mechanisms by which PLX4720 treatment enhances immune activity and these could reveal previously unappreciated mechanisms of immunosuppression in the tumor microenvironment.
Melanoma cells have been shown to promote accumulation of Tregs and possibly CD11b+/Gr-1+ myeloid cells by secreting large amount of CCL2 and VEGF (18, 22), and accumulation of these immunomodulatory cells could dampen the activity and helper functions of T cells in tumors (8, 32, 33). Interestingly, our results demonstrated that CD40L and IFNγ blockade during PLX4720 treatment differentially affected the infiltration of intratumoral Tregs and CD11b+/Gr-1+ myeloid cells, respectively, indicating that the accumulation of these cells in tumors is likely governed by distinct pathways. Since CD11b+/Gr-1+ myeloid cells are immature myeloid cells and IFNγ can promote their differentiation into macrophages both in vitro and in vivo (38, 49), PLX4720 therapy might reduce the size of the CD11b+/Gr-1+ myeloid cells population by interfering with their recruitment or promoting their differentiation in an IFNγ-dependent manner. On the other hand, CD40L blockade can ameliorate autoimmune disease and rejection of cardiac allograft by inducing Treg formation and suppressing T and B cell activation and antibody production (36). Therefore it is possible that the loss of CD40L signaling on T cells as tumors progress might facilitate Treg formation and impede T cell activation within tumors. We also found that administration of agonistic anti-CD40 mAb alone could suppress tumor progression in this model of melanoma. Agonistic anti-CD40 antibody has been shown to be a new therapeutic option for a variety of cancers in preclinical studies and humanized anti-CD40 antibodies are under phase I trial (13, 27, 29). Activation of intratumoral macrophages and/or rejuvenation of exhausted or tolerized T cells might underlie the anti-tumor effects of this drug, but it is not clear if agonistic anti-CD40 antibody could potentiate the therapeutic efficacy of other immunotherapies. Since anti-CD40 treatment impedes BrafV600E/Pten melanoma growth in a T cell-independent manner, it is possible other immunotherapies that enhance TIL recruitment or function may act in synergy with anti-CD40 mAb treatment and provide alternative forms of treatment for BrafV600E inhibitor-resistant melanomas.
Collectively, we found that PLX4720 treatment created a immunostimulatory tumor microenvironment, in part, by augmenting CD40:CD40L and IFNγ signaling. A future goal will be to evaluate the therapeutic efficacy of treatments that enhance CD40L and IFNγ signaling in combination with BrafV600E inhibitors or other immunotherapies. After prolonged treatment, tumor cells acquire resistance to BrafV600E inhibitors with additional mutations in MAPK pathways and other signaling cascades. However, tumor cells may gain resistance to immune attack by these additional mutations. For example, a recent study showed that activation of MAPK pathway can elevate PD-L1 expression in BrafV600E inhibitor-resistant melanoma cells, which could dampen T cell activity and anti-tumor immunity (50). Interestingly, our results showed that BrafV600E inhibition augmented PD-1 expression on infiltrating T cells, and a similar observation has been reported in humans (21). These findings raise the possibility that intratumoral CD4 T cells might become more sensitive to PD-1:PD-L1 signaling pathways after vemurafenib treatment, which would support combining PD-1 blockade with BrafV600E inhibitor treatment to inhibit tumor growth or possibly delay resistance to BrafV600E inhibitors. It would be an important issue for future studies to examine whether CD40:CD40L and IFNγ signaling would be diminished in BrafV600E inhibitor-resistant melanoma and whether immunotherapies that provide these immune stimuli could be utilized to treat BrafV600E inhibitor-resistant melanomas.
Supplementary Material
Acknowledgements
We thank all members of the Kaech and Bosenberg labs, Drs. K. Politi, and P. West for insightful discussions and experimental advice.
Financial Support: This work was supported in part by NIH R01AI074699 (S.M. Kaech), Yale Cancer Center (S.M. Kaech), Yale SPORE in Skin Cancer 5 P50 CA121974 (Halaban R, PI), Melanoma Research Alliance Development Award (S.M. Kaech and M.W. Bosenberg), Howard Hughes Medical Institute (S.M. Kaech), Melanoma Research Foundation (M.W. Bosenberg), and the National Cancer Center (P-C. Ho).
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
REFERENCES
- 1.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. The New England journal of medicine. 2012;367:1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jang S, Atkins MB. Which drug, and when, for patients with BRAF-mutant melanoma? The lancet oncology. 2013;14:e60–9. doi: 10.1016/S1470-2045(12)70539-9. [DOI] [PubMed] [Google Scholar]
- 3.Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, Jr., et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nature genetics. 2009;41:544–52. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, et al. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nature reviews Drug discovery. 2012;11:873–86. doi: 10.1038/nrd3847. [DOI] [PubMed] [Google Scholar]
- 5.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. The New England journal of medicine. 2012;366:707–14. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalos M, June CH. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity. 2013;39:49–60. doi: 10.1016/j.immuni.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braumuller H, Wieder T, Brenner E, Assmann S, Hahn M, Alkhaled M, et al. T-helper-1-cell cytokines drive cancer into senescence. Nature. 2013;494:361–5. doi: 10.1038/nature11824. [DOI] [PubMed] [Google Scholar]
- 8.Shiao SL, Ganesan AP, Rugo HS, Coussens LM. Immune microenvironments in solid tumors: new targets for therapy. Genes & development. 2011;25:2559–72. doi: 10.1101/gad.169029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. The Journal of clinical investigation. 2011;121:2350–60. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–84. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 11.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. The New England journal of medicine. 2013;369:122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Callahan MK, Wolchok JD, Allison JP. Anti-CTLA-4 antibody therapy: immune monitoring during clinical development of a novel immunotherapy. Seminars in oncology. 2010;37:473–84. doi: 10.1053/j.seminoncol.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–6. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bak SP, Barnkob MS, Bai A, Higham EM, Wittrup KD, Chen J. Differential requirement for CD70 and CD80/CD86 in dendritic cell-mediated activation of tumor-tolerized CD8 T cells. Journal of immunology. 2012;189:1708–16. doi: 10.4049/jimmunol.1201271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keller AM, Schildknecht A, Xiao Y, van den Broek M, Borst J. Expression of costimulatory ligand CD70 on steady-state dendritic cells breaks CD8+ T cell tolerance and permits effective immunity. Immunity. 2008;29:934–46. doi: 10.1016/j.immuni.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 16.Roberts DJ, Franklin NA, Kingeter LM, Yagita H, Tutt AL, Glennie MJ, et al. Control of established melanoma by CD27 stimulation is associated with enhanced effector function and persistence, and reduced PD-1 expression of tumor infiltrating CD8(+) T cells. Journal of immunotherapy. 2010;33:769–79. doi: 10.1097/CJI.0b013e3181ee238f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer research. 2010;70:5213–9. doi: 10.1158/0008-5472.CAN-10-0118. [DOI] [PubMed] [Google Scholar]
- 18.Liu C, Peng W, Xu C, Lou Y, Zhang M, Wargo JA, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:393–403. doi: 10.1158/1078-0432.CCR-12-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:1386–94. doi: 10.1158/1078-0432.CCR-11-2479. [DOI] [PubMed] [Google Scholar]
- 20.Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, et al. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer research. 2012;72:3928–37. doi: 10.1158/0008-5472.CAN-11-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:1225–31. doi: 10.1158/1078-0432.CCR-12-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A, et al. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. The Journal of clinical investigation. 2013;123:1371–81. doi: 10.1172/JCI66236. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Hooijkaas A, Gadiot J, Morrow M, Stewart R, Schumacher T, Blank CU. Selective BRAF inhibition decreases tumor-resident lymphocyte frequencies in a mouse model of human melanoma. Oncoimmunology. 2012;1:609–17. doi: 10.4161/onci.20226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. The Journal of clinical investigation. 2011;121:4746–57. doi: 10.1172/JCI58814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Damsky WE, Curley DP, Santhanakrishnan M, Rosenbaum LE, Platt JT, Gould Rothberg BE, et al. beta-catenin signaling controls metastasis in Braf-activated Pten-deficient melanomas. Cancer cell. 2011;20:741–54. doi: 10.1016/j.ccr.2011.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nature reviews Immunology. 2011;11:723–37. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:1035–43. doi: 10.1158/1078-0432.CCR-12-2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swain SL, McKinstry KK, Strutt TM. Expanding roles for CD4(+) T cells in immunity to viruses. Nature reviews Immunology. 2012;12:136–48. doi: 10.1038/nri3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang L, Chen X, Liu X, Kline DE, Teague RM, Gajewski TF, et al. CD40 ligation reverses T cell tolerance in acute myeloid leukemia. The Journal of clinical investigation. 2013;123:1999–2010. doi: 10.1172/JCI63980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kornbluth RS, Kee K, Richman DD. CD40 ligand (CD154) stimulation of macrophages to produce HIV-1-suppressive beta-chemokines. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:5205–10. doi: 10.1073/pnas.95.9.5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nature reviews Immunology. 2008;8:958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kerkar SP, Restifo NP. Cellular constituents of immune escape within the tumor microenvironment. Cancer research. 2012;72:3125–30. doi: 10.1158/0008-5472.CAN-11-4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baitsch L, Fuertes-Marraco SA, Legat A, Meyer C, Speiser DE. The three main stumbling blocks for anticancer T cells. Trends in immunology. 2012;33:364–72. doi: 10.1016/j.it.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 34.Van Deusen KE, Rajapakse R, Bullock TN. CD70 expression by dendritic cells plays a critical role in the immunogenicity of CD40-independent, CD4+ T cell-dependent, licensed CD8+ T cell responses. Journal of leukocyte biology. 2010;87:477–85. doi: 10.1189/jlb.0809535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Higham EM, Wittrup KD, Chen J. Activation of tolerogenic dendritic cells in the tumor draining lymph nodes by CD8+ T cells engineered to express CD40 ligand. Journal of immunology. 2010;184:3394–400. doi: 10.4049/jimmunol.0903111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang X, Sun W, Guo D, Cui Z, Zhu L, Lin L, et al. Cardiac allograft acceptance induced by blockade of CD40-CD40L costimulation is dependent on CD4+CD25+ regulatory T cells. Surgery. 2011;149:336–46. doi: 10.1016/j.surg.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 37.Propper DJ, Chao D, Braybrooke JP, Bahl P, Thavasu P, Balkwill F, et al. Low-dose IFN-gamma induces tumor MHC expression in metastatic malignant melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2003;9:84–92. [PubMed] [Google Scholar]
- 38.Yang WC, Ma G, Chen SH, Pan PY. Polarization and reprogramming of myeloid-derived suppressor cells. Journal of molecular cell biology. 2013;5:207–9. doi: 10.1093/jmcb/mjt009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khalili JS, Liu S, Rodriguez-Cruz TG, Whittington M, Wardell S, Liu C, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:5329–40. doi: 10.1158/1078-0432.CCR-12-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hooijkaas AI, Gadiot J, van der Valk M, Mooi WJ, Blank CU. Targeting BRAFV600E in an inducible murine model of melanoma. The American journal of pathology. 2012;181:785–94. doi: 10.1016/j.ajpath.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 41.Allalunis-Turner MJ, Siemann DW. Recovery of cell subpopulations from human tumour xenografts following dissociation with different enzymes. British journal of cancer. 1986;54:615–22. doi: 10.1038/bjc.1986.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EK, et al. Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. The Journal of experimental medicine. 2010;207:651–67. doi: 10.1084/jem.20091921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. The New England journal of medicine. 2008;358:2698–703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rakhra K, Bachireddy P, Zabuawala T, Zeiser R, Xu L, Kopelman A, et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer cell. 2010;18:485–98. doi: 10.1016/j.ccr.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baudy AR, Dogan T, Flores-Mercado JE, Hoeflich KP, Su F, van Bruggen N, et al. FDG-PET is a good biomarker of both early response and acquired resistance in BRAFV600 mutant melanomas treated with vemurafenib and the MEK inhibitor GDC-0973. EJNMMI research. 2012;2:22. doi: 10.1186/2191-219X-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer cell. 2013;23:302–15. doi: 10.1016/j.ccr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hall A, Meyle KD, Lange MK, Klima M, Sanderhoff M, Dahl C, et al. Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the (V600E)BRAF oncogene. Oncotarget. 2013;4:584–99. doi: 10.18632/oncotarget.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annual review of immunology. 2013;31:259–83. doi: 10.1146/annurev-immunol-032712-095956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Angulo I, Rullas J, Campillo JA, Obregon E, Heath A, Howard M, et al. Early myeloid cells are high producers of nitric oxide upon CD40 plus IFN-gamma stimulation through a mechanism dependent on endogenous TNF-alpha and IL-1alpha. European journal of immunology. 2000;30:1263–71. doi: 10.1002/(SICI)1521-4141(200005)30:5<1263::AID-IMMU1263>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 50.Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:598–609. doi: 10.1158/1078-0432.CCR-12-2731. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.