

Abstract
Extracellular flux (XF) analysis has become a mainstream method to measure bioenergetic function in cells and tissues. While this technique is commonly used to measure energetics in intact cells, we outline here a detailed XF protocol for measuring respiration in permeabilized cells. Cells are permeabilized using saponin, digitonin, or recombinant perfringolysin O (XF PMP reagent) and provided with specific substrates to measure complex I- or II-mediated respiratory activity, Complex III+IV respiratory activity, or Complex IV activity. Medium- and long-chain acylcarnitines or glutamine may also be provided for measuring fatty acid oxidation or glutamine oxidation, respectively. This protocol allows for such measurements using a minimal number of cells compared with other protocols, without the need for mitochondrial isolation. The results are highly reproducible, and mitochondria remain well coupled. Collectively, this protocol provides comprehensive and detailed information regarding mitochondrial activity and efficiency, and, following preparative steps, takes approximately 6 hours to complete.
Keywords: mitochondria, respiration, metabolism, oxidative phosphorylation, bioenergetics
INTRODUCTION
Metabolism comprises the critical, life-sustaining chemical reactions occurring within cells and tissues. Mitochondria, in particular, are critical hubs for such biochemical transformations. Although best known for their capacity to produce energy through oxidative phosphorylation, they are also important for the synthesis of biomolecules and for regulating calcium homeostasis, cell survival decisions, and redox signaling. Not surprisingly, these roles are highly integrated, such that perturbation in the machinery responsible for energy production, for example, could have substantial effects on related processes such as cataplerosis or apoptosis. Therefore, the ability to identify and quantify changes in specific components that constitute mitochondrial function is increasingly important for understanding relationships between bioenergetics and changes in cell phenotype.
Development of the protocol
Measurements of mitochondrial function became common nearly half a century ago through the work of Britton Chance and G.R. (Ron) Williams1,2. Their methods to isolate intact and active mitochondrial preparations (derived from modifications of methods from Schneider3 and Lardy and Wellman4) paved the way for seminal discoveries in mitochondrial biology, including Peter Mitchell’s chemiosmotic theory of oxidative phosphorylation5 and the elucidation of the tricarboxylic acid (TCA) cycle6. As such, the measurement of mitochondrial function continues to be widely used to identify mechanistic bases of disease and for understanding the modes of toxicity of environmental factors and the mechanisms of drug action.
The specific aspects of mitochondrial biology that can be revealed in the purified organelle include assessment of respiratory activity, efficiency and capacity7–9, reactive oxygen species (ROS) generation10,11, mitochondrial permeability transition (mPTP)12–15, protein import into the mitochondrion16,17, and the mitochondrial proteome18–20. While assays using isolated mitochondria have bestowed remarkable knowledge of how disease affects each of these end points, this approach has several limitations. First, these methods require relatively large quantities of cells or tissues21, and standard techniques, e.g., differential centrifugation, commonly retrieve a low fraction of the total mitochondrial content. In tissues such as muscle, this latter shortcoming has been suggested to lead to bias due to selective representation of the mitochondrial pool22–24. Second, the isolation process could decrease mitochondrial integrity25, with such damage possibly more evident in mitochondria isolated from injured tissues22. Third, mitochondria isolated from cells and tissues are essentially stripped from their intracellular locale. This has been shown to result in changes in mitochondrial morphology, sensitization to mPTP, alterations in mitochondrial respiration, and increased ROS production24,25. Hence, methods of measuring mitochondria in their unperturbed environment are likely more useful for identifying veritable changes in mitochondrial function due to disease conditions and interventions.
Extracellular flux (XF) analysis has become a mainstream, high-throughput means to measure bioenergetic function in cells and tissues. This technique has overcome the limitations of classic oxygen electrodes and allows for measurement of mitochondrial energetics in up to 24 or 96 samples (in XF24 or XF96 analyzers, respectively) at one time in intact, adherent cells26,27. Data acquired using this technique can confer knowledge of changes in oxygen consumption due to ATP turnover, proton leak, and maximal respiratory capacity9,28–34; however, in intact cell and tissue35 measurements, the ability to identify site-specific changes such as those due to damage to components of the respiratory chain or anaplerotic enzymes are diminished. Whereas isolated mitochondria allow for such measurements, they have the aforementioned limitations of the need of large amounts of starting material and relatively poor yield, deleterious changes to the mitochondrial architecture and function, and selection bias. To bridge the gap between intact cell bioenergetic measurements and isolated mitochondria experiments, a comprehensive XF assay to measure respiration in permeabilized cells has been developed. This assay, or modifications thereof, has been used to understand the effects of mitochondrial fission on cellular bioenergetics36, identify novel metabolic actions of thiazolidinediones37 and understand the effects of drugs on mitochondrial toxicity in neurons38. The permeabilizers used in this assay have also been shown to work in other platforms, such as in suspended cells using an oxygen electrode39,40.
The fundamental principles underlying the permeabilized cell methodology have been outlined elsewhere40–43, with the major improvements in this protocol being: the application of the permeabilized cell methodology in the high-throughput XF24/96 system; a more thorough interrogation of ETC components, fatty acid oxidation, and anaplerotic metabolism; and the ability to select from a variety of permeabilizing agents, the properties of which may differ slightly. Briefly, treatment of cells with compounds that form complexes with cholesterol, which is abundant in the plasma membrane and much lower in organelles such as mitochondria, results in selective plasmalemma permeabilization. In this protocol, we outline the use of three different permeabilizing agents: saponin (SAP), digitonin (DIG), and recombinant perfringolysin O (rPFO; XF PMP reagent). Proper treatment with these agents causes loss of plasma membrane integrity, resulting in the leakage of cytosolic solutes from the cell, with little to no effect on mitochondrial membranes. Addition of select oxidizable substrates, e.g., TCA cycle intermediates, acylcarnitines, and amino acids, as well as ADP, uncouplers, and inhibitors of respiratory complexes or membrane translocators allows for a comprehensive interrogation of mitochondrial function.
In permeabilized cells, the architecture of organelles such as the endoplasmic reticulum and mitochondria as well as the cytoskeleton remains largely intact44. Whereas isolated mitochondria assume the globular or fragmented morphology, mitochondria in permeabilized cells retain their vermiform appearance and tubular network25. This is especially important because several studies show structure-function relationships in mitochondria that relate largely to fission and fusion activities and mitochondrial architecture45–48. Additionally, measuring respiratory activity in permeabilized cells using XF analysis allows for coordinated experiments to be designed in intact cells and permeabilized cells. This could greatly increase the resolution for pinpointing consequential sites of mitochondrial damage and allows for a more complete understanding of the impact of site-specific changes in the context of intact cell bioenergetics and phenotype. Using the XF24 analyzer, these measurements use as little as 20,000 cells per well, which, compared with isolated mitochondria experiments using Clark-type electrodes, decreases the cell number requirement by up to 10,000-fold (optimal yield and quality of isolated mitochondria requires 2–200 × 106 cells or more21,38). The XF96 platform requires even less cells, as shown previously37.
Applications of the method
This method may be applied to most cells that adhere to polystyrene XF tissue culture microplates and should be applicable to the wide range of cells that have been tested in intact XF assays (see list at: http://www.seahorsebio.com/learning/cell-line.php). To date, it has been shown to work in vascular smooth muscle cells, C2C12 cells, differentiated C2C12 myotubes, human skeletal muscle myotubes, neonatal rat cardiomyocytes, H9C2 cardiomyocytes, differentiated H9C2 cardiomyocytes, HEK 293a cells, HepG2 cells, RAW 264.7 macrophages L6 cells, rat cortical neurons, and brown adipocyte precursors36–38. The method has the potential to be modified for use in measuring activity in tissue slices and muscle fibers41. With regards to application, this method should aid in understanding how conditions related to diabetes, neurodegeneration, and cardiovascular disease affect mitochondrial function and bioenergetics. It is envisaged that this technique may also help describe the unique metabolic phenotypes of cancer cells and stem cells, and could be useful in elucidating how metabolism integrates with and regulates cell differentiation and phenotype. In addition, this methodology may be useful to diagnose systemic mitochondrial disease using cells isolated from patients40,49.
Application of this method allows for interrogation and measurement of the following in permeabilized cells (see Fig. 1):
Figure 1. Key steps in mitochondrial metabolism and oxidative phosphorylation.

In this protocol, specific complexes and respiratory activities can be interrogated using endogenous and synthetic substrates. (a) Oxidation of pyruvate in the tricarboxylic acid (TCA) cycle in the mitochondrial matrix promotes the reduction of NAD+ to NADH. Electrons from NADH enter complex I and are transferred through CoQ to complexes III and IV. Oxidation of succinate to fumarate causes reduction of FAD to FADH2, the electrons of which enter the electron transport chain (ETC) and are transferred to complexes III and IV. Fatty acids delivered to the mitochondrial matrix through the actions of carnitine palmitoyl transferase I (CPT I) and CPT II enter into β-oxidation and also generate NADH and FADH2. Duroquinol, a synthetic substrate, donates electrons to complex III. TMPD, in the presence of ascorbate, can donate electrons to cytochrome c to reduce complex IV, which then reduces oxygen to water. The overall proton gradient across the inner mitochondrial membrane (IMM) created by these redox reactions drives the phosphorylation of ADP to ATP by the ATP synthase. The complex-specific inhibitors shown in red are used to dissect the contribution of individual complexes or to measure mitochondrial coupling; OMM, outer mitochondrial membrane; UCP, uncoupling protein. (b) Plasma membrane permeabilization allows for controlled delivery of endogenous substrates (such as pyruvate, succinate, acylcarnitines and glutamine) into mitochondria. Oxidation of these substrates produces reducing equivalents, NADH and FADH2, which enter the ETC through complexes I and II, respectively. Shown in bold are those substrates and reagents that are used with this permeabilization protocol.
Complex I-mediated respiratory activity
Providing pyruvate or glutamate activates dehydrogenases, which reduce nicotinamide adenine dinucleotide (NAD+), i.e., to NADH. NADH then feeds electrons into Complex I (NADH-ubiquinone oxidoreductase) that transfer through the Q cycle to Complex III, cytochrome c, and finally Complex IV, which reduces O2 to H2O. It should be noted that giving glutamate or pyruvate alone may result in much lower oxygen consumption rates (e.g., see50) than when combined with malate. In the absence of malate, accumulation of acetyl CoA could inhibit pyruvate dehydrogenase (PDH) via feedback inhibition; adding small amounts of malate therefore allows for oxaloacetate production and condensation with acetyl CoA, allowing normal PDH flux. In addition, pyruvate or glutamate given alone may result in loss of some TCA cycle intermediates, e.g., through the dicarboxylate carrier.
In higher organisms, malate, by itself, will not support high rates of respiration in isolated mitochondria or in permeabilized cells and tissues. Oxaloacetate formed from malate cannot be metabolized further without a source of acetyl CoA, i.e., pyruvate, or in the case of added glutamate, it can be metabolized to oxoglutarate, similar to metabolism with acetyl CoA and citrate synthase. Also, TCA cycle intermediates are depleted in the presence of malate alone: mitochondrial citrate and oxoglutarate can be depleted by antiport with malate through tricarboxylate and oxoglutarate carrier exchanges, respectively. Similarly, succinate is lost through the dicarboxylate carrier. It should be noted that Complex II is generally not involved in respiration when Complex I substrates are given. This is primarily due to the fact that high malate concentrations both increase oxaloacetate and equilibrate with fumarate, which inhibits SDH activity, and oxoglutarate and succinate are lost into the medium due to carrier exchange.
Complex II-mediated respiratory activity
Complex II (SDH) is a membrane-bound enzyme that is also part of the TCA cycle. Addition of succinate results in its transport into mitochondria through the dicarboxylate carrier. Succinate is then oxidized to fumarate, with concomitant reduction of FAD to FADH2, and electrons are delivered into the Q cycle; however, rotenone must be present to prevent reverse electron transfer to complex I (which results in ROS production) and to prevent accumulation of oxaloacetate, which is a more potent inhibitor of SDH than malonate. In the presence of rotenone, Complex I is inhibited, and malate is extruded from mitochondria through the dicarboxylate carrier, thereby preventing oxaloacetate accumulation.
Oxygen consumption deriving from Complex III and IV activities
Complex III (Ubiquinol:cytochrome c oxidoreductase) is a central component of the respiratory chain, catalyzing transfer of electrons from ubiquinol to oxidized cytochrome c. Like Complex I, this electron transfer reaction is coupled to proton translocation51. In intact cells and tissues, electrons feeding into Complex III are derived from the oxidation of mitochondrial substrates (e.g., pyruvate and fatty acids leading to formation of NADH and FADH2), with electrons donated initially to Complex I and the Q cycle. Thus, using such substrates makes the identification of specific sites of damage more difficult because of the multiple enzymes and electron transfer reactions involved.
In permeabilized cells, Complex III+IV activity and integrity can be assessed using durohydroquinone (duroquinol)40,52–55. Duroquinol donates electrons directly to Complex III, and the rate of its oxidation requires electron transport through Complexes III and IV; hence, the use of this substrate allows for assessment of differences in respiratory activity derived from these latter electron transfer reactions.
Complex IV activity
The final step in the electron transport chain catalyzes the transfer of four electrons from reduced cytochrome c to O2, forming H2O, with the concomitant pumping of protons into the intermembrane space. Complex IV (cytochrome c oxidase) activity has classically been measured using spectroscopic techniques. However, the activity of Complex IV may also be measured in permeabilized cells (and isolated mitochondria) by recording the oxygen consumption rate (OCR) when non-physiological electron-donating compounds, such as tetramethyl-p-phenylene diamine (TMPD) are used56. Here, ascorbate is added as a reductant to regenerate TMPD from its oxidized form (Wurster’s blue). TMPD donates electrons to cytochrome c, which then reduces Complex IV. Complex IV then reduces O2, forming water.
Medium- and long-chain fatty acid oxidation
Fatty acids (FAs) are an important source of energy, with mitochondrial FA oxidation (β-oxidation) being the principle route by which they are oxidized as fuel. Cells and tissues typically derive oxidizable fat via transport across the plasma membrane. Once inside the cell, FAs are activated by esterification to CoA-SH, which is catalyzed by acyl CoA synthetases. However, for transport into the mitochondrion, the fatty acyl-CoA molecules typically must be esterified to carnitine, which is accomplished through the actions of carnitine palmitoyltransferase-I (CPT-I). A carnitine/acylcarnitine translocase then shuttles the acylcarnitine into the mitochondrion, where CPT-II then regenerates the acyl CoA molecule, which is the substrate for the FA oxidation machinery57. However, short-chain FAs, such as butyrate, do not require the acyl carnitine transferase mechanism58,59. β-oxidation involves the actions of multiple chain length-specific enzymes that catalyze the cyclic release of acetyl CoA. These reactions not only promote ATP production via acetyl CoA production, but also by the direct generation of the reducing equivalents FADH2 and NADH, the electrons of which are then passed to the Q cycle by electron transfer flavoprotein oxidoreductase and to Complex I, respectively60–62. In permeabilized cells, measurements of fat oxidation may be obtained by providing carnitine-conjugated FAs. In this protocol, we outline the use of octanoylcarnitine (medium-chain acylcarnitine; C8) and palmitoylcarnitine (long-chain acylcarnitine; C16). Although unconjugated octanoate has been used to assess respiratory activity, notable uncoupling may occur63. Hence, this protocol only applies to the measurement of respiration in permeabilized cells in the presence of acylcarnitines. In addition to the acylcarnitines, “sparker” malate is added, as this has been shown to increase mitochondrial β-oxidation64,65. This malate-induced increase in β-oxidation activity is likely due to an increased ability to derive NADH from TCA cycle reactions.
Glutamine-supported respiration
Glutamine is the most abundant amino acid in the body and plays an important role in bioenergetics and biosynthesis. “Glutamine anaplerosis,” as it has been recently termed66,67, is initiated by glutaminase, which generates glutamate. Glutamate is then catabolized to 2-oxoglutarate (α-ketoglutarate) by glutamate dehydrogenase or transaminases. Glutamine-derived 2-oxoglutarate helps in maintaining TCA cycle intermediates, is critical for the production of building blocks such as nucleotides, lipids and amino acids68, and is an important source of energy for proliferating cells—especially cancer cells66,69,70. In permeabilized cell assays, the addition of glutamine and potentially other anaplerotic pathway precursors71 (e.g., branched chain amino acids) could be used to identify differences in anaplerosis-derived energy production. As with glutamate-supported respiration and FA oxidation (vide supra), malate is typically added to enhance glutamine-mediated respiration. Changes in glutamine-supported respiration may indicate alterations in the transamination machinery or Complex I-mediated respiration.
Key indices and rate-limiting steps of mitochondrial function
The order of agents used in classical oxygen electrode experiments, initially shown by Chance and Williams, allows for the understanding of several respiratory “states.” These mitochondrial states may also be examined in permeabilized cells using XF technology. Conventionally, oxygen consumption is measured in isolated mitochondria alone in the presence of only Pi (State 1). Substrates such as pyruvate+malate or succinate are then added to measure ADP-independent respiratory activity (State 2), which is generally ascribed to proton leak. ADP, once added in the presence of respiratory substrates, results in rapid oxygen consumption and the formation of ATP (State 3), and once all the ADP is depleted respiration typically returns to rates similar to that of State 2 (State 4). Of these indices, State 3 and State 2 or 4 respiration are most important and allow for the calculation of the respiratory control ratio (RCR) for assessing mitochondrial coupling9. This protocol modifies the order of measurement of the respiratory states, first measuring State 3 respiration, and then State 4° respiration, which is induced by addition of oligomycin, an inhibitor of ATP synthase (Complex V). Importantly, inhibitors may also be used to identify key steps important to the overall rate of respiration. For example, inhibition of the pyruvate or succinate transporter can be used to investigate substrate transport across the membrane; specific dehydrogenases may be examined using enzyme-specific inhibitors (e.g., malonate for SDH); adenine nucleotide transport may be inhibited with atractylate; and estimations of proton leak may be assessed by the oligomycin-independent rate of oxygen consumption7,37,56. Collectively, the integration of each of these factors—Complex I-derived respiration; Complex II-mediated respiration; duroquinol-supported oxygen consumption; Complex IV activity; FA oxidation; glutamine oxidation; and the use of inhibitors—allows for a comprehensive assessment of respiratory function in permeabilized cells, which, by deduction, can be used to identify key and site-specific differences in mitochondrial activity between cell types, phenotypes, or treatments.
Comparison with other methods
This protocol bridges the gap between measurements of respiration in isolated mitochondria and intact cells9. As mentioned above, the XF-permeabilized cell protocol uses far less biological material for examining mitochondrial function than traditional isolated mitochondria assays, prevents damage and alterations in mitochondrial morphology due to isolation, prevents mitochondrial subpopulation selection bias, and is a remarkable increase in throughput, allowing up to 24 or 96 samples to be measured simultaneously (depending on the XF analyzer model). Compared with other permeabilized cell protocols40,41, this method uses 12–100-fold less cells per sample. In addition, experiments may be designed such that intact cell measurements and permeabilized cell measurements are obtained concurrently. This is important because it would allow for a clear assessment of the biological significance of differences in mitochondrial energetic parameters on intact cell bioenergetics. Other high-throughput methods, relying on Pt-porphyrin-based O2 sensing probes in a plate reader format, have been described for the measurement of intact and permeabilized cell mitochondrial function72,73. Such assays have the advantage of flexibility with respect to the use of common microtiter plates, allowing for measurement of up to 384 samples72,74; however, this methodology is limited in that mineral oil must overlay each sample to limit oxygen diffusion, which makes the addition of chemicals or compounds and subsequent repeated measures on the same samples difficult. With XF technology, a semi-sealed chamber that limits oxygen diffusion is made intermittently26,75, which allows for the accurate measurement of oxygen consumption as well as provides the user with the ability to inject compounds of interest during the sample assay. Moreover, this technology is becoming more ubiquitous, with more than 600 papers published using XF methodology to date (based on PubMed database search).
Previous protocols for examining mitochondrial function in permeabilized cells have relied primarily on two permeabilizing agents, SAP and DIG38,40,41,43; however, it is possible that mitochondria in some cells will be damaged by these compounds37. This protocol also includes the use of mutant rPFO (PMP reagent, Seahorse Biosciences)37. PFO is a cholesterol-dependent cytolysin secreted by Clostridium perfringins and it forms pores in the plasma membrane allowing the passage of solutes and proteins up to 200 kDa in size76–79. rPFO appears to have a much broader window for use in permeabilized cell experiments compared with reagents such as SAP and DIG37. Data suggest that PFO facilitates pore formation by a cholesterol-dependent mechanism, in which its binding to membranes occurs when cholesterol concentration exceeds a certain threshold80–82. Intracellular organelles likely do not possess enough cholesterol to facilitate pore formation by rPFO, and, for this reason, rPFO appears to have a much broader window for use in permeabilized cell experiments compared with reagents such as SAP and DIG. Nevertheless, this protocol demonstrates how to define useful ranges for these more cost-effective permeabilizing reagents.
Limitations
While XF permeabilized cell respirometry has several advantages over other methods, limitations remain. Respirometry using an oxygen electrode generally uses relatively small amounts of ADP to promote transient state 3 respiration; hence, once ADP is depleted, State 4 respiration ensues. This allows for the determination of “the number of moles of ADP phosphorylated to ATP per 2e− flowing through a defined segment of an electron transfer to oxygen,” i.e., the P/O ratio56. However, in this XF protocol, high concentrations of ADP (1–4 mM) should be present upon permeabilization, and ADP is not depleted; therefore, the P/O ratio cannot be calculated. However, if need be, one can estimate the P/O ratio by using lower concentrations of ADP (e.g., 0.25 or 0.5 mM, similar to that shown by Rogers et al83). Another rather obvious limitation is accessibility to the XF analyzer. While not as plentiful as microplate readers, there are now numerous laboratories in multiple countries that use this instrument routinely, as evinced by the growing number of XF publications. In addition, there are currently 25 Core Research Facilities with XF capabilities (see: http://www.seahorsebio.com/learning/core-facilities.php), which could be called upon to help perform experiments if the investigator does not have access to the equipment.
Experimental design
Optimal cell density
The workflow in Fig. 2 shows a general time frame for permeabilized cell assays. Prior to examining mitochondrial function in a number of samples by XF analysis or doing comprehensive analyses, it is important to first determine the optimal seeding density for mitochondrial function assays. As shown in Fig. 3a, cells may be seeded at 10,000–100,000 cells per well and oxygen consumption in permeabilized cells can be measured. For the XF 96, the following densities were shown to be optimal: C2C12 myoblasts, 1.5 × 104 cells/well; primary skeletal muscle myotubes, 2.0 × 104 cells/well; L6 myotubes, 1.0 × 104 cells/well; neonatal rat cardiomyocytes myocytes (NRCMs), 5.0 × 104 cells/well; brown adipose tissue precursors, 8.0 × 104 cells/well; and cortical neurons, 4.0 × 104 cells/well. Optimal cell seeding densities for the XF24 are commonly 2.5-fold higher than that for the XF9637.
Figure 2. Experimental procedure flowchart.

General steps in the XF permeabilized assay protocol. Timing of steps may be modified as required.
Figure 3. Cell density optimization.

Optimizing cell seeding density is an important step prior to functional analyses of mitochondria in permeabilized cells. (a) Microplate layout for seeding density optimization. Cells are seeded in 100 μl final volume. Wells marked with an “X” are background (temperature) control wells and should remain unseeded. (b) Oxygen consumption rate (OCR) traces of permeabilized cells seeded at different densities. Here, succinate (Succ; 10 mM), rotenone (Rot; 1 μM), ADP (1 mM), and saponin (Sap; 25 μg/ml) were co-injected in port A. Oligomycin (Oligo; 1 μg/ml) was injected in port B, and antimycin A (10 μM) was injected in port C. Cell seeding densities that provide basal rates of 150–200 pmol O2/min and maximal rates of 200–600 pmol O2/min are often desirable. Rates below 100 pmol O2/min or above 800 pmol O2/min could require modifications in the measure times of the XF analyzer protocol. (c, d) Effects of seeding density on state 3 and 4 respiration and RCR, respectively. In general, seeding densities that provide state 3 and 4 OCRs in the linear range and maximal RCRs should be used. In this example, 40–50K cells per well would be considered optimal.
Choice of permeabilizer
An example of data derived from this initial experiment is shown in Fig. 3b, where rat aortic smooth muscle cells were seeded at various densities and the OCR was measured before permeabilization and after addition of permeabilizing agent and mitochondrial substrate. As shown by the OCRs after antimycin A and rotenone injection (e.g., see Fig. 3b), non-mitochondrial OCR is very low, as cytosolic oxidases are lost (diluted) upon permeabilization. We36 and others38,40 have found that 25 μg/ml SAP or 25 μg/ml DIG are good initial concentrations to use for permeabilized cell experiments. As shown in Fig. 3c,d, the data can be plotted to identify how cell seeding density may impact State 3, State 4°, and RCR. Next, it is essential to ensure that the permeabilizer is optimal. Recent studies have shown that permeabilizers such as DIG have a narrow effective concentration and may cause cytochrome c release from mitochondria, whereas rPFO is not injurious to mitochondria, even at concentrations that are 10× the dose required to permeabilize cells37. However, multiple other studies have effectively used reagents such as SAP and DIG to measure bioenergetics in permeabilized cells38,40,41,43. To determine which permeabilizer may be best for the experimental conditions and cell types, initial dose response studies performed with SAP, DIG, and/or rPFO can be used to identify optimal concentrations and to assess whether the permeabilizers damage mitochondria within the timeframe of a typical XF assay. Concentration-dependent responses of the permeabilizers may be performed following the protocol, in the presence of a substrate such as succinate (+rotenone). An example of this is shown in Fig. 4. Once the optimal concentration of the permeabilizer is determined, assessment of mitochondrial damage, e.g., by measuring cytochrome c release, may also be performed (see Box 1). However, adding back cytochrome c, e.g., by injecting it via one of the ports as shown in ref. 37, can be used to provide a quick answer.
Figure 4. Permeabilization of plasma membrane permits controlled delivery of substrates into mitochondria.

XF traces of OCR in detergent-permeabilized cells: (a, d) Concentration-dependent effects of saponin (SAP) and digitonin (DIG), respectively, co-injected with ADP and succinate+rotenone to stimulate state 3 respiration. (b) Quantification of state 3 and 4° OCRs from (a). (c) RCR values calculated from (a). (e) Quantification of state 3 and 4° OCRs from (d). (f) RCR values calculated from (d). Concentrations of permeabilizer that demonstrate maximal state 3 respiration and the highest RCR values are typically chosen for use. In this example, 25 μg/ml saponin or digitonin would be considered optimal.
Box 1. Assessment of cytochrome c release caused by permeabilizing agents.
TIMING 48 h
To prevent damage of the outer mitochondria membrane (OMM), it is important to optimize permeabilizer concentrations. In addition to functional assays shown in Fig. 4, damage to the OMM may be assessed by measuring the loss of cytochrome c from the mitochondria by Western blotting. For this, cells are exposed to the permeabilizing agents at several concentrations, and cytochrome c, in addition to cytosolic protein markers, are examined (Fig. 10).
The following outline is a step-by-step protocol (modified from37,88) to assess mitochondrial integrity by Western blotting after exposure to permeabilizing agent.
-
1
Seed cells in 6-well plates and culture to confluency. A seeding density of 5 × 105 cells per well of a 6-well plate should provide enough cells needed for Western blotting the following day. Cells could be seeded in 12-well plates or XF24 plates to minimize the amount of cells and reagents needed.
-
2
Warm a solution of MAS-BSA buffer to 37°C in a water bath.
-
3
Make a concentrated stock solution of permeabilizer (typically 25 mg/ml stock for SAP or DIG or stock solution provided as PMP reagent) in pre-warmed MAS-BSA buffer.
-
4
Dilute the stock permeabilizer to the various concentrations to be tested. Concentrations between 0–250 μg/ml for SAP or DIG and 0–10 nM rPFO (XF PMP) are suggested.
-
5
Gently rinse each well of the confluent 6-well plates once with 2 ml warmed MAS-BSA buffer and then aspirate.
-
6
Add the various detergent concentrations made in step 4 above into the designated wells and incubate the plates in the 37°C incubator for 25 min. This duration of incubation should correspond to the length of time the cells will be exposed to permeabilizer during the XF assay.
-
7
Aspirate the MAS-BSA-detergent solution and gently rinse 2× with RT PBS.
-
8
Add 100 μl of protein lysis buffer into each well and proceed to the next step for protein estimation (for XF wells, add 20 μl lysis buffer).
PAUSE POINT Plates can be stored at −80°C for later use.
-
9
Scrape cells from each well and transfer contents into 1.7 ml Eppendorff tubes.
-
10
Centrifuge samples at 4°C for 10 min at 14,000 g using a bench top centrifuge.
-
11
Transfer the supernatants into new, clearly labeled tubes.
-
12
Use an aliquot (usually 5 μl) of each crude cell protein preparation to estimate protein concentration. This can be done using a standard method such as shown in Box 2. Note: if using XF24 plates, use all 20 μl for SDS-PAGE
-
13
Pipette between 5–20 μg of each crude cell protein preparation into appropriately labeled eppendorff tubes.
-
14
Make 2× concentrated gel electrophoresis sample (Laemmli) buffer (see reagent set up) containing ~100 mM DTT.
-
15
Add Laemmli sample buffer into the crude cell protein preparations to yield a final concentration of 1× sample buffer and ~50 mM DTT. The maximum volume of the resulting mixture should not exceed 40 μl.
-
16
Boil mixture on a heating block for 5 min at 100°C.
-
17
After allowing samples to cool, pipette them into each lane of a 10.5–14% polyacrylamide gel and electrophorese.
-
18
Follow standardized Western blotting protocol (e.g., see http://www.cellsignal.com/support/protocols/Western_BSA.html) and then blot for the indicated proteins.
Experimental matrices
After identifying the optimal cell density and choosing a permeabilizer, the cells may then be subjected to comprehensive mitochondrial analysis. It is commonly helpful to design experimental matrices to address the following: (1) respiratory complex activity; (2) glutamine oxidation; and (3) fatty acid oxidation. Of course, the layout of the experiment will depend on the question being asked. For example, to address how complex I- and II-mediated respiratory function are affected between two different cell types or treatments, the XF24 matrix in Fig. 5a would be useful. Other experimental matrices that may be useful for examining mitochondrial respiration and complex activity, fatty acid oxidation, and anaplerotic metabolism are shown in Fig. 5b–d. Inhibitors that are complexed with interrogation of specific components of the respiratory chain are shown in Table 1. Additionally, intact cell respiratory measurements and permeabilized cell measurements can be combined on the same plate. An experimental template that we commonly use to plan and design experiments is available in Suppl. Fig. 1. This experimental matrix could of course be expanded for use with an XF96 system, where multiple complexes and treatment groups could be examined.
Figure 5. XF24 plate design and some experimental layouts for permeabilized cell assays.

Examples of experimental matrices: (a) Experimental matrix for complex I (I; e.g, pyruvate+malate) and complex II (II; succinate)-mediated respiration in cells ± treatment. Different cell (pheno)types may also be examined using a similar matrix. (b) Measurement of respiration in permeabilized cells provided with pyruvate/malate (I), succinate (II), duroquinol (III), or TMPD/ascorbate (IV), which will collectively give a measure of the integrity of Complexes I–IV of the respiratory chain. (c) Matrix for measuring the concentration-dependent effects of a compound or treatment on complex II-mediated respiration. Similarly, this experimental design may be used for measuring the concentration-dependent effects of a compound or treatment on other indices of mitochondrial activity (e.g., Complex I-mediated respiration, Complex IV activity, glutamine oxidation, etc.). (d) Matrix for measuring fatty acid oxidation and glutamine oxidation in a single cell plate.
Table 1.
Substrates and inhibitors for examining mitochondrial respiratory activity
| Substrates | Description | Inhibitors complexed with assay |
|---|---|---|
| Pyruvate (or glutamate) & Malate | Use for measuring Complex I-mediated respiration | Rotenone |
| Succinate | Use for measuring Complex II-mediated respiration | Rotenone and/or malonate or antimycin A |
| Duroquinol | Artificial substrate used for measuring Complex III+IV respiratory activities | Azide |
| Tetamethylphenylenediamine (TMPD) & Ascorbate | Artificial substrate used to measure Complex IV activity. Ascorbate helps maintain TMPD in the reduced form | Azide |
| Palmitoyl & octanoyl-L-carnitine | Acylcarnitines for measuring respiratory activity supported by fatty acid oxidation | Rotenone and/or antimycin A |
| Glutamine & Malate | Use for measuring respiratory activity supported by glutaminolysis | Rotenone and/or antimycin A* |
| ADP | Use for measuring state 3 respiratory activity | Atractyloside# |
|
| ||
| Other important compounds | ||
|
| ||
| Carbonyl cyanide 4-(trifluoromethoxy)phenyl hydrazone (FCCP) | Uncoupler used to measure maximal respiratory activity | |
| Oligomycin | ATP synthase inhibitor; used to assess State 4° | |
Controls and replicates
It is important to note that experimental control groups are helpful for determining whether the experiment “worked” and to assess assay-to-assay variability. For example, when using the XF permeabilized cell assay to measure fatty acid or glutamine oxidation, it is helpful to include substrates known to give a robust response, e.g., succinate or pyruvate and malate, in 2 or 3 wells. This allows the user to determine whether the cells respond to substrates as predicted as well as facilitates comparison between experiments. Additionally, exclusion of substrates in one or a few wells is useful as a “negative” control and helps to establish a baseline or “State 1” rate. Typically 3–5 replicates per group (per individual experiment) are optimal. This can be adjusted depending on the well-to-well intra-assay variability; the % coefficient of variation (CV) should be <20%. It is strongly recommended that each experiment be repeated at least three separate times and that the investigator does not rely on one experiment with technical replicates to draw definitive conclusions. Lastly, it is helpful to make up all storable reagent stock solutions the day before the assay (Table 2).
Table 2.
Stock reagent preparation and storage
| Compound | FW | Stock Solution (stored at − 20°C) | 10× port solution$ | Final conc. after injection |
|---|---|---|---|---|
| Pyruvate/Malate | 88.06/134.09 | 440.3 mg pyruvate + 335.2 mg malate added to 10 ml MAS (500 mM/250 mM)* | 300 μl added to 2.37 ml MAS-BSA$ | 5 mM/2.5 mM |
| Succinate | 118.09 | 590 mg succinic acid added to 10 ml MAS (500 mM)* | 600 μl added to 2.07 ml MAS- BSA$@ | 10 mM |
| Duroquinol | 166.22 | 831 mg duroquinol added to 10 ml DMSO (500 mM) | 30 μl added to 2.64 ml MAS-BSA$ | 0.5 mM |
| TMPD/Ascorbate | 164.25/176.12 | 821.25 mg TMPD added to 10 ml ethanol (500 mM) 352 mg Ascorbate added to 10 ml MAS (200 mM)* | 30 μl TMPD stock + 300 μl Ascorbate stock added to 2.34 ml MAS-BSA$ | 0.5 mM/2 mM |
| Palmitoyl-L-carnitine chloride | 436.07 | 229.3 μl DMSO added to 5 mg palmitoyl-L-carnitine chloride (50 mM) | 30 μl added to 2.64 ml MAS-BSA$ | 50 μM |
| Octanoyl-L-carnitine hydrochloride | 323.85 | 1.544 ml DMSO added to 25 mg octanoyl-L-carnitine hydrochloride (50 mM) | 60 μl added to 2.61 ml MAS-BSA$ | 100 μM |
| L-Glutamine/Malate | 146.15/134.09 | 584 mg glutamine + 67 mg malate added to 10 ml MAS (400 mM/50 mM)* | 300 μl added to 2.37 ml MAS-BSA$ | 4 mM/0.5 mM |
| ADP$ | 501.32 | 501 mg ADP added to 10 ml MAS (100 mM)* | 300 μl added to 10× port A solution | 1 mM |
| Rotenone# | 394.42 | 1.267 ml DMSO added to 5 mg rotenone (10 mM) | 3 μl added to 2.997 ml MAS-BSA | 1 μM |
| Malonate | 104.06 | 41.6 mg malonate added to 10 ml MAS (40 mM)* | 30 μl added to 2.97 ml MAS-BSA | 40 μM |
| Antimycin A (AA)# | 534.60 | 1.27 ml of DMSO added to 25 mg antimycin A (36 mM) | 16.7 μl added to 2.98 ml MAS-BSA | 20 μM |
| Oligomycin# | 791.06 | 500 μl of DMSO added to 5 mg Oligomycin (10 mg/ml) | 3 μl added to 2.99 ml MAS-BSA | 1 μg/ml |
| Potassium azide# | 81.12 | 162 mg potassium azide added to 10 ml MAS (200 mM)* | Pipette 3 ml into new tube | 20 mM |
| FCCP# | 254.17 | 787 μl DMSO added to 10 mg FCCP (50 mM) | 1 μl added to 4.99 ml MAS-BSA | 1 μM |
| Saponin$ | 414.62 | 25 mg added to 1 ml MAS* | Use stock solution | 25 μg/ml |
| Digitonin#$ | 1,229.31 | 25 mg added to 1 ml MAS* | Use stock solution | 25 μg/ml |
| rPFO (PMP)$ | 10 μM (supplied by Seahorse) | Add to MAS-BSA (port concentration = 10–50 nM) | 1–5 nM |
Adjust pH to 7.2 with KOH. Note that a substantial volume of concentrated KOH may need to be used. For example, for making 10 ml of 500 mM succinate solution, add 580 mg of succinate to 5 ml of MAS buffer, adjust pH to 7.2 by adding 5 M KOH (dropwise), and then q.s. to 10 ml with MAS buffer.
CAUTION: acute toxicity hazard. Take proper precautions.
For complete 10× port A solution, add 300 μl ADP stock solution + 30 μl SAP, DIG, or appropriate amount of rPFO (PMP®) stock solution.
For measuring Complex II-mediated respiration, include 3 μl of rotenone stock in 10× port A solution.
MATERIALS
REAGENTS
! CAUTION: The ETC inhibitors, rotenone, antimycin A, oligomycin, potassium azide, and the uncoupler, FCCP, can be acutely toxic. Digitonin, a permeabilizer, can also be acutely toxic. Personal protective equipment should be worn at all times while handling; wear gloves, protective clothing, face and eye shields, and respiratory protection. Thoroughly wash skin and workspace following preparation. Additionally, inhibitors and uncoupler may be light sensitive; stock preparations should be stored in the dark.
Example cell line
-
Rat aortic smooth muscle cells (RASMCs; Lonza, cat. no. R-ASM-580); alternatively, the cells may be isolated as described in Salabei et al.84.
! CAUTION If isolating cells from animals, appropriate institutional regulatory board guidelines must be followed.
Reagent list
Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies–Invitrogen, cat. no. 10567-014)
Fetal bovine serum (FBS; Atlanta Biologicals, cat. no. S11550H)
Penicillin-Streptomycin (5000 U/mL; Life Technologies–Invitrogen, cat. no. 15070-063)
Phosphate-buffered saline (PBS; Invitrogen, cat. no. 10010-049)
Trypsin-EDTA (0.25% w/v; Invitrogen, cat. no. 25200-056)
L-Glutamine powder (Cellgro, cat. no. 61-030-RM)
XF calibrant solution (Seahorse Bioscience, cat. no. 100840-000)
Octanoyl-L-Carnitine hydrochloride (Advent Bio, cat. no. 43360)
Digitonin (Sigma Aldrich, cat. no. D141) ! CAUTION Saponin (Sigma Aldrich, cat. no. 47036)
rPFO (XF PMP reagent; Seahorse Bioscience, cat. no. 102504-100)
Palmitoyl-L-carnitine chloride (Sigma Aldrich, cat. no. P1645)
N,N,N′,N′-Tetramethyl-p-phenylenediamine (TMPD; Sigma Aldrich, cat. no. T7394)
L-Ascorbic acid (Sigma Aldrich, cat. no. A5960)
Malonic acid (Sigma Aldrich, cat. no. 68714)
Oligomycin (Sigma Aldrich, cat. no. O4876) ! CAUTION Antimycin A (Sigma Aldrich, cat. no. A8674) ! CAUTION
Rotenone (Sigma Aldrich, cat. no. R8875) ! CAUTION Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP; Sigma Aldrich, cat. no. C2920) ! CAUTION
Succinic acid (Sigma Aldrich, cat. no. S3674)
Pyruvic acid (Sigma Aldrich, cat. no. 107360)
Potassium azide (Sigma Aldrich, cat. no. 740411) ! CAUTION
Adenosine diphosphate (ADP; Sigma Aldrich, cat. no. A5285)
Malic acid (Sigma Aldrich, cat. no. M0875)
Duroquinol (TCI America, cat. no. T0822)
HEPES (Alfa Aesar, cat. no. A14777)
NP-40 (Sigma Aldrich, cat. no. 74385)
EGTA (Sigma Aldrich, cat. no. E4378)
EDTA (Sigma Aldrich, cat. no. E6758)
SDS (Sigma Aldrich, cat. no. L4579)
D-Mannitol (Alfa Aesar, cat. no. A14030)
Sucrose (Calbiochem, cat. no. 573113)
KH2PO4, (Calbiochem, cat. no. 529568)
MgCl2 (Sigma Aldrich, cat. no. M8266)
Fatty-Acid Free BSA (Roche Diagnostics, cat. no. 03117405001)
Protease inhibitor (Sigma Aldrich, cat. no. P8340)
Phosphatase inhibitor (ThermoScientific, cat. no. 1862495)
Dimethyl Sulfoxide (Sigma Aldrich, cat. no. D2438)
Potassium Hydroxide (Sigma Aldrich, cat. no. P1767)
Glycerol (Calbiochem, cat. no. 56-81-5)
Tris Base, ULTROL grade (Calbiochem, cat. no. 648311)
Bromophenol Blue (Sigma Aldrich, cat. no. B0126)
DC Protein Assay Reagent A (Bio-Rad, cat. no. 500-0113)
DC Protein Assay Reagent S (Bio-Rad, cat. no. 500-0115)
DC Protein Assay Reagent B (Bio-Rad, cat. no. 500-0114)
10.5–14% Bis–Tris gel (Bio-Rad, cat. no. 345-9949)
Disposable culture tubes (Fischer Scientific, cat. no. 14-961-26)
Disposable plastic cuvettes (Fischer Scientific, cat. no. 14-955-127)
PVDF membrane (GE Healthcare, cat. no. RPN303LFP)
ECL® reagents (ThermoScientific, cat. no. 32132)
Anti COX IV antibody (Cell Signaling Technologies, cat. no. 4844).
Anti Cytochrome c antibody (Cell Signaling Technologies, cat. no. 4272).
Anti GAPDH antibody (Cell Signaling Technologies, cat. no. 3683).
Anti Aldehyde dehydrogenase 2 antibody (ALDH2) (LSBio, cat. no. C138016)
Anti α-tubulin antibody (Sigma-Aldrich, cat. no. T6074)
EQUIPMENT
Cell culture plates (10 cm petri dishes) (TPP)
Cell culture flasks (T75 flasks) (TPP)
Centrifuge tubes (VWR)
Conical tubes (TPP)
Disposable pipettes, pipette tips, and filter units (VWR)
XF24 Extracellular Flux Analyzer (Seahorse Bioscience)
XF24 FluxPaks (Seahorse Bioscience, cat. no. 100850-001)
XF24 Cell culture plates (Seahorse Bioscience, cat. no. 100777-004)
Cell culture centrifuge
Cell culture hood
Carbon dioxide incubator (Thermoscientific)
Non-carbon dioxide incubator (Thermoscientific)
Hemocytometer for cell counting (Bright-Line)
pH meter (VWR)
Water bath (VWR)
REAGENT SETUP
Cell culture growth medium
For primary rat aortic smooth muscle cells, DMEM (containing 5 mM glucose), 10% FBS (vol/vol), and 1% penicillin / streptomycin (vol/vol) is used as the growth medium. Follow recommended protocols and guidelines for other primary cells and cell lines. May be stored at 4°C for 1 month.
Mannitol and sucrose (MAS) buffer
Make MAS buffer (in diH2O) containing 70 mM sucrose, 220 mM Mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, and 1 mM EGTA. Adjust pH to 7.2 using 0.1 M KOH and filter sterilize. May be stored at 4°C for 2 months.
Protein lysis buffer
Make protein lysis buffer (in diH2O) containing 25 mM HEPES, 1 mM EDTA, 1 mM EGTA, 0.1% SDS (wt/vol), 1% NP-40 (vol/vol), 1× protease and phosphatase inhibitor cocktails. Adjust pH to 7.0 with NaOH. May be stored at room temperature (RT, 25°C). It is advised to make fresh lysis buffer every 6 months.
Gel electrophoresis sample buffer
Make 5× concentrated Laemmli buffer (in diH2O) containing 400 mM Tris-HCl, 50% glycerol (vol/vol), 0.1% bromophenol blue (wt/vol), and 10% SDS (wt/vol); adjust pH to 6.8 with HCl. May be stored at RT. It is advised to make fresh Laemmli buffer every 6 months.
Respiratory substrates and inhibitors
See Table 2. May be stored as aliquots at −20°C for 2 months. Duroquinol, however, should be made up fresh every 1–2 weeks.
PROCEDURE
Seed cells in the XF cell microplate: TIMING 6–17 h (total time to assay)
! CAUTION: Use sterile technique throughout steps 1–3.
CRITICAL: First, the optimal seeding density must be determined. For this, seed 10–100×103 cells per well, using, for example, the template shown in Fig. 3a. Typically, between 20,000 and 60,000 cells per well is found to be optimal, but this will depend on cell type.
-
1
Grow the cells in a T75 culture dish to achieve desired amount of cells.
-
2
Before trypsinization, rinse cells once with 10 ml sterile PBS (pH 7.4, kept at RT).
-
3
Add 2–5 ml of trypsin-EDTA solution and swirl to make sure cells are covered with trypsin solution. Place flask in the incubator (5% CO2) for 3 min at 37°C.
-
4
Add 8 ml of cell culture growth medium (warmed to 37°C in the water bath).
-
5
Transfer cell suspension to a 15 ml conical tube and centrifuge at 300–500×g for 5 min at RT.
-
6
Aspirate the supernatant and resuspend cells in 1–3 ml of cell culture growth medium.
-
7
Count the cells using a hemocytometer and adjust the concentration of counted cells to 10× the desired cell number to be seeded in each well. For example, if seeding 40,000 cells per well, adjust concentration to 4 × 105 cells per ml culture medium. Prepare enough volume at this cell concentration to seed the entire plate (or desired number of wells).
-
8
Seed cells into the XF24 culture plate by pipetting 100 μl of the cell suspension into each well. Leave unseeded the four wells indicated with an “X” in Fig. 3a. These wells serve as temperature control/background wells. After the cells attach, which generally takes ~4 h, add 250 μl growth medium to each well.
CRITICAL STEP: Seeding density must be accurately determined for each cell type. Generally, the goal is to have a monolayer of cells that achieves 75–95% confluency at the time of assay. Higher density seeding could lead to extremely high oxygen consumption, which may obfuscate rate determinations, or create a semi-hypoxic environment. Also, seeding a homogenous layer of cells is critical for achieving optimal OCRs with low variability. Orient the pipette as in Fig. 6a and disperse cells quickly into the well. Direct application of cells to bottom, e.g., as shown in Fig. 6b, commonly causes them to cluster in the middle or around the sides. After seeding all wells, incubate cells for ~5 min and then examine them using a light microscope (×10 magnification). If cells are not evenly distributed, rock or tap the plate gently to redistribute cells, or seed another plate.
-
9
(Optional depending on the experiment). The following day carefully aspirate each well and replace media with pre-warmed DMEM (containing 0–0.5% FBS and 1% Penicillin-Streptomycin) and incubate for 24 h to synchronize the cell cycle of dividing cells. Concentrations of FBS or other growth factors required to synchronize the cells will depend on the cell type.
Figure 6. Pipette orientation and placement during cell seeding, media addition, and media aspiration.

Cell seeding should be done with pipette tip placed on the corner of the well (a) and not directly in the center of the well (b). For media aspiration (c) or addition (d), pipette tip should be placed as indicated.
Rehydration of an XF24 FluxPak: TIMING ≥ 3 h
-
10
XF FluxPaks comprise a cartridge and a utility plate. At least 3 h prior to assay, hydrate the cartridge by adding ~1 ml of XF calibrant solution into each well of the utility plate. Place the cartridge back onto the utility microplate. Typically, it is useful to hydrate a cartridge the night before an experiment.
-
11
Incubate the plate in a non-CO2 incubator at 37°C.
CRITICAL STEP This hydration step is required for proper functioning of the sensors at time of OCR measurements. Cartridges may be used within 3–4 h of hydration, but may be used up to 72 h after addition of calibrant solution. Note that the incubator must be humidified if hydrating for longer periods of time to prevent evaporation of calibrant solution.
Set up XF24 experiment protocol, make injection solutions, and calibrate cartridge: TIMING 1 h
-
12
Immediately prior to the experiment, weigh 200 mg of fatty acid-free BSA and add it to 50 ml of MAS buffer (yielding a 4 mg/ml MAS-BSA solution).
-
13
Warm the MAS-BSA solution to 37°C in the water bath. Make sure all BSA is dissolved and then, if needed, adjust the pH of the warmed solution to 7.2 with NaOH or HCl.
CRITICAL STEP: Inclusion of BSA in the MAS buffer is essential for maintaining well-coupled mitochondria following permeabilization. See Suppl. Fig. 2 for expected differences in mitochondrial coupling if BSA must be omitted from the protocol.
-
14
Set up an experimental protocol in the XF analyzer following the manufacturer instructions. A typical XF running protocol for the permeabilized cell assay is shown in Fig. 7.
-
15
Pipette ~15 ml of the warmed MAS-BSA solution into a clean conical tube and use it to make 10× stock concentrations of the compounds to be loaded into the ports of the reagent delivery cartridges (see Table 2). The remaining 35 ml of the warmed MAS-BSA buffer must be kept at 37°C until step 18–21.
-
16
Load 10× solutions into their respective ports: 75.0 μl of the substrate/ADP/permeabilizer mix into port A, 83.3 μl of oligomycin into port B, and 92.6 μl of antimycin A/rotenone (or other appropriate inhibitor) into port C. See Table 1 for appropriate substrates and inhibitors for examining mitochondrial activity and specific respiratory complexes.
! CAUTION The respiratory inhibitors and uncouplers are toxic.
CRITICAL STEP: Maximal respiratory capacity may be measured if FCCP is included with substrates in the assay, i.e., in Port A. We and others38 have found that the FCCP response after permeabilization is significantly improved when FCCP is added simultaneously with both permeabilization agent and substrates compared with it being added later in the assay. Using this method, we find that the FCCP-stimulated rate is ~20% higher than State 3 respiration in smooth muscle cells; however, this is likely to differ between cell types.
-
17
Insert the hydrated XF cartridge into the XF analyzer and hit “start” to run the calibration step. This should take between 6–30 min depending on your administration settings.
! CAUTION Remember to take off the clear cartridge lid before inserting the cartridge into the XF24 analyzer. Also, ensure that the orientation of the cartridge is correct.
Figure 7. Example of a permeabilized cell assay protocol using an XF 24 analyzer.

“Mix” and “wait” periods can be changed as desired. However, it is advisable to keep the “measure” periods to 2–3 min. Two min is typically the minimum time required to generate a rate.
Replace media with MAS-BSA buffer: TIMING 5–10 min
-
18
Approximately 5–10 min prior to the end of the calibration protocol, remove the XF culture plate from the incubator and gently aspirate all but 50 μl of DMEM culture medium.
-
19
Gently add 700 μl warmed MAS-BSA buffer to each well.
-
20
Then, aspirate all but 50 μl from each well.
-
21
Add 625 μl of MAS-BSA buffer per well (for a total of ~675 μl per well).
-
22
Place cells in a non-CO2 incubator until the calibration is complete.
CRITICAL STEP All media aspirations and additions should be performed with the pipette tip placed on the side wall of the well (Fig. 6c, d, respectively). This will prevent the cells from dislodging during these steps.
CRITICAL STEP It should be noted that MAS buffer, while balanced for osmolarity for intact cells, is not balanced in this respect for ionic strength. Therefore, time between replacement of the medium by MAS buffer and permeabilization should be minimized as much as possible, or, as was performed in ref. 36, 37 the cells may be permeabilized immediately upon the media change.
Run XF assay: TIMING 30–80 min
-
23
After the cartridge has finished calibrating, hit “ok”, remove the calibrant plate and discard. The upper half of the FluxPak, i.e., the cartridge, is retained in the instrument.
-
24
Place the XF cell culture plate on the platform, and hit “continue”. This command will run the remainder of the experimental protocol that was designed in step 14 above.
! CAUTION Ensure proper orientation of the XF culture plate. Also, be sure to remove the lid from the culture plate before loading into the instrument.
-
25
Immediately after the run has completed, remove the cell culture plate and hit “continue” to end the program.
TROUBLESHOOTING
Protein concentration determination and normalization of data: TIMING 1–2 h
-
26
Quickly but carefully aspirate solution from each well and discard.
-
27
Add 1 ml of PBS (at RT) to each well, and then aspirate and discard.
CRITICAL STEP After addition of PBS, use a light microscope to quickly verify whether the cell “ghosts” remain attached to the bottom of the wells. This may be helpful in explaining any discrepancies in protein estimation.
-
28
Repeat 27 once more.
-
29
Add 20 μl of cell lysis buffer per well.
PAUSE POINT The cell plate may be frozen at −20°C until protein estimation.
-
30
Follow steps in Box 2 for protein concentration estimation and normalization of data.
TROUBLESHOOTING
-
31
After normalization, State 3, State 4° and RCR values may be calculated. As a guide to the reader, we provide a sample data set in Fig. 8. Permeabilized cells not provided with substrate may be used as a control and a measure of State 1 respiration (which should be at or near the non-mitochondrial rate). Calculate these values as follows, using the points indicated on Fig. 8:
State 3 OCR = (point 4) – (point 8)
State 4 OCR = (point 6) – (point 8)
RCR = [(point 4) – (point 8)] ÷ [(point 6) – (point 8)]
Box 2. Normalization of OCR values.
TIMING 1–2 h
Data normalization is commonly critical for comparing OCR values. This is very important in experiments where the cell numbers are expected to change non-uniformly across the plate or when different cell types are used.
OCR values may be reported in the following units:
pmol O2/min
pmol O2/min/μg protein
pmol O2/min/number of cells
pmol O2/min/band density
For experiments in which the number of cells seeded per well is expected not to change or to evenly change across the plate, the resulting OCR values could be reported in pmol O2/min (or in % baseline values, depending on the experimental conditions). Also, for comparing RCRs, the data do not need to be normalized, as the value is a ratio between the State 3 and State 4° rates. In most cases, however, it is appropriate to normalize the OCR values to total protein and report as pmol O2/min/μg protein. For experiments where OCR values will be reported as pmol O2/min/μg protein, we have provided a step-by-step protocol for protein estimation using the Lowry’s method below. Note that the protein in each well could alternatively be used to run Western blots for specific intracellular proteins. For this, a protein assay is not performed and the entire protein content of each well is used for immunoblotting for proteins such as COX IV or a matrix protein such as aldehyde dehydrogenase 2 or citrate synthase. The band intensity could then be used to normalize the OCR rates, and the OCR values would be expressed in pmol O2/min/band intensity of the specific protein. This could be useful to delineate whether changes in OCR might be due to differences in mitochondrial abundance.
Lowry’s assay
Make a solution of 1 mg/ml BSA solution. If stored at –20°C, this solution can be reused multiple times.
Set up BSA solutions for generating a standard curve as follows: In 5 Fisherbrand disposable culture tubes, pipette volumes of 1 mg/ml BSA solution to give final concentrations of 0.2–1 mg/ml BSA (diluted in water or PBS) in a total volume of 25 μl per tube.
In a 15 ml conical tube, prepare Biorad’s solution A+S as follows: 80 μl of solution S into 4 ml solution A, and then vortex.
Pipette 125 μl of A+S solution into each well of the microplate from Step 29, after adding the cell lysis buffer (see Step 29 of the Procedure). Also, add 125 μl of A+S solution into each tube containing the BSA standard.
Add 1 ml of DC Protein Reagent B into each well of the microplate and into each tube containing the BSA standard. After addition of solution B, pipette the contents of each well up and down and vortex the tubes containing the BSA standards.
Keep the plate and tubes at RT for 15 min.
Set up the spectrophotometer; absorbance should be acquired at 750 nm wavelength.
Transfer the content of each well into a Fisherbrand plastic cuvette.
Blank the spectrophotometer with the content from one of the unseeded wells (i.e., a well marked with an X in Fig. 3).
Calculate concentration of protein using standard curve.
To determine the total amount of protein per well, multiply the protein concentration of each well by 20 (since this was the total volume per well after addition of protein lysis buffer). For example, if the protein concentration in a well is 1 μg/μl, the total protein in that well would be 20 μg.
Proceed to normalize data to total protein amounts.
Figure 8. Indices of mitochondrial function.

A typical XF analyzer trace of a permeabilized cell assay. Here Rates 1–9 are shown; these can be used to calculate State 3 and 4 OCR and RCR as described in Step 31 of the Procedure. After baseline measurements are recorded, a SAP/Succinate/Rot/ADP mixture is injected into the wells to induce state 3 respiration. After two state 3 measurement recordings, oligomycin is injected to induce state 4 respiration. Antimycin A, which inhibits mitochondrial respiration, is subsequently injected.
Timing
Steps 1–8, seeding cells; 6–17 h
Step 9 (optional), cell synchronization; 16–24 h
Steps 10–11, rehydration of XF24 FluxPak; ≥ 3 h
Steps 12–17, setting up experimental protocol, making injection solutions and calibrating cartridge; 1 h
Steps 18–22, replacing media; 5–10 min
Steps 23–25, running XF assay; 30–80 min
Steps 26–30, protein concentration determination and normalization of data; 1–2 h
Step 31, calculations; 1–2 h
ANTICIPATED RESULTS
Optimizing the plasma membrane permeabilizing agent
Damage to mitochondria by SAP or DIG, which may be monitored by loss of cytochrome c or diminishment in State 3 respiration, is usually evident only at concentrations of > 25 μg/ml SAP or DIG (e.g., see Box 1). Examples of the effects of SAP and DIG concentration on mitochondrial activity in smooth muscle cells is shown in Fig. 4. Since the cholesterol content of the cell membrane varies with cell types, preliminary experiments, such as that shown in Fig. 4, are advised to identify the appropriate concentration of permeabilizing agent. For rPFO, concentrations from 1–10 nM have been shown to be adequate, with 1–3 nM suitable in most cell types37. In smooth muscle cells, we find similar State 3 and State 4° rates with SAP compared with rPFO (Suppl. Fig. 3).
Interrogation of mitochondrial function
As mentioned in the Introduction, this protocol is useful for evaluating complex I- and II-mediated respiratory activity, as well as Complex III and IV integrity. As shown in Fig. 9, measuring each of these indices of mitochondrial function could be expected to yield important information on the electron transport chain. Oxygen consumption in the presence of substrate and ADP (i.e., State 3 respiration) can be expected to increase progressively as electron donation moves “upstream” in the respiratory chain (Fig. 9a, b). Respiratory activity in the presence of oligomycin (i.e., State 4°) increases more so, such that electron input at Complex I yields the highest RCR and electron input at cytochrome c yields the lowest “RCR” (Fig. 9c). In Fig. 9d–f, we demonstrate typical OCR traces and indices of mitochondrial function when octanoyl-L-carnitine, palmitoyl-L-carnitine, and glutamine are provided as substrates.
Figure 9. Anticipated changes in respiration with provision of diverse respiratory substrates in the XF24 permeabilized cell assay.

OCR traces and indices of mitochondrial respiration: (a) Comprehensive assessment of the respiratory chain: after 3 baseline measurements, saponin (SAP; 25 μg/ml) was co-injected with ADP and the Complex I–IV substrates: pyruvate+malate (pyr/mal); succinate+rotenone (Succ/Rot); duroquinol (Duro); or TMPD+ascorbate (TMPD/Asc). (b) Quantification of state 3 and 4° OCRs from (a). (c) RCR values calculated from (a). (d) Fatty acid and glutamine-supported respiration in permeabilized cells: after 3 baseline measurements, SAP (25 μg/ml) was co-injected with ADP and glutamine, palmitoyl-L-carnitine, or octanoyl-L-carnitine. Note that port A also contains malate (5 mM; final concentration after injection is 0.5 mM). (e) Quantification of state 3 and 4 OCRs from (d). (f) RCR values calculated from (d). Collectively, these measurements provide insights into how mitochondria in permeabilized cells respond to substrates specific for Complex I- and Complex II-mediated respiration as well as Complex III+IV and Complex IV activities (panels a–c). In addition, glutamine and fatty acid oxidation may also be measured using this assay (panels d–f).
Note that the decrease in RCR as electron input moves up the chain can be explained by basic principles. The chemiosmotic theory5 predicts that the rate of flow of electrons is a function of the respiratory intermediates and the proton motive force (Δp) across the inner mitochondrial membrane. The higher OCRs when duroquinol or TMPD/ascorbate are given can thus be explained by the relatively lower Δp, as the NADH/O reaction translocates 10H+/2e− and succinate reactions translocate 6H+/2e−, whereas duroquinol and TMPD reactions have lower H+/2e− stoichiometries56.
Caution should be used concerning the utilization of RCR as a primary metric of mitochondrial coupling in permeabilized cells. For example, as shown in Fig. 4d, the RCR of cells permeabilized with 100 ug/ml DIG is similar to that of 25 ug/ml; however, it is apparent that the state 3 rate is diminished by the higher concentration of DIG, indicating a significant amount of mitochondrial damage. Therefore, states 3 and 4 should be the primary indices for assessing mitochondrial function in permeabilized cells. The RCR may be used as an additional measure of coupling, especially in experiments in which a given cell type is permeabilized in an identical manner and the effect of a treatment or toxin is assessed.
Practically, these assessments can be used to identify critical sites of damage or where differences in electron transport may lie. For example, should a similar % decrease in OCR be identified with each substrate given, i.e., those donating e− to Complexes I (e.g. pyruvate), II (succinate), III (duroquinol), and IV (TMPD), compared with a control group, then it could be surmised that the defect lies in the latter portions of the respiratory chain (cytochrome c/cytochrome oxidase). Changes in Complex II-mediated respiration alone would suggest differences in this input into the Q cycle. Respirasome assemblies are increasingly recognized to be important regulators of mitochondrial energetics, and these measurements, combined with other methods such as Blue-Native PAGE85, may also bequeath important information regarding how respirasome activities may be perturbed86,87. Changes in glutamine oxidation-related enzymes or enzymes involved in β-oxidation may similarly be identified.
Supplementary Material
Figure 10. Assessment of mitochondrial damage by cytochrome c release from mitochondria.
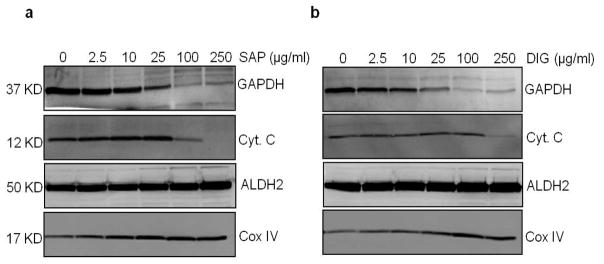
Western blots of glyceraldehyde-3-phosphate dehydrogenase (GAPDH; cytosolic protein), cytochrome c (cyt c; intermembrane space protein involved in electron transfer), aldehyde dehydrogenase 2 (ALDH2; matrix protein), and cytochrome oxidase (COXIV subunit; inner mitochondrial membrane protein) in cells permeabilized with 0–250 μg/ml SAP (a) or DIG (b). Amount of protein loaded for Western blotting are as follows: 5 μg protein for Cox IV; and 20 μg protein for cyt c, GAPDH, and ALDH2. This test may be used to help further determine the optimal concentration of permeabilizing agent or to choose between permeabilizers. A concentration or permeabilizer that results in loss of cytosolic proteins and solutes but does not result in release of cyto c is typically optimal for use in XF24/96 permeabilized cell assays. Here, a 25 μg/ml concentration of saponin or digitonin resulted in release of cytosol-localized GAPDH but did not cause loss of cyto c.
Table 3.
Troubleshooting table
| Step | Problem | Possible cause(s) | Possible solution(s) |
|---|---|---|---|
| 25 | Low OCR readings | Cell density not optimal | Check cell seeding density and uniformity prior to assay by light microscopy. Repeat cell density studies to confirm correct cell density |
| Permeabilizer type or concentration not optimized | Try rPFO (PMP) or perform saponin or digitonin titration studies | ||
| pH of MAS-BSA buffer is not in the physiological range (~pH 7.2) | Examine pH during the run using the “pH” tab in the XF analyzer software. Properly pH all solutions | ||
| Lack of proper cartridge rehydration | Rehydrate properly as outlined in steps 10 and 11 of the procedure | ||
| Low mitochondrial abundance in cell type of interest | Increase seeding density | ||
| 25 | High OCR readings (e.g., above 1000 pmol/min) | Cell density not optimal; O2 depleted during measurement | Use lower density of cells or decrease measure time |
| 25 | High OCR variability between technical replicates (%CV > 20%) | Cell density is not uniform between wells | Examine in microscope to identify seeding homogeneity between wells; if problems are identified, seed a new plate (steps 1–8) |
| Cells in different stages of the cell cycle | Synchronize cells by serum deprivation (step 9) | ||
| Oxygen consumption too high, leading to improper calculation of OCR | Use lower density of cells or decrease measure time | ||
| 25 | Poor coupling | Absence of BSA in MAS buffer | Add BSA to MAS buffer (0.2– 0.4%, wt/vol) |
| 25 | No response to TMPD/ascorbate | Auto-oxidation of TMPD. If TMPD is blue, oxidation is likely | Make TMPD fresh, and always dilute TMPD in MAS buffer containing ascorbate (2 mM final) |
| 25 | No changes in OCR upon injection of duroquinol | Duroquinol is oxidized | Store properly under Argon upon receipt Duroquinol should be made fresh every 1–2 weeks |
| 25 | No increase in OCR after injection of port A | A component of the substrate/ADP/permeabilizer mixture was not added | If permeabilizer is omitted, OCR should remain near baseline values If substrate is omitted, OCR immediately drops to zero If ADP is omitted, the OCR upon oligomycin will likely be identical to the pre-oligomycin OCR |
| Problems with substrate or buffer constituents | Make new stock solutions | ||
| 30 | Inconsistent protein values | Presence of BSA in MAS buffer may skew protein measurements | Gently rinse cells thoroughly (at least 2× with 1 ml RT PBS) before proceeding to addition of protein lysis buffer |
| Detachment of cells from bottom of well in the course of the experiment | Normalize OCR to cell seeding number, provided no significant increases in cell numbers are expected after seeding | ||
| Cell detachment caused by the presence of the permeabilizing agent | Decrease the time after injection of permeabilizing agent, e.g., one measurement after injection of each port Try rPFO (PMP) instead of SAP or DIG |
Acknowledgments
The authors acknowledge support by the National Institutes of Health (grants GM103492 and HL078825).
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare that they have no competing financial interests.
AUTHOR CONTRIBUTIONS
JKS, AAG, and BGH were involved in all parts of manuscript preparation.
References
- 1.Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. II. Difference spectra. J Biol Chem. 1955;217:395–407. [PubMed] [Google Scholar]
- 2.Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. VI. The effects of adenosine diphosphate on azide-treated mitochondria. J Biol Chem. 1956;221:477–489. [PubMed] [Google Scholar]
- 3.Schneider WC. Intracellular distribution of enzymes; the oxidation of octanoic acid by rat liver fractions. J Biol Chem. 1948;176:259–266. [PubMed] [Google Scholar]
- 4.Lardy HA, Wellman H. Oxidative phosphorylations; role of inorganic phosphate and acceptor systems in control of metabolic rates. J Biol Chem. 1952;195:215–224. [PubMed] [Google Scholar]
- 5.Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191:144–148. doi: 10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- 6.Williams GR. Dynamic Aspects of the Tricarboxylic Acid Cycle in Isolated Mitochondria. Can J Biochem. 1965;43:603–615. doi: 10.1139/o65-070. [DOI] [PubMed] [Google Scholar]
- 7.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Divakaruni AS, Brand MD. The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda) 2011;26:192–205. doi: 10.1152/physiol.00046.2010. [DOI] [PubMed] [Google Scholar]
- 9.Hill BG, et al. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol Chem. 2012;393:1485–1512. doi: 10.1515/hsz-2012-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lambert AJ, Brand MD. Reactive oxygen species production by mitochondria. Methods Mol Biol. 2009;554:165–181. doi: 10.1007/978-1-59745-521-3_11. [DOI] [PubMed] [Google Scholar]
- 11.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hunter DR, Haworth RA, Southard JH. Relationship between configuration, function, and permeability in calcium-treated mitochondria. J Biol Chem. 1976;251:5069–5077. [PubMed] [Google Scholar]
- 13.Brookes PS, et al. Concentration-dependent effects of nitric oxide on mitochondrial permeability transition and cytochrome c release. J Biol Chem. 2000;275:20474–20479. doi: 10.1074/jbc.M001077200. [DOI] [PubMed] [Google Scholar]
- 14.West MB, et al. Cardiac myocyte-specific expression of inducible nitric oxide synthase protects against ischemia/reperfusion injury by preventing mitochondrial permeability transition. Circulation. 2008;118:1970–1978. doi: 10.1161/CIRCULATIONAHA.108.791533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brenner C, Moulin M. Physiological roles of the permeability transition pore. Circ Res. 2012;111:1237–1247. doi: 10.1161/CIRCRESAHA.112.265942. [DOI] [PubMed] [Google Scholar]
- 16.Rapaport D, Neupert W, Lill R. Mitochondrial protein import. Tom40 plays a major role in targeting and translocation of preproteins by forming a specific binding site for the presequence. J Biol Chem. 1997;272:18725–18731. doi: 10.1074/jbc.272.30.18725. [DOI] [PubMed] [Google Scholar]
- 17.Freeman KB, Yatscoff RW, Ridley RG. Experimental approaches to the study of the biogenesis of mammalian mitochondrial proteins. Biochem Cell Biol. 1986;64:1108–1114. doi: 10.1139/o86-146. [DOI] [PubMed] [Google Scholar]
- 18.Bailey SM, Landar A, Darley-Usmar V. Mitochondrial proteomics in free radical research. Free Radic Biol Med. 2005;38:175–188. doi: 10.1016/j.freeradbiomed.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 19.Wong R, Aponte AM, Steenbergen C, Murphy E. Cardioprotection leads to novel changes in the mitochondrial proteome. Am J Physiol Heart Circ Physiol. 2010;298:H75–91. doi: 10.1152/ajpheart.00515.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kasumov T, et al. Assessment of cardiac proteome dynamics with heavy water: slower protein synthesis rates in interfibrillar than subsarcolemmal mitochondria. Am J Physiol Heart Circ Physiol. 2013;304:H1201–1214. doi: 10.1152/ajpheart.00933.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- 22.Piper HM, et al. Development of ischemia-induced damage in defined mitochondrial subpopulations. J Mol Cell Cardiol. 1985;17:885–896. doi: 10.1016/s0022-2828(85)80102-4. [DOI] [PubMed] [Google Scholar]
- 23.Picard M, et al. Mitochondrial functional impairment with aging is exaggerated in isolated mitochondria compared to permeabilized myofibers. Aging Cell. 2010;9:1032–1046. doi: 10.1111/j.1474-9726.2010.00628.x. [DOI] [PubMed] [Google Scholar]
- 24.Picard M, Taivassalo T, Gouspillou G, Hepple RT. Mitochondria: isolation, structure and function. J Physiol. 2011;589:4413–4421. doi: 10.1113/jphysiol.2011.212712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Picard M, et al. Mitochondrial structure and function are disrupted by standard isolation methods. PLoS One. 2011;6:e18317. doi: 10.1371/journal.pone.0018317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferrick DA, Neilson A, Beeson C. Advances in measuring cellular bioenergetics using extracellular flux. Drug Discov Today. 2008;13:268–274. doi: 10.1016/j.drudis.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 27.Nadanaciva S, et al. Assessment of drug-induced mitochondrial dysfunction via altered cellular respiration and acidification measured in a 96-well platform. J Bioenerg Biomembr. 2012;44:421–437. doi: 10.1007/s10863-012-9446-z. [DOI] [PubMed] [Google Scholar]
- 28.Hill BG, Dranka BP, Zou L, Chatham JC, Darley-Usmar VM. Importance of the bioenergetic reserve capacity in response to cardiomyocyte stress induced by 4-hydroxynonenal. Biochem J. 2009;424:99–107. doi: 10.1042/BJ20090934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hill BG, Higdon AN, Dranka BP, Darley-Usmar VM. Regulation of vascular smooth muscle cell bioenergetic function by protein glutathiolation. Biochim Biophys Acta. 2010;1797:285–295. doi: 10.1016/j.bbabio.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dranka BP, Hill BG, Darley-Usmar VM. Mitochondrial reserve capacity in endothelial cells: The impact of nitric oxide and reactive oxygen species. Free Radic Biol Med. 2010;48:905–914. doi: 10.1016/j.freeradbiomed.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dranka BP, et al. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic Biol Med. 2011;51:1621–1635. doi: 10.1016/j.freeradbiomed.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sansbury BE, Jones SP, Riggs DW, Darley-Usmar VM, Hill BG. Bioenergetic function in cardiovascular cells: the importance of the reserve capacity and its biological regulation. Chem Biol Interact. 2011;191:288–295. doi: 10.1016/j.cbi.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sansbury BE, et al. Responses of hypertrophied myocytes to reactive species: implications for glycolysis and electrophile metabolism. Biochem J. 2011;435:519–528. doi: 10.1042/BJ20101390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Readnower RD, Brainard RE, Hill BG, Jones SP. Standardized bioenergetic profiling of adult mouse cardiomyocytes. Physiol Genomics. 2012;44:1208–1213. doi: 10.1152/physiolgenomics.00129.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sansbury BE, et al. Overexpression of endothelial nitric oxide synthase prevents diet-induced obesity and regulates adipocyte phenotype. Circ Res. 2012;111:1176–1189. doi: 10.1161/CIRCRESAHA.112.266395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salabei JK, Hill BG. Mitochondrial fission induced by platelet-derived growth factor regulates vascular smooth muscle cell bioenergetics and cell proliferation. Redox Biol. 2013;1:542–55. doi: 10.1016/j.redox.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Divakaruni AS, et al. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc Natl Acad Sci U S A. 2013;110:5422–5427. doi: 10.1073/pnas.1303360110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clerc P, Polster BM. Investigation of mitochondrial dysfunction by sequential microplate-based respiration measurements from intact and permeabilized neurons. PLoS One. 2012;7:e34465. doi: 10.1371/journal.pone.0034465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krippner A, Matsuno-Yagi A, Gottlieb RA, Babior BM. Loss of function of cytochrome c in Jurkat cells undergoing fas-mediated apoptosis. J Biol Chem. 1996;271:21629–21636. doi: 10.1074/jbc.271.35.21629. [DOI] [PubMed] [Google Scholar]
- 40.Ye F, Hoppel CL. Measuring oxidative phosphorylation in human skin fibroblasts. Anal Biochem. 2013;437:52–58. doi: 10.1016/j.ab.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 41.Kuznetsov AV, et al. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat Protoc. 2008;3:965–976. doi: 10.1038/nprot.2008.61. [DOI] [PubMed] [Google Scholar]
- 42.Fiskum G, Craig SW, Decker GL, Lehninger AL. The cytoskeleton of digitonin-treated rat hepatocytes. Proc Natl Acad Sci U S A. 1980;77:3430–3434. doi: 10.1073/pnas.77.6.3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Safiulina D, Kaasik A, Seppet E, Peet N, Zharkovsky A. Method for in situ detection of the mitochondrial function in neurons. J Neurosci Methods. 2004;137:87–95. doi: 10.1016/j.jneumeth.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 44.Saks VA, et al. Permeabilized cell and skinned fiber techniques in studies of mitochondrial function in vivo. Mol Cell Biochem. 1998;184:81–100. [PubMed] [Google Scholar]
- 45.Bach D, et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem. 2003;278:17190–17197. doi: 10.1074/jbc.M212754200. [DOI] [PubMed] [Google Scholar]
- 46.Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci U S A. 2009;106:11960–11965. doi: 10.1073/pnas.0904875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sarin M, et al. Alterations in c-Myc phenotypes resulting from dynamin-related protein 1 (Drp1)-mediated mitochondrial fission. Cell Death Dis. 2013;4:e670. doi: 10.1038/cddis.2013.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hagenbuchner J, Kuznetsov AV, Obexer P, Ausserlechner MJ. BIRC5/Survivin enhances aerobic glycolysis and drug resistance by altered regulation of the mitochondrial fusion/fission machinery. Oncogene. 2013;32:4748–57. doi: 10.1038/onc.2012.500. [DOI] [PubMed] [Google Scholar]
- 49.Chacko BK, et al. Methods for defining distinct bioenergetic profiles in platelets, lymphocytes, monocytes, and neutrophils, and the oxidative burst from human blood. Lab Invest. 2013;93:690–700. doi: 10.1038/labinvest.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Puchowicz MA, et al. Oxidative phosphorylation analysis: assessing the integrated functional activity of human skeletal muscle mitochondria--case studies. Mitochondrion. 2004;4:377–385. doi: 10.1016/j.mito.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 51.Hatefi Y. The mitochondrial electron transport and oxidative phosphorylation system. Annu Rev Biochem. 1985;54:1015–1069. doi: 10.1146/annurev.bi.54.070185.005055. [DOI] [PubMed] [Google Scholar]
- 52.Lesnefsky EJ, et al. Aging decreases electron transport complex III activity in heart interfibrillar mitochondria by alteration of the cytochrome c binding site. J Mol Cell Cardiol. 2001;33:37–47. doi: 10.1006/jmcc.2000.1273. [DOI] [PubMed] [Google Scholar]
- 53.Marres CA, de Vries S. Reduction of the Q-pool by duroquinol via the two quinone-binding sites of the QH2: cytochrome c oxidoreductase. A model for the equilibrium between cytochrome b-562 and the Q-pool. Biochim Biophys Acta. 1991;1057:51–63. doi: 10.1016/s0005-2728(05)80083-0. [DOI] [PubMed] [Google Scholar]
- 54.Ray S, Dutta S, Halder J, Ray M. Inhibition of electron flow through complex I of the mitochondrial respiratory chain of Ehrlich ascites carcinoma cells by methylglyoxal. Biochem J. 1994;303 (Pt 1):69–72. doi: 10.1042/bj3030069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gudz TI, Tserng KY, Hoppel CL. Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J Biol Chem. 1997;272:24154–24158. doi: 10.1074/jbc.272.39.24154. [DOI] [PubMed] [Google Scholar]
- 56.Nicholls DG, Ferguson SJ. Bioenergetics 3. 3. Academic Press; 2002. [Google Scholar]
- 57.Kerner J, Hoppel C. Fatty acid import into mitochondria. Biochim Biophys Acta. 2000;1486:1–17. doi: 10.1016/s1388-1981(00)00044-5. [DOI] [PubMed] [Google Scholar]
- 58.Morrow RJ, Neely ML, Paradise RR. Functional utilization of palmitate, octanoate, and glucose by the isolated rat heart. Proc Soc Exp Biol Med. 1973;142:223–229. doi: 10.3181/00379727-142-36993. [DOI] [PubMed] [Google Scholar]
- 59.Lewandowski ED, Chari MV, Roberts R, Johnston DL. NMR studies of beta-oxidation and short-chain fatty acid metabolism during recovery of reperfused hearts. Am J Physiol. 1991;261:H354–363. doi: 10.1152/ajpheart.1991.261.2.H354. [DOI] [PubMed] [Google Scholar]
- 60.Eaton S. Control of mitochondrial beta-oxidation flux. Prog Lipid Res. 2002;41:197–239. doi: 10.1016/s0163-7827(01)00024-8. [DOI] [PubMed] [Google Scholar]
- 61.Marin-Garcia J, Goldenthal MJ. Fatty acid metabolism in cardiac failure: biochemical, genetic and cellular analysis. Cardiovasc Res. 2002;54:516–527. doi: 10.1016/s0008-6363(01)00552-1. [DOI] [PubMed] [Google Scholar]
- 62.Houten SM, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis. 2010;33:469–477. doi: 10.1007/s10545-010-9061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kingsley-Hickman PB, Sako EY, Ugurbil K, From AH, Foker JE. 31P NMR measurement of mitochondrial uncoupling in isolated rat hearts. J Biol Chem. 1990;265:1545–1550. [PubMed] [Google Scholar]
- 64.Bjorntorp P. The oxidation of fatty acids combined with albumin by isolated rat liver mitochondria. J Biol Chem. 1966;241:1537–1543. [PubMed] [Google Scholar]
- 65.Harper RD, Saggerson ED. Some aspects of fatty acid oxidation in isolated fat-cell mitochondria from rat. Biochem J. 1975;152:485–494. doi: 10.1042/bj1520485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 67.Csibi A, et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell. 2013;153:840–854. doi: 10.1016/j.cell.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kovacevic Z, McGivan JD. Mitochondrial metabolism of glutamine and glutamate and its physiological significance. Physiol Rev. 1983;63:547–605. doi: 10.1152/physrev.1983.63.2.547. [DOI] [PubMed] [Google Scholar]
- 69.Reynolds MR, et al. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene. 2013 doi: 10.1038/onc.2012.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weinberg F, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem. 2002;277:30409–30412. doi: 10.1074/jbc.R200006200. [DOI] [PubMed] [Google Scholar]
- 72.Papkovsky DB. Phosphorescent oxygen-sensitive probes. Springer; 2012. [Google Scholar]
- 73.Jonckheere AI, et al. High-throughput assay to measure oxygen consumption in digitonin-permeabilized cells of patients with mitochondrial disorders. Clin Chem. 2010;56:424–431. doi: 10.1373/clinchem.2009.131441. [DOI] [PubMed] [Google Scholar]
- 74.Will Y, Hynes J, Ogurtsov VI, Papkovsky DB. Analysis of mitochondrial function using phosphorescent oxygen-sensitive probes. Nat Protoc. 2006;1:2563–2572. doi: 10.1038/nprot.2006.351. [DOI] [PubMed] [Google Scholar]
- 75.Gerencser AA, et al. Quantitative microplate-based respirometry with correction for oxygen diffusion. Anal Chem. 2009;81:6868–6878. doi: 10.1021/ac900881z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rossjohn J, et al. Structures of perfringolysin O suggest a pathway for activation of cholesterol-dependent cytolysins. J Mol Biol. 2007;367:1227–1236. doi: 10.1016/j.jmb.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ramachandran R, Heuck AP, Tweten RK, Johnson AE. Structural insights into the membrane-anchoring mechanism of a cholesterol-dependent cytolysin. Nat Struct Biol. 2002;9:823–827. doi: 10.1038/nsb855. [DOI] [PubMed] [Google Scholar]
- 78.Heuck AP, Moe PC, Johnson BB. The cholesterol-dependent cytolysin family of gram-positive bacterial toxins. Subcell Biochem. 2010;51:551–577. doi: 10.1007/978-90-481-8622-8_20. [DOI] [PubMed] [Google Scholar]
- 79.Sanyal S, Claessen JH, Ploegh HL. A viral deubiquitylating enzyme restores dislocation of substrates from the endoplasmic reticulum (ER) in semi-intact cells. J Biol Chem. 2012;287:23594–23603. doi: 10.1074/jbc.M112.365312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ohno-Iwashita Y, Iwamoto M, Ando S, Iwashita S. Effect of lipidic factors on membrane cholesterol topology--mode of binding of theta-toxin to cholesterol in liposomes. Biochim Biophys Acta. 1992;1109:81–90. doi: 10.1016/0005-2736(92)90190-w. [DOI] [PubMed] [Google Scholar]
- 81.Heuck AP, Hotze EM, Tweten RK, Johnson AE. Mechanism of membrane insertion of a multimeric beta-barrel protein: perfringolysin O creates a pore using ordered and coupled conformational changes. Mol Cell. 2000;6:1233–1242. doi: 10.1016/s1097-2765(00)00119-2. [DOI] [PubMed] [Google Scholar]
- 82.Johnson BB, et al. Modifications in perfringolysin O domain 4 alter the cholesterol concentration threshold required for binding. Biochemistry. 2012;51:3373–3382. doi: 10.1021/bi3003132. [DOI] [PubMed] [Google Scholar]
- 83.Rogers GW, et al. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One. 2011;6:e21746. doi: 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Salabei JK, et al. PDGF-mediated autophagy regulates vascular smooth muscle cell phenotype and resistance to oxidative stress. Biochem J. 2013;451:375–388. doi: 10.1042/BJ20121344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wittig I, Braun HP, Schagger H. Blue native PAGE. Nat Protoc. 2006;1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- 86.Rosca MG, et al. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res. 2008;80:30–39. doi: 10.1093/cvr/cvn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lapuente-Brun E, et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science. 2013;340:1567–1570. doi: 10.1126/science.1230381. [DOI] [PubMed] [Google Scholar]
- 88.Alfonso-Pecchio A, Garcia M, Leonardi R, Jackowski S. Compartmentalization of mammalian pantothenate kinases. PLoS One. 2012;7:e49509. doi: 10.1371/journal.pone.0049509. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.