

Abstract
Aims
Although mitochondrial abnormalities have been reported within paraspinal muscles in patients with axial weakness and neuromuscular disease as well as with ageing, the basis of respiratory deficiency in paraspinal muscles is not known. This study aimed to determine the extent and basis of respiratory deficiency in paraspinal muscles from cases undergoing surgery for degenerative spinal disease and post mortem cases without a history of spinal disease, where age-related histopathological changes were previously reported.
Methods
Cervical and lumbar paraspinal muscles were obtained peri-operatively from 13 patients and from six post mortem control cases (age range 18–82 years) without a neurological disease. Sequential COX/SDH (mitochondrial respiratory chain complex IV/complex II) histochemistry was performed to identify respiratory-deficient muscle fibres (lacking complex IV with intact complex II activity). Real-time polymerase chain reaction, long-range polymerase chain reaction and sequencing were used to identify and characterize mitochondrial DNA (mtDNA) deletions and determine mtDNA copy number status. Mitochondrial respiratory chain complex subunits were detected by immunohistochemistry.
Results
The density of respiratory-deficient fibres increased with age. On average, 3.96% of fibres in paraspinal muscles were respiratory-deficient (range 0–10.26). Respiratory deficiency in 36.8% of paraspinal muscle fibres was due to clonally expanded mtDNA deletions. MtDNA depletion accounted for further 13.5% of respiratory deficiency. The profile of immunohistochemically detected subunits of complexes was similar in respiratory-deficient fibres with and without mtDNA deletions or mtDNA depletion.
Conclusions
Paraspinal muscles appeared to be particularly susceptible to age-related mitochondrial respiratory chain defects. Clonally expanded mtDNA deletions and focal mtDNA depletion may contribute towards the development of age-related postural abnormalities.
Keywords: ageing, mitochondrial DNA deletion, paraspinal muscle
Introduction
The paraspinal muscles are essential for truncal movement and maintaining the erect posture. Clinical presentation of isolated paraspinal muscle weakness (axial myopathy) includes dropped head and bent spine syndromes as well as camptocormia [1–5]. Paraspinal muscle weakness may also be part of a diffuse process as described in polymyositis, inclusion body myositis, myasthenia gravis, amyotrophic lateral sclerosis and primary mitochondrial disorders due to mutations in mitochondrial DNA (mtDNA) [6–8]. Histopathological studies of paraspinal muscles from axial myopathies and a number of diffuse muscle disorders identified mitochondrial abnormalities in a proportion of patients [1,2,4,5,7]. Respiratory-deficient fibres [lacking mitochondrial respiratory chain complex IV or cytochrome c oxidase (COX) and with intact complex II or succinate dehydrogenase (SDH) activity, also termed COX-deficient] and mitochondrial abnormalities at an ultrastructural level were reported within paraspinal muscles in patients with degenerative cervical and lumbar spine and post mortem controls without a history of degenerative spine disease [1,9,10]. Wharton et al. reported the histopathological changes within cervical paraspinal muscles in the surgical and post mortem cases to be comparable and age-related [10]. However, the extent and basis of respiratory deficiency within paraspinal muscles in such cases is not known.
The mitochondrial respiratory chain complexes are made up of multiple subunits encoded by nuclear DNA and mtDNA [11]. Given that there are multiple copies of mtDNA in single cells, the proportion of any mtDNA mutation – as a percentage of total mtDNA copies (heteroplasmy) – needs to exceed a threshold level to cause respiratory deficiency [11]. High heteroplasmy levels of inherited and sporadic mtDNA deletions are well recognized within respiratory-deficient limb skeletal muscle fibres in certain primary mitochondrial disorders [11,12]. Somatic mtDNA deletions are also recognized with ageing in limb skeletal muscles [13–15]. Irrespective of the mode of mtDNA deletion generation, high levels of mtDNA deletion and mtDNA depletion frequently affect the activity as well as subunits of mtDNA encoded complexes (complex I, complex III, complex IV and complex V) [11]. Complex II, which is encoded entirely by nuclear DNA, is spared in conditions due to or associated with mtDNA mutations (deletions or point mutations) and mtDNA depletion. MtDNA mutations and copy number abnormalities may be investigated at a single cell level using several well-established molecular biology techniques. A proportion of cases with histochemical evidence of mitochondrial dysfunction in limb skeletal muscle and supposed primary mitochondrial disease, however, remained unexplained [16]. In cases where a mitochondrial genetic abnormality of the respiratory chain is not apparent, the profile of mitochondrial respiratory chain subunits detected by immunohistochemistry may guide towards a possible nuclear genetic defect [16,17].
The aim of this study was to determine the density and the basis of respiratory-deficient fibres in paraspinal muscles from patients undergoing spinal surgery as well as autopsy cases without a history of degenerative spine or neuromuscular disease. We used histochemical, immunohistochemical and molecular biology techniques to study paraspinal muscles from 19 cases (age range 18–82 years). The density of respiratory-deficient fibres in paraspinal muscles increased with age and was greater than previously reported within limb skeletal muscles in controls [13–15]. Approximately a third of respiratory-deficient fibres harboured clonally expanded mtDNA deletions while total mtDNA copy number was depleted in approximately a tenth of all respiratory-deficient fibres. Immunohistochemical profile of respiratory chain subunits in respiratory-deficient fibres with and without mtDNA deletions or mtDNA depletion was similar. Paraspinal muscles appear to be particularly susceptible to mitochondrial respiratory chain defects and somatic mtDNA deletions, which may contribute towards the age-related postural abnormalities.
Materials and methods
Patients and tissue processing
Nineteen patients were used in this study (Table 1) with a median age of 62 years (range 18–82 years). Either deep cervical (semispinalis cervicis) or lumbar paraspinal muscles (multifidus) were sampled peri-operatively and deep lumbar deep paraspinal muscles were obtained from the post mortem cases. Post mortem paraspinal muscle samples from cases without degenerative spine disease or neuromuscular disorders were obtained from UK Multiple Sclerosis and Parkinson's Tissue Bank. None of the patients had a neurodegenerative disorder including Parkinson's disease or multiple sclerosis or a family history indicative of a neuromuscular, primary mitochondrial disorder. Physical signs suggestive of a primary mitochondrial disorder were absent in all cases. All patients were mobile with muscle strength of at least grade 4 (Medical Research Council Scale) and none had spine immobilized prior to surgery. Newcastle and North Tyneside 1 Research Ethics Committee (09/H/0906/675) approved this study. Samples were snap frozen in liquid isopentane at −150°C and stored at −80°C. Tissue blocks were orientated transversely and serially sectioned on a cryostat onto both glass and PEN membrane slides (Leica, Bannockburn, IL, USA) at a thickness of 15 µm.
Table 1.
Clinical details
| Patient | Age (gender) | Surgical procedure | Muscle sampled | Duration of symptoms |
|---|---|---|---|---|
| 1 | 18 (F) | Scoliosis surgery | Lumbar | NA |
| 2 | 18 (F) | Scoliosis surgery | Lumbar | NA |
| 3 | 42 (F) | L5/S1 decompression | Lumbar | 1.5 years |
| 4 | 42 (M) | C6/C7 decompression | Cervical | 9 months |
| 5 | 51 (F) | C4/C5 decompression | Cervical | 4 years |
| 6 | 51 (M) | C5/C7 fusion | Cervical | 9/12 |
| 7 | 52 (F) | C1 decompression | Cervical | NA |
| 8 | 53 (M) | L3/L4 decompression | Lumbar | NA |
| 9* | 57 (F) | – | Lumbar | – |
| 10* | 62 (F) | – | Lumbar | – |
| 11* | 64 (M) | – | Lumbar | – |
| 12 | 64 (M) | L3/L4 decompression | Lumbar | 4.5 years |
| 13 | 65 (M) | L4/L5 decompression | Lumbar | 1.2 years |
| 14* | 71 (M) | – | Lumbar | – |
| 15 | 72 (M) | C3/C4 foraminectomy | Cervical | 6 months |
| 16 | 74 (M) | L4/L5 decompression | Lumbar | 3 years |
| 17* | 77 (M) | – | Lumbar | – |
| 18 | 80 (M) | Occipitocervical fusion | Cervical | 2 months |
| 19* | 82 (M) | – | Lumbar | – |
The asterisk (*) denotes post mortem cases. The mean age of surgical cases was not statistically difference compared with the mean age of post mortem cases (P = 0.069). The control cases (post mortem and surgical) did not have clinical or pathological evidence of a central nervous system disorder. NA, not available.
Histochemistry
Cryosections were subjected to haematoxylin and eosin (H&E) staining, ATPase (pH 4.3) staining and sequential COX/SDH histochemistry. For ATPase (pH 4.3) histochemistry, sections were incubated in a 0.2 M acetate solution (pH 4.3) for 30 min and washed in water followed by incubation in an ATPase medium (pH 9.5) for 45 min at 37°C. Sections were washed in solutions of 1% calcium chloride, 2% cobalt nitrate and incubated in a 1% solution of ammonium sulphide for 2 min. Sections were then mounted in Canada Balsam. COX/SDH was performed as previously described [18]. For the purposes of mtDNA copy number analysis, membrane slide sections were incubated in SDH media and serial sections were used to identify respiratory-deficient muscle fibres [19].
Immunohistochemistry
Immunohistochemistry was performed on serial glass sections to ascertain status of mitochondrial respiratory chain complex subunits and histological characterization. Cryosections were air dried for 1 h before being fixed in a cold solution of 4% paraformaldehyde for 10 min and subjected to immunohistochemistry as previously described [20]. The primary antibodies were applied overnight at 4°C (Table S1). Sections were washed in Tris-buffered saline and developed using the Menapath X-Cell Plus HRP Polymer Detection System (A. Menarini Diagnostics Ltd, Wokingham, UK) as per manufacturer's guidelines.
Laser microdissection and long-range polymerase chain reaction
Individual COX-deficient muscle fibres and groups of COX-positive muscle fibres were laser microdissected (Leica) for long-range polymerase chain reaction (PCR) analysis. Fibres were lysed and long-range PCR was used to identify mtDNA deletions within the major arc of the mitochondrial genome using the Expand Long Template PCR System (Roche, Indianapolis, IN, USA), as previously described [20]. DNA products were separated using a 0.7% agarose gel containing ethidium bromide and viewed under UV light. Blood DNA from an age-matched individual without any medical diagnoses was used as wild-type control.
Real-time PCR
Membrane slides with SDH histochemistry alone were used to laser microdissect COX-positive and COX-deficient fibres for real-time PCR analysis. Images of COX/SDH staining were taken from serial sections and used as a reference to identify COX-positive and COX-deficient fibres. The area of the dissected fibre (µm2) was noted for the purposes of copy number analysis using the laser microdissection software (Leica). Real-time PCR was used to determine both levels of mtDNA deletions and mtDNA copy number (of total and wild type). A real-time PCR assay, which determines the simultaneous and equally efficient amplification of the MTND1 and MTND4 regions, was used to investigate levels of mtDNA deletions, as previously described [20,21]. Briefly, 2 µl of DNA from a single muscle fibre was added to a mastermix of Taqman® universal PCR mastermix (Applied Biosystems, Life Technologies, Carlsbad, CA, USA), 100 nM probes for a region in MTND1 (5′-CCATCACCCTCTACATCACCGCCC-3′) and MTND4 (5′-CCGACATCATTACCGGGTTTTCCTCTTG-3′), 300 nM forward and reverse primers to amplify a region in both MTND1 (forward: 5′-CCCTAAAACCCGCCACATCT-3′; reverse: 5′-GAGCGATGGTGAGAGCTAAGGT-3′) and MTND4 (forward: 5′-CCATTCTCCTCCTATCCCTCAAC-3′; reverse: 5′-CACAATCTGATGTTTTGGTTAAACTATATTT-3′) and dH2O. Cycle conditions consisted of a cycle of 2 min at 50°C and 10 min at 95°C followed by 40 cycles of 15 s at 95°C and 1 min at 60°C.
Determining mtDNA heteroplasmy level and mtDNA copy number
MtDNA deletion levels were calculated using a multiplex real-time PCR assay and the ΔCt method, based on the observation that a region in MTND4, located in the major arc, is deleted in 96% of cases with multiple mtDNA deletions, while a region in MTND1, in the minor arc of the mitochondrial genome, is rarely deleted [21]. The change in cycle threshold or ΔCt (CtND1–ND4) and the calculated relative copy number (R = 2−ΔCt), taking background fluorescence into account, were used to determine the percentage level of deleted mtDNA [(1 −R) × 100][22]. For all samples, ΔCt values were obtained in triplicate and were used to calculate mtDNA deletion levels. Within each experiment, control samples of known mtDNA deletion heteroplasmy levels as well as a wild type blood control were run to quality control the assay.
For determining mtDNA copy number, a standard curve was generated for both MTND1 and MTND4 using samples with known copy number values. A standard curve was used with all samples from lysed muscle fibres tested. Total mtDNA copy number was determined from the MTND1 Ct values, using the standard curve, while the wild-type mtDNA copy number was determined from the MTND4 Ct values. MtDNA copy number values were expressed as per µm2 prior to determining the ratio of mtDNA copies between COX-deficient and COX-positive fibres for each individual case (mean copy number of 10 random COX-positive fibres) to exclude interindividual variability [19]. MtDNA depletion in respiratory-deficient fibres was defined as ratio of mtDNA copies below the lower limit of 95% confidence interval of mtDNA copy ratios in COX-positive fibres.
MtDNA sequencing
To confirm the presence of clonally expanded mtDNA deletions and characterize mtDNA deletion breakpoints, PCR products were extracted using a Qiagen (Valencia, CA, USA) gel extraction kit as per manufacturer's guidelines. Cycle sequencing was carried out as previously described using an ABI 3130xl Genetic Analyser (Applied Biosystems) [20]. Sequences were subsequently compared with the revised Cambridge Reference Sequence (rCRS) with SeqScape software (Applied Biosystems).
Microscopy and quantification of histochemical and immunohistochemical findings
To characterize respiratory-deficient muscle fibres, respiratory-deficient, COX-positive and intermediate fibres were captured using Zeiss Axioimager 2 with Axiovision 4.8 software (Zeiss, Cambridge, UK) at a magnification of ×20. The density of respiratory-deficient fibres as a percentage of total muscle fibres was determined per case by I.Z., who was blinded to case details and mutational analysis. In order to determine the profile of mitochondrial respiratory chain complex subunits in paraspinal muscle fibres, fibres with intact COX activity and respiratory-deficient fibres were located using serial sections subjected to COX/SDH histochemistry. Furthermore, respiratory-deficient fibres were located according to heteroplasmy level of mtDNA deletions (<60% or >60%) and mtDNA depletion using serial sections; we were able to determine mitochondrial respiratory chain subunit profiles, heteroplasmy levels of mtDNA deletions and mtDNA copy number within the same muscle fibres as serial cryostat sections were cut onto both glass and membrane slides. Fibres without either high heteroplasmy level of mtDNA deletions or mtDNA depletion were identified as unexplained fibres in images of serial sections. Immunoreactivity in individual fibres was determined using densitometry on greyscale images captured at identical settings. To negate any interindividual variability, the mean densitometry of 10 random COX-positive muscle fibres was determined in each case to use as a reference to establish a ratio for each serial COX-deficient fibre.
Statistical analysis
All graphs and statistical comparisons were performed using Graph Pad Prism 4 (GraphPad, San Diego, CA, USA). Parametric test (t-test) was used to compare two groups. When more than two groups were compared anova was used. P-values of <0.05 were considered statistically significant.
Results
The density of respiratory-deficient fibres in paraspinal muscle and correlation with age
Respiratory-deficient muscle fibres were detected in all but the two youngest cases (Figure 1). In the remaining 17 cases, the density of respiratory-deficient fibres varied from 0.71% to 10.28% with a mean of 3.96% (with the total number of fibres analysed per case ranging from 635 to 1123) (Figure 1). Cases over 60 years of age harboured respiratory-deficient fibres at a mean density of 5.22% compared with 2.56% in <60-year-olds. Furthermore, there was a significant correlation between the density of respiratory-deficient fibres in paraspinal muscles and age. We did not observe a significant difference in the density of respiratory-deficient fibres between peri-operative and autopsy samples nor between muscle samples from cervical and lumbar regions (Figure S1). Histopathological findings in paraspinal muscles included predominance of type I fibres, atrophy of type II fibres and central cores, as previously reported [10], as well as type IIc fibres (Figure S2).
Figure 1.
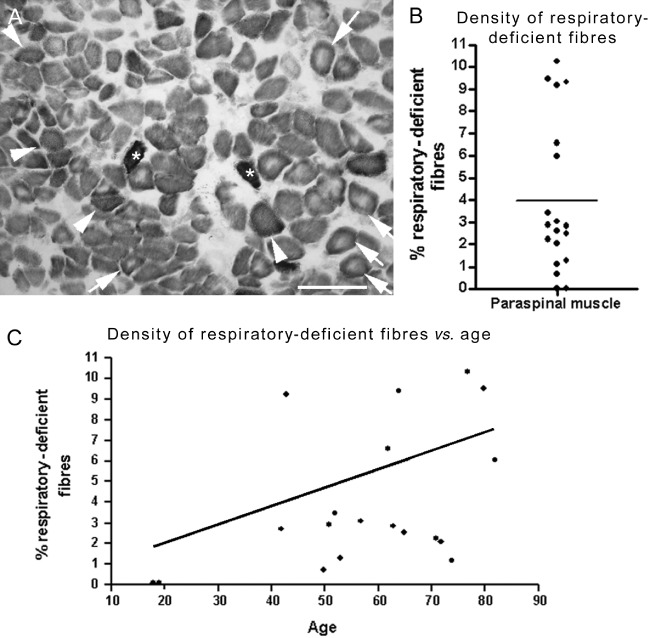
The density of respiratory-deficient fibres in paraspinal muscle. (A) There were numerous respiratory-deficient fibres (lacking complex IV activity and with intact complex II activity stained blue, arrowheads) apparent in paraspinal muscles following sequential COX/SDH (mitochondrial respiratory chain complex IV/complex II) histochemistry. In addition, we observed respiratory-deficient and complex II (SDH) hyper-reactive fibres indicative of subsarcolemmal mitochondrial accumulation (asterisks) and paraspinal muscle fibres contained central cores, as previously noted (arrows and see Figure S1) [10]. Scale bar: 200 µm. (B–C) On average, 3.96% of muscle fibres in paraspinals were respiratory-deficient (B). The mean density of respiratory-deficient fibres in cases over 60 years of age was 5.22% compared with 2.56% in less than 60 years of age. There was a statistically significant (P = 0.04, r2 = 0.23) positive correlation between the density of respiratory-deficient fibres and age (C).
High heteroplasmy level of clonally expanded mtDNA deletions within respiratory-deficient paraspinal muscle fibres
Two populations of respiratory-deficient fibres were apparent when levels of somatic mtDNA deletions were determined in individual laser captured muscle fibres by real-time PCR (Figure 2). Approximately 37% of all respiratory-deficient fibres in paraspinal muscles contained high levels (>60% mutation load) of mtDNA deletion. In contrast, none of the muscle fibres with intact COX activity contained high level of mtDNA deletion. The percentage of respiratory-deficient fibres harbouring high heteroplasmy levels of mtDNA deletions (>60%) varied between cases, ranging from zero to 81.25% when respiratory-deficient fibres were analysed per case (Figure 2C). Histopathological features did not distinguish cases where mtDNA deletion led respiratory deficiency was predominant from cases where high heteroplasmy levels of mtDNA deletions were sparse.
Figure 2.
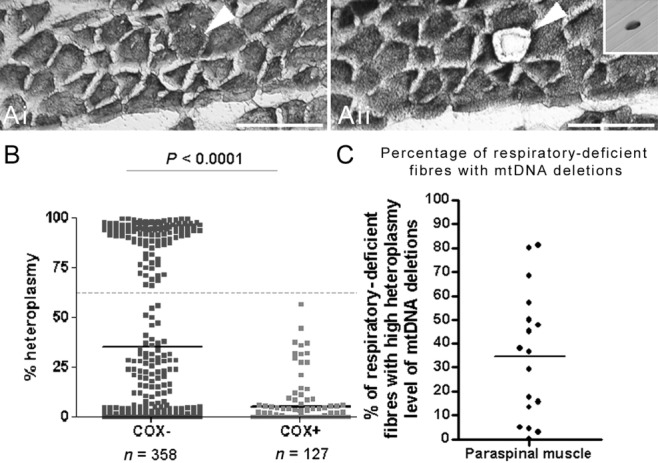
Level of mitochondrial DNA (mtDNA) deletion in individual paraspinal muscle fibres. (A) Respiratory-deficient fibres (arrowhead) were detected using sequential COX/SDH (mitochondrial respiratory chain complex IV/complex II) histochemistry on cryosectioned muscles on membrane slides (Ai–Aii). Individual muscle fibres were laser captured into Eppendorf tubes (Aii insert) from serial sections subjected to SDH histochemistry for molecular characterization. Scale bar: 200 µm. (B–C) Real-time polymerase chain reaction on DNA extracted from individual respiratory-deficient (blue, COX-deficient or COX−) fibres and fibres with intact COX activity (brown, COX-positive) showed significantly (P < 0.0001) higher heteroplasmy level of mtDNA deletions in COX− compared with COX+ fibres (B). Interestingly, ascertainment of mtDNA heteroplasmy levels appeared to identify two populations of COX-deficient fibres. Approximately 37% of COX-deficient fibres harboured >60% heteroplasmy levels of mtDNA deletions. When respiratory deficiency was analysed per case, there were cases where approximately 80% of all respiratory-deficient fibres harboured high heteroplasmy levels (>60%) as well as cases where mtDNA deletions did not explain the biochemical defect (C). COX−: respiratory-deficient or COX-negative; COX+: with intact complex IV activity or COX-positive.
As next step, we used long-range PCR and breakpoint sequencing to characterize mtDNA deletions observed within paraspinal muscle fibres (Figure 3). The high heteroplasmy level of mtDNA deletion in individual respiratory-deficient fibres appeared to be due to expansion of one clone of an mtDNA deletion or clonal expansion, as single mtDNA deletions were detected in several respiratory-deficient fibres. MtDNA deletions were also detected when fibres with intact COX activity, where the level of mtDNA deletion was lower than the threshold (<60%), were pooled. Sequencing of gel-extracted DNA following long-range PCR identified and characterized the breakpoints of mtDNA deletions within respiratory-deficient fibres (Figure 3C and Table S2).
Figure 3.
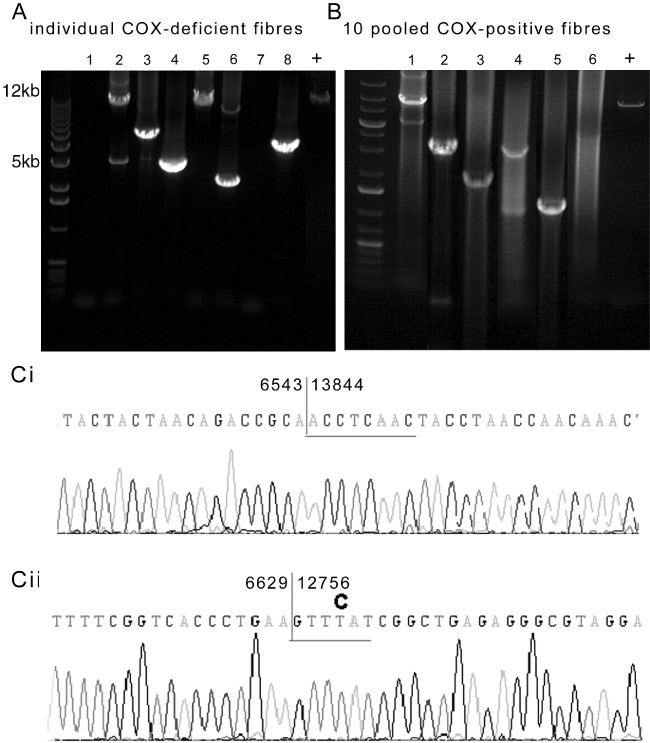
Confirmation of mitochondrial DNA (mtDNA) deletions and identification of breakpoints. (A–B) When DNA extracted from individual respiratory-deficient paraspinal muscle fibres was subjected to long-range polymerase chain reaction (PCR), multiple mtDNA deletions were detected. However, more than one or two mtDNA deletions within an individual fibre was rarely identified, suggestive of clonally expanded mtDNA deletions (A). When DNA was extracted from 10 pooled fibres with intact mitochondrial respiratory chain complex IV or cytochrome c oxidase (COX) activity, multiple mtDNA deletions were also detected (B); however, real-time data shown in Figure 2 did not indicate expansion of mtDNA deletions to high heteroplasmy levels in such fibres. COX-deficient: fibres lacking complex IV and with intact complex II activity. COX-positive: fibres with complex IV activity. + indicates full-length amplified product from wild-type DNA. Lanes 1–8 (A) show amplified products of DNA extracted from individual fibres from different cases and lanes 1–6 (B) show 10 pooled fibres, from different cases. (C) Sequencing of individual mtDNA deletions extracted following long-range PCR confirmed the mtDNA deletions and identified breakpoints. MtDNA deletions were flanked by either a perfect (Ci) or an imperfect (Cii) repeat sequence as previously reported in ageing [23].
Depletion of mtDNA in a proportion of respiratory-deficient paraspinal muscle fibres
Besides clonally expanded mtDNA deletions, mtDNA depletion is a well-recognized cause of respiratory deficiency in skeletal muscle. Overall, respiratory-deficient fibres contained significantly more mtDNA copies (wild-type and deleted molecules) than fibres with intact COX activity (Figure 4), as previously reported in limb skeletal muscle and extraocular muscles (EOM) [19]. The wild-type copy number, however, was significantly lower in respiratory-deficient fibres with high heteroplasmy levels compared with fibres with intact COX activity. Based on the lower limit of 95% confidence interval of the total copy number ratio in fibres with intact COX activity as the threshold, 13.5% of all respiratory-deficient fibres demonstrated mtDNA depletion in the absence of high levels of mtDNA deletion.
Figure 4.
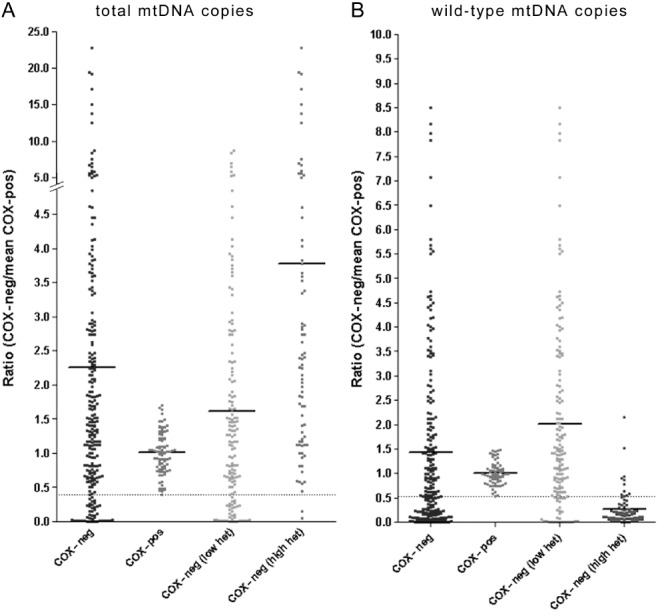
Total and wild-type mtDNA copies in individual paraspinal muscle fibres. (A) Ratio of total mitochondrial DNA (mtDNA) copies in respiratory-deficient paraspinal muscle fibres [wild-type and deleted based on MTND1 values in real-time polymerase chain reaction (PCR)], corrected for case to case variability by using the mean copy number of 10 fibres with intact mitochondrial respiratory chain complex IV or cytochrome c oxidase (COX) activity (COX-positive) in each case, indicated a significantly greater total mtDNA copy number compared with COX-positive fibres (A). When the lower limit of 95% confidence interval of total copy number ratios in COX-positive fibres were used to identify fibres with mtDNA depletion (A, below the dotted line), we detected a number of respiratory-deficient fibres (13.5% of all respiratory-deficient fibres) where total mtDNA copies were depleted in the absence of high level of mtDNA deletion. (B) Ratio of wild-type mtDNA copies in respiratory-deficient paraspinal muscle fibres (based on MTND4 values in real-time PCR), corrected for variability in wild-type mtDNA copies within fibres with intact COX activity (COX-positive), indicated a significantly decrease in wild-type mtDNA copies in respiratory-deficient fibres with high heteroplasmy levels compared with COX-positive fibres (B). The increase in total mtDNA copies and decrease in wild-type mtDNA copies in respiratory-deficient fibres were consistent with pathogenecity of high levels of mtDNA deletion, previously reported [12,19]. When the lower limit of 95% confidence interval of wild type copy number ratios in COX-positive fibres were used to identify fibres with mtDNA depletion (B, below the dotted line), a number of respiratory-deficient fibres (11.4% of all respiratory-deficient fibres) with depletion of wild type mtDNA copies and without high level of mtDNA deletion was detected.
Profile of immunohistochemically detected respiratory chain complex subunits within respiratory-deficient fibres in paraspinal muscle
Next we investigated whether the respiratory-deficient fibres without high levels of mtDNA deletion or mtDNA depletion (unexplained) differed at the level of mitochondrial respiratory chain complex subunits from fibres where the respiratory deficiency was explained above. Following identification of fibres harbouring high heteroplasmy level of mtDNA deletions or mtDNA depletion, immunoreactivity of complex I-20kDa, complex II-70kDa and subunit-I of complex IV (COX-I) within individual fibres in serial sections was assessed by densitometry (Figure 5). Overall, complex I-20kDa and COX-I subunits were significantly decreased within respiratory-deficient fibres compared with fibres with intact COX activity. There were respiratory-deficient fibres intensely stained with complex II-70kDa consistent with COX-deficient fibres that were hyper-reactive for SDH. The patterns of complex I and complex IV subunits in the unexplained population of respiratory-deficient fibres were similar to the population explained by mtDNA deletions or mtDNA depletion.
Figure 5.
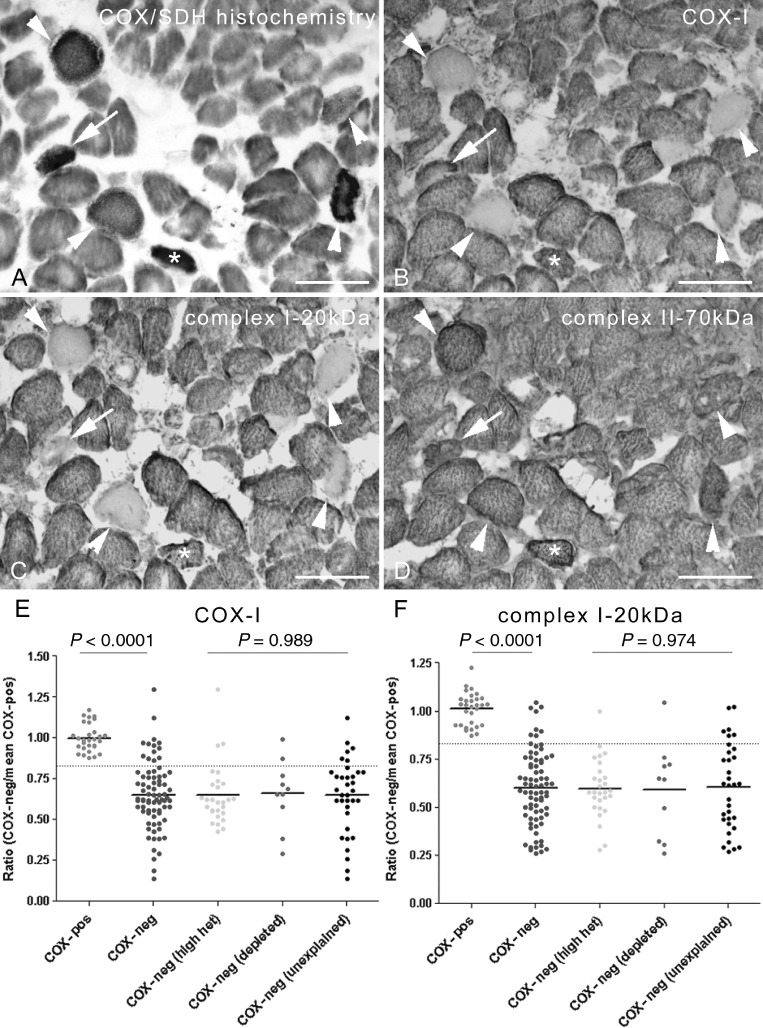
The profile of immunohistochemically detected mitochondrial respiratory chain complex subunits in paraspinal muscle. (A–D) The status of mitochondrial respiratory chain complex subunits was determined in relation to biochemical activity of paraspinal muscle fibres by subjecting serial cryostat sections to histochemistry (A) and immunohistochemistry (B–D). A proportion of respiratory-deficient fibres lacked complex I-20kDa (B, arrowheads) and subunit-I of complex IV or COX-I (c, arrowheads). In contrast, there were respiratory-deficient fibres where COX-I and complex I-20kDa were detectable by immunohistochemistry (B–C, asterisk). Some fibres lacked complex I-20kDa while retaining COX-I (B–C, arrows). In keeping with intact complex II activity, complex II-70kDa was detected within respiratory-deficient fibres (D). Intense staining of some of the respiratory-deficient fibres within complex II-70kDa indicates mitochondrial proliferation (D). Scale bar: 125 µm. (E) When immunoreactivity was analysed by densitometry, respiratory-deficient fibres demonstrated a significant decrease in complex I-20kDa and COX-I compared with fibres with intact complex IV activity (brown). Interestingly, the status of subunits within respiratory-deficient fibres where clonally expanded mitochondrial DNA (mtDNA) deletions at >60% heteroplasmy level or mtDNA depletion did not explain the biochemical detect (unexplained) was similar to fibres where the biochemical detect was due to mtDNA deletions or mtDNA depletion (E–F). COX/SDH, mitochondrial respiratory chain complex IV/complex II.
Discussion
In this study we have established clonally expanded mtDNA deletions and mtDNA depletion as causes of respiratory deficiency in paraspinal muscles from patients undergoing surgery for degenerative spinal disease and post mortem cases without a history of spinal disease. In addition, we identified the density of respiratory-deficient fibres in paraspinal muscles to be greater than previously reported with ageing in limb skeletal muscle (<1.7%) [13–15]. The significant correlation between respiratory deficiency and age as well as clonally expanded mtDNA deletions, which are well recognized in ageing limb skeletal muscle [11,23], indicated that the respiratory deficiency within paraspinal muscles is an age-related phenomenon.
In contrast to limb skeletal muscles, the basis of respiratory-deficient fibres within paraspinal muscles in elderly subjects and cases with neuromuscular disorders was not investigated [1,9,10,13–15,24]. There are fundamental differences between limb skeletal and paraspinal muscles. With respect to embryonic origin and development, the myogenic precursor cells that give rise to paraspinal muscles are derived from the dorsal-medial portion of dermomyotome whereas cells in ventral-lateral portion migrate and form the limb muscles [25]. Paraspinal muscle differentiation during initial stages of development gives rise to mononucleated cells whereas multinucleated cells are the product of differentiation in limb skeletal muscles [26]. There is a predominance of type I fibres in paraspinal muscles in contrast to limb skeletal muscles [10,27]. From a functional perspective, paraspinal muscles are isometric compared with phasic or dynamic movements of limb skeletal muscle [27]. The above features may make paraspinal muscles more susceptible to age-related mtDNA deletions than limb skeletal muscles.
The mtDNA deletions found in the present study are no doubt occurring as somatic mutation as the cases did not show a family history or clinical phenotype suggestive of primary mitochondrial disease. Clonally expanded mtDNA deletions, which may occur through random genetic drift, are recognized within limb skeletal muscle and the central nervous system in elderly subjects [14]. Although the mechanism of mtDNA deletion formation is not fully elucidated, they are thought to form following double-strand breaks mediated by DNA repair processes [28]. Once mtDNA deletions are formed, the level of mtDNA deletion needs to exceed a critical mutation threshold before a biochemical defect results. The process by which a single species of mtDNA deletion expands to high heteroplasmy level is termed clonal expansion [11,29,30]. Evidence from in vivo models of primary mitochondrial disorders suggests that factors such as replicative advantage of deleted mtDNA molecules and metabolic rate may influence the rate of clonal expansion in single cells [30]. The fact that long-range PCR showed mtDNA deletions within fibres with intact complex IV activity, although not expanded to high levels, highlights the potential of the paraspinal muscle to harbour mtDNA deletions. The cases with clonally expanded mtDNA deletions in their fourth or fifth decade of life at biopsy indicated an early mutational event in paraspinal muscles before clonal expansion occurred, as proposed by Elson et al. [29]. The intrinsic properties of and functional demand on paraspinal muscles may then drive clonal expansion of mtDNA deletions and cause respiratory deficiency; mtDNA deletions were abundant within respiratory-deficient fibres in EOM, which share some of the intrinsic properties of paraspinals [12,19]. The increase in total copy number and a decrease in wild-type copies of mtDNA in respiratory-deficient fibres with greater than 60% heteroplasmy level are consistent with the pathogenicity of clonally expanded mtDNA deletions in paraspinal muscles. Interestingly, quantitative loss of mtDNA copy number only accounted for a small proportion of respiratory chain deficiency. Depletion of mtDNA and impaired transcription of mitochondrial genes are well recognized in denervated and disused or immobilized muscles [31,32]. The small proportion of respiratory-deficient fibres with mtDNA depletion is consistent with ongoing use of paraspinal muscles in subjects that underwent spinal surgery.
Clonally expanded mtDNA deletions and mtDNA depletion explained the observed biochemical defect in approximately half of respiratory-deficient fibres within paraspinal muscles. Previous reports that investigated age-related mitochondrial defects in limb skeletal muscle did not attempt to explain the respiratory failure comprehensively; studies either explained up to 10% of respiratory-deficient fibres using techniques that were not sensitive enough and only screened for specific mtDNA mutations (for example, the 4977-bp ‘common deletion’) or did not characterize a large number of respiratory-deficient fibres [13–15]. As a result, the extent to which mtDNA deletions and depletion explain the age-related respiratory deficiency in limb skeletal muscles is unclear. Based on the level of mtDNA deletion in fibres with intact COX activity, the threshold necessary for a biochemical defect in paraspinal muscle fibres appeared to be less than reported in limb skeletal muscle and EOM [12]. Future studies of paraspinal muscles from primary mitochondrial cases may shed light on the heteroplasmy threshold necessary for a biochemical defect in paraspinal muscles [7]. The studies that included immunohistochemical profiling of skeletal muscles, used immunohistochemistry to support a pathogenic role for mtDNA mutations rather than as a guide to the underlying molecular defect where mtDNA mutations were not apparent. A study of cases with suspected primary mitochondrial disorders involving COX-deficiency by Rahman et al. identified intact subunits in respiratory-deficient fibres as a potential indicator of nuclear DNA defects whereas loss of subunits was consistent with mtDNA mutations [16,17]. MtDNA point mutations may also form the basis of the biochemical defect in a subset of unexplained respiratory-deficient fibres, given the fact the majority of these unexplained respiratory-deficient fibres lacked both COX-I and complex I-20kDa. Future studies exploring mtDNA for point mutations may explain the biochemical defect at least in a small subset of respiratory-deficient fibres, similar to the findings in EOM [19]. Post-transcriptional modifications to mitochondrial subunits may also exist in paraspinal muscles. With the greater understanding of the nuclear DNA and mitochondrial cross-talk the opportunities to completely explain the basis of respiration deficiency in paraspinal muscles is likely to increase [33].
In summary, mitochondrial defects including respiratory-deficient fibres harbouring high levels of mtDNA deletion were more frequent in paraspinal muscles than previously recognized in ageing limb skeletal muscles. Clonally expanded mtDNA deletions and mtDNA depletion may contribute towards the age-related postural abnormalities and certain axial myopathies in which mitochondrial respiratory chain abnormalities are reported.
Acknowledgments
This study was supported by the Wellcome Trust (Intermediate Clinical Fellowship to D.M.), Newcastle Healthcare Charity (to D.M.), The Wellcome Trust Centre for Mitochondrial Research and the Newcastle University Centre for Brain Ageing and Vitality funded by Biotechnology and Biological Sciences Research Council, Engineering and Physical Sciences Research Council, Economic and Social Research Council and Medical Research Council as part of the cross-council Lifelong Health and Wellbeing Initiative (G0700718). We are grateful to Dr Laurie Bindoff for helpful discussion on mitochondrial abnormalities in paraspinal muscles. We thank Dr Djordje Gveric from UK Multiple Sclerosis and Parkinson's Tissue Bank for providing post mortem tissue and Mr Christopher Gerber from Department of Neurosurgery, Newcastle General Hospital for providing the peri-operative samples.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Histopathological features ofparaspinal muscle. (A–F)H&E stain (A) showed variable fibre diameterand spaces presumably represent replacement adipose tissuefollowing fibre loss. COX/SDH histochemistry identifiedrespiratory-deficient fibres (B, arrowheads) aswell as fibres with intact COX activity (B,containing brown DAB product). Central cores were apparent in manyfibres, as previously reported(B–C)[10]. A serial section (C),which reacted for SDH histochemistry, showed intact complex IIactivity (arrowheads) in respiratory-deficient fibres (comparedwith the same fibre in B) as well as fibres whereCOX/SDH histochemistry identified complex IV activity (stainedbrown in B). Immunohistochemistry for slow(D) and fast (E) myosin heavychain identified a predominance of type I muscle fibres and alsoatrophy of type II muscle fibres (E, arrowheads).ATPase pH 4.3 reactivity confirmed the predominance of type Ifibres and identified type IIc fibres in paraspinal muscles(F, arrowheads). DAB: 3,3′-Diaminobenzidine. Scale bar: 200 μm.
Figure S2. Comparison of respiratory deficiencyand high levels of mtDNA deletion in individual fibres betweensurgical and post mortem cases and cervical and lumbarparaspinal muscles. (A–B)When the density of respiratory-deficient fibres within paraspinalmuscles in peri-operative samples and from post mortem caseswere compared we did not identify a significant difference betweenthe two groups (A). The percentage ofrespiratory-deficient fibres harbouring high heteroplasmy level ofmtDNA deletions was also not significantly different betweenperi-operative samples and tissue derived from post mortemcases, which is consistent previous observations(B) [10]. Furthermore, we didnot identify a significant difference in the density ofrespiratory-deficient fibres or the percentage ofrespiratory-deficient fibres harbouring high heteroplasmy level ofmtDNA deletions between cervical and lumbar deep paraspinal muscles(C–D).
Table S1. Details of primary antibodies.
Table S2. MtDNA deletion breakpoints identified in respiratory-deficient paraspinal muscle fibres.
References
- 1.Delisle MB, Laroche M, Dupont H, Rochaix P, Rumeau JL. Morphological analyses of paraspinal muscles: comparison of progressive lumbar kyphosis (camptocormia) and narrowing of lumbar canal by disc protrusions. Neuromuscul Disord. 1993;3:579–82. doi: 10.1016/0960-8966(93)90120-9. [DOI] [PubMed] [Google Scholar]
- 2.Schabitz WR, Glatz K, Schuhan C, Sommer C, Berger C, Schwaninger M, Hartmann M, Hilmar Goebel H, Meinck HM. Severe forward flexion of the trunk in Parkinson's disease: focal myopathy of the paraspinal muscles mimicking camptocormia. Mov Disord. 2003;18:408–14. doi: 10.1002/mds.10385. [DOI] [PubMed] [Google Scholar]
- 3.Serratrice G. Axial myopathies: an elderly disorder. Acta Myol. 2007;26:11–13. [PMC free article] [PubMed] [Google Scholar]
- 4.Serratrice G, Pouget J, Pellissier JF. Bent spine syndrome. J Neurol Neurosurg Psychiatry. 1996;60:51–4. doi: 10.1136/jnnp.60.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshiyama Y, Takama J, Hattori T. The dropped head sign in parkinsonism. J Neurol Sci. 1999;167:22–5. doi: 10.1016/s0022-510x(99)00129-x. [DOI] [PubMed] [Google Scholar]
- 6.Hund E, Heckl R, Goebel HH, Meinck HM. Inclusion body myositis presenting with isolated erector spinae paresis. Neurology. 1995;45:993–4. doi: 10.1212/wnl.45.5.993. [DOI] [PubMed] [Google Scholar]
- 7.Sakiyama Y, Okamoto Y, Higuchi I, Inamori Y, Sangatsuda Y, Michizono K, Watanabe O, Hatakeyama H, Goto Y, Arimura K, Takashima H. A new phenotype of mitochondrial disease characterized by familial late-onset predominant axial myopathy and encephalopathy. Acta Neuropathol (Berl) 2011;121:775–83. doi: 10.1007/s00401-011-0818-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wakata N, Konno S, Nomoto N, Sugimoto H, Nemoto H, Kurihara T, Kishi M. Myasthenia gravis with concomitant severe paraspinal muscle degeneration and mitochondrial DNA4977 deletion. Intern Med. 2007;46:747–50. doi: 10.2169/internalmedicine.46.6400. [DOI] [PubMed] [Google Scholar]
- 9.Ramsbacher J, Theallier-Janko A, Stoltenburg-Didinger G, Brock M. Ultrastructural changes in paravertebral muscles associated with degenerative spondylolisthesis. Spine (Phila Pa 1976) 2001;26:2180–4. doi: 10.1097/00007632-200110150-00003. discussion 5. [DOI] [PubMed] [Google Scholar]
- 10.Wharton SB, Chan KK, Pickard JD, Anderson JR. Paravertebral muscles in disease of the cervical spine. J Neurol Neurosurg Psychiatry. 1996;61:461–5. doi: 10.1136/jnnp.61.5.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larsson NG. Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem. 2010;79:683–706. doi: 10.1146/annurev-biochem-060408-093701. [DOI] [PubMed] [Google Scholar]
- 12.Greaves LC, Yu-Wai-Man P, Blakely EL, Krishnan KJ, Beadle NE, Kerin J, Barron MJ, Griffiths PG, Dickinson AJ, Turnbull DM, Taylor RW. Mitochondrial DNA defects and selective extraocular muscle involvement in CPEO. Invest Ophthalmol Vis Sci. 2010;51:3340–6. doi: 10.1167/iovs.09-4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brierley EJ, Johnson MA, Lightowlers RN, James OF, Turnbull DM. Role of mitochondrial DNA mutations in human aging: implications for the central nervous system and muscle. Ann Neurol. 1998;43:217–23. doi: 10.1002/ana.410430212. [DOI] [PubMed] [Google Scholar]
- 14.Bua E, Johnson J, Herbst A, Delong B, McKenzie D, Salamat S, Aiken JM. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet. 2006;79:469–80. doi: 10.1086/507132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fayet G, Jansson M, Sternberg D, Moslemi AR, Blondy P, Lombes A, Fardeau M, Oldfors A. Ageing muscle: clonal expansions of mitochondrial DNA point mutations and deletions cause focal impairment of mitochondrial function. Neuromuscul Disord. 2002;12:484–93. doi: 10.1016/s0960-8966(01)00332-7. [DOI] [PubMed] [Google Scholar]
- 16.Rahman S, Lake BD, Taanman JW, Hanna MG, Cooper JM, Schapira AH, Leonard JV. Cytochrome oxidase immunohistochemistry: clues for genetic mechanisms. Brain. 2000;123:591–600. doi: 10.1093/brain/123.3.591. [DOI] [PubMed] [Google Scholar]
- 17.De Paepe B, Smet J, Lammens M, Seneca S, Martin JJ, De Bleecker J, De Meirleir L, Lissens W, Van Coster R. Immunohistochemical analysis of the oxidative phosphorylation complexes in skeletal muscle from patients with mitochondrial DNA encoded tRNA gene defects. J Clin Pathol. 2009;62:172–6. doi: 10.1136/jcp.2008.061267. [DOI] [PubMed] [Google Scholar]
- 18.Johnson MA, Bindoff LA, Turnbull DM. Cytochrome c oxidase activity in single muscle fibers: assay techniques and diagnostic applications. Ann Neurol. 1993;33:28–35. doi: 10.1002/ana.410330106. [DOI] [PubMed] [Google Scholar]
- 19.Yu-Wai-Man P, Lai-Cheong J, Borthwick GM, He L, Taylor GA, Greaves LC, Taylor RW, Griffiths PG, Turnbull DM. Somatic mitochondrial DNA deletions accumulate to high levels in aging human extraocular muscles. Invest Ophthalmol Vis Sci. 2010;51:3347–53. doi: 10.1167/iovs.09-4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campbell GR, Ziabreva I, Reeve AK, Krishnan KJ, Reynolds R, Howell O, Lassmann H, Turnbull DM, Mahad DJ. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann Neurol. 2011;69:481–92. doi: 10.1002/ana.22109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He L, Chinnery PF, Durham SE, Blakely EL, Wardell TM, Borthwick GM, Taylor RW, Turnbull DM. Detection and quantification of mitochondrial DNA deletions in individual cells by real-time PCR. Nucleic Acids Res. 2002;30:e68. doi: 10.1093/nar/gnf067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krishnan KJ, Bender A, Taylor RW, Turnbull DM. A multiplex real-time PCR method to detect and quantify mitochondrial DNA deletions in individual cells. Anal Biochem. 2007;370:127–9. doi: 10.1016/j.ab.2007.06.024. [DOI] [PubMed] [Google Scholar]
- 23.Reeve AK, Krishnan KJ, Turnbull D. Mitochondrial DNA mutations in disease, aging, and neurodegeneration. Ann N Y Acad Sci. 2008;1147:21–9. doi: 10.1196/annals.1427.016. [DOI] [PubMed] [Google Scholar]
- 24.Brierley EJ, Johnson MA, James OF, Turnbull DM. Effects of physical activity and age on mitochondrial function. Q J Med. 1996;89:251–8. doi: 10.1093/qjmed/89.4.251. [DOI] [PubMed] [Google Scholar]
- 25.Ordahl CP, Le Douarin NM. Two myogenic lineages within the developing somite. Development. 1992;114:339–53. doi: 10.1242/dev.114.2.339. [DOI] [PubMed] [Google Scholar]
- 26.Keynes RJ, Stern CD. Mechanisms of vertebrate segmentation. Development. 1988;103:413–29. doi: 10.1242/dev.103.3.413. [DOI] [PubMed] [Google Scholar]
- 27.Regev GJ, Kim CW, Thacker BE, Tomiya A, Garfin SR, Ward SR, Lieber RL. Regional myosin heavy chain distribution in selected paraspinal muscles. Spine (Phila Pa 1976) 2010;35:1265–70. doi: 10.1097/BRS.0b013e3181bfcd98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krishnan KJ, Reeve AK, Samuels DC, Chinnery PF, Blackwood JK, Taylor RW, Wanrooij S, Spelbrink JN, Lightowlers RN, Turnbull DM. What causes mitochondrial DNA deletions in human cells? Nat Genet. 2008;40:275–9. doi: 10.1038/ng.f.94. [DOI] [PubMed] [Google Scholar]
- 29.Elson JL, Samuels DC, Turnbull DM, Chinnery PF. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am J Hum Genet. 2001;68:802–6. doi: 10.1086/318801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nicholas A, Kraytsberg Y, Guo X, Khrapko K. On the timing and the extent of clonal expansion of mtDNA deletions: evidence from single-molecule PCR. Exp Neurol. 2009;218:316–19. doi: 10.1016/j.expneurol.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen YW, Gregory CM, Scarborough MT, Shi R, Walter GA, Vandenborne K. Transcriptional pathways associated with skeletal muscle disuse atrophy in humans. Physiol Genomics. 2007;31:510–20. doi: 10.1152/physiolgenomics.00115.2006. [DOI] [PubMed] [Google Scholar]
- 32.Vielhaber S, Kunz D, Winkler K, Wiedemann FR, Kirches E, Feistner H, Heinze HJ, Elger CE, Schubert W, Kunz WS. Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain. 2000;123:1339–48. doi: 10.1093/brain/123.7.1339. [DOI] [PubMed] [Google Scholar]
- 33.McFarland R, Taylor RW, Turnbull DM. A neurological perspective on mitochondrial disease. Lancet Neurol. 2010;9:829–40. doi: 10.1016/S1474-4422(10)70116-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.