

Abstract
Fragile X syndrome (FXS) is an inherited disorder that results in intellectual disability and a characteristic behavioral profile that includes autism spectrum disorder, attention deficit hyperactivity disorder, sensory hypersensitivity, hyperarousal, and anxiety. The epigenetic silencing of FMR1 and the consequent absence of its protein product, FMRP, is the most common cause of fragile X. The development of animal models of fragile X syndrome 20 years ago has produced a considerable increase in our understanding of the consequences of the absence of FMRP on the structure and function of the nervous system. Some of the insights gained have led to proposals of treatment strategies that are based on cellular and molecular changes observed in animals lacking FMRP. One such proposal is treatment with lithium, a drug with a long history of clinical efficacy in psychiatry and a drug with newly described uses in degenerative disorders of the nervous system. Lithium treatment has been studied extensively in both mouse and fruit fly models of FXS, and it has been shown to reverse numerous behavioral, physiological, cellular, and molecular phenotypes. A report of a pilot clinical trial on a limited number of adult FXS patients indicated that measurable improvements in behavior and function were seen after 2 months of lithium treatment. A double-blind clinical trial of lithium treatment in FXS patients is now needed.
Keywords: Fragile X syndrome, FMR1, FMRP, intellectual disability, autism, dendritic spines, glycogen synthase kinase-3 (GSK-3)
Fragile X syndrome (FXS) is the leading known genetic cause of intellectual disability and monogenic cause of autism with a prevalence of 1/4000 in males and 1/6000 in females.1 In most cases, FXS is caused by expansion of a sequence of CGG trinucleotide repeats in the 5′-untranslated region of the FMR1 gene. In humans, the number of CGG repeats is highly polymorphic. Normal individuals have between 5 and 54 repeats. When the number of repeats expands to more than 55 but less than 200, it is referred to as fragile X premutation. When the repeat length is greater than 200, the gene is hypermethylated and transcription is silenced with consequent loss of the gene product, fragile X mental retardation protein (FMRP). This is known as FXS or the full fragile X mutation.2 Individuals with FXS have a characteristic facial morphology and macroorchidism. They are also prone to seizure disorders. Behavioral phenotypes include hyperactivity, attention deficit, intellectual disability, learning deficits, hyperarousal, anxiety, and autism.3
Animal Models of Fragile X Syndrome
The development of animal models of FXS has led to considerable advances in our understanding of the pathophysiology of the disease. The most widely studied model is the Fmr1 knockout (KO) mouse advanced by the Dutch-Belgian Fragile X Consortium in 1994.4 There are also models of the disease in rats, Drosophila, and zebrafish.5,6 For the purpose of this review, remarks will be limited to the mouse and Drosophila models. Initially, work on these models was directed at establishing the phenotype in animals. Fmr1 KO mice are susceptible to audiogenic seizures7 likely in parallel to the tendency of children with FXS to have seizures. Fmr1 KO mice are hyperactive4 and have deficits on tests of social interaction8 and certain types of learning and memory.4,9−11 These characteristics are similar to symptoms seen in patients with FXS. Tests of anxiety-like behavior indicate that general anxiety is decreased in Fmr1 KO mice compared to wild type (WT).12,8 In the disease in humans, however, general as well as social anxiety is elevated. It is possible that the measures of general anxiety in rodents (behavior in the center of an open field arena and behavior in elevated plus and zero mazes) cannot distinguish between reduced anxiety and impulsivity or lack of executive function. Impulsivity is also reported to be characteristic of Fmr1 KO mice.13 The hallmark neuropathological phenotype of FXS is the change in dendritic spines, and this change is seen in both patients and in Fmr1 KO mice. In the absence of FMRP, dendritic spines at excitatory synapses tend to be of a more immature type, long, thin, and filopodial-like; they also tend to be increased in density.14 In Fmr1 KO mice, effects on synaptic plasticity have been reported including alterations in the course of developmental plasticity as indicated by changes in the critical period for thalamocortical synapses.15,16 Developmental delay is typical in patients with delays in motor skills and motor coordination.
Treatments for Fragile X Syndrome
Until recently, pharmacological therapies for treatment of FXS have been symptom-based. With increases in our understanding of the cellular and molecular basis of dysfunction in the animal models is the hope that rationale therapies addressing specific core targets can be developed. The absence of FMRP is the primary change and likely at the biochemical core of the disease. FMRP is a polyribosome-associated RNA-binding protein, suggesting a role in regulating translation. In in vitro model systems for the study of mRNA translation, FMRP negatively regulates the translation of its target mRNAs.17,18 FMRP has been shown to reversibly stall ribosomes specifically on its target mRNAs during elongation.19 FMRP also recruits cytoplasmic FMRP-interacting protein (CYFIP1), consequently blocking formation of the eIF4F complex and preventing translation initiation.20 Translation is also inhibited by the recruitment by FMRP of RNA-induced silencing complex (RISC).21 Consistent with this role for FMRP as a suppressor of translation, elevated rates of protein synthesis measured in the intact nervous system have been demonstrated in Fmr1 KO mice.22 Studies in hippocampal slices from Fmr1 KO mice confirm increased incorporation rates in vitro.15,23−25 Given the centrality of the effect on protein synthesis, a treatment for FXS that can reverse this phenotype in animals offers considerable promise.
mGluR Theory of Fragile X Syndrome
One observation that appears to be linked to the effect on protein synthesis is the finding that the response to group I metabotropic glutamate receptor (Gp1 mGluR) activation in CA1 hippocampal neurons was exaggerated in Fmr1 KO mice.26 The response was a long-term depression (LTD) coupled with internalization of 2-amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl)propanoic acid (AMPA) receptors, both of which were increased in Fmr1 KO mice. This finding together with the finding that activation of Gp1 mGluRs increased the synthesis of FMRP in synaptoneurosomes prepared from brains of WT mice27 led to the mGluR theory of FXS.28 It had been shown that activation of Gp1 mGluRs induces hippocampal LTD that requires rapid translation of pre-existing mRNA.29 In Fmr1 KO mice, this response was exaggerated, but no longer dependent on protein synthesis.30 This latter finding suggests that, in the absence of FMRP, “plasticity proteins” necessary for induction of LTD are already present in sufficient concentrations. Increased levels of several “plasticity proteins” including activity-regulated cytoskeletal protein (Arc), microtubule associated protein 1β (Map1β), and α calcium/calmodulin-dependent kinase II (αCAMKII) have been reported in Fmr1 KO mice.31 The mGluR theory proposed that in WT mice synthesis of FMRP in response to activation of Gp1 mGluRs acts as a brake on the protein synthesis-LTD response preparing the synapse for the capacity to respond to the next signal. In the absence of FMRP, as in FXS, the synaptic response may lack an “off” switch.
mGluR-LTD requires a cascade of signaling events that culminate in increased protein synthesis likely via mammalian target of rapamycin (mTOR) activation and/or extracellular signal-related kinase (ERK) signaling. Both the phosphatidylinositol 3-kinase (PI3K) and ERK pathways are involved in this response and have been proposed as therapeutic targets.32,23 Another approach to therapeutic intervention in FXS is through an understanding of the signaling pathways acting on FMRP and regulated by FMRP. Through an enzyme-mediated change in its phosphorylation status, FMRP activity is affected by activation of mGluRs, AMPARs, γ-aminobutyric acid (GABA) receptors, N-methyl-d-aspartate (NMDA) receptors, and tyrosine kinase (TrkB) receptors. For example, within 1 min of Gp1 mGluR stimulation, FMRP is dephosphorylated by protein phosphatase 2A (PP2A)33 and translation is released from FMRP’s suppression. After 1 min, the rephosphorylation of FMRP catalyzed by ribosomal protein S6 kinase 1 (S6K1)34 results in the reinstatement of a translation-suppressed state. Activity-dependent phosphorylation of FMRP by S6K1 requires signaling from mTOR, ERK1/2, and PP2A.
Based on the mGluR theory, it was proposed that reducing Gp1 mGluR activation might reverse phenotypes in Fmr1 KO mice. Many studies have been published addressing this proposition in experimental animals confirming that both genetic reduction of mGluR515 and pharmacological reduction of Gp1 mGluR response by treatment with negative allosteric modulators of mGluR5, for example, 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP), correct many of the behavioral and pathophysiological phenotypes in the mouse (reviewed in ref (35)) and Drosophila(36,37) models. Preliminary results of clinical trials of negative allosteric modulators of mGluR5 in adults with FXS have yielded some indications of promise on tests of prepulse inhibition38 and, in a subset of patients, on the Aberrant Behavior Checklist–Community Edition.39
Lithium as a Therapeutic for Fragile X Syndrome
Lithium has been proposed as a therapeutic for FXS, and there is mounting evidence to support its potential. Lithium has a long history in psychiatric medicine. It was one of the first drugs to be used in the treatment of mental illness. In the 19th century, lithium was used to treat mania that was thought to result from the toxic effects of gout and the accumulation of uric acid. Its use was expanded for the treatment of mania in general following the observation that lithium had a tranquilizing effect in guinea pigs and in patients with mania (reviewed in ref (40)). Today, lithium serves as a mood stabilizer primarily in the treatment of bipolar disorder, where it plays a role in normalizing mania and depression, both acute and long-term phases. The use of lithium as a therapeutic has been tempered by its toxicity occurring just beyond its narrow therapeutic range. In patients on lithium therapy, blood levels must be monitored and optimal concentrations maintained (0.4–1.2 mequiv/L). Toxic effects of lithium are largely due to its inhibitory effect on vasopressin so patients with toxic levels of lithium are prone to dehydration. Whereas the potential toxicity of lithium has limited its use, careful monitoring of blood levels is effective in preventing toxic reactions.
In rodents, lithium treatment alters many behavioral attributes such as aggression,41 depression,42 amphetamine-induced hyperlocomotion43 and reserpine-induced hypolocomotion.44 Chronic lithium also enhances learning45 and spatial memory in rats.46,47 Electrophysiologically, chronic lithium increases long-term potentiation (LTP) in hippocampal neurons48 and alleviates stress-induced LTP impairment.49 Lithium also exerts neuroprotective effects.50 The effects of lithium are mediated by several known mechanisms such as down-regulation of the phospholipase C signaling pathway,51 reduction of NMDA receptor-initiated signaling52 and inhibition of GSK-3.53,54
Lithium Treatment in the Drosophila Model of Fragile X
The first tests of lithium treatment in FXS were in the Drosophila model. Drosophila provide a powerful genetic model system for biomedical research, and a model for FXS was developed in Drosophila that is based on loss-of-function mutants of dfmr1.5 Studies of dfmr1 mutants reveal that their neuronal and behavioral phenotypes parallel symptoms observed in FXS patients. The dfmr1 mutants have abnormal circadian rhythms,55 courtship behavior,55 cognitive function,56 impairments on social interaction and long-term olfactory memory.57,58 Neurite branching patterns are aberrant,56,59 and interestingly, dfmr1 mutants have developmental defects in mushroom bodies in brain.60 Mushroom bodies are the best-characterized insect brain region involved in many forms of learning and memory.
In a test of viability, dfmr1 mutant Drosophila reared on food containing increased levels of glutamate61 died during development, consistent with the mGluR theory that FMRP loss results in excess glutamate signaling. In this lethal phenotype, lithium treatment improved viability. An assay of courtship behavior in flies was used to test the effects of lithium treatment on experience-dependent plasticity. Young adult (5 day), male Drosophila display a characteristic sequence of behaviors when presented with a virgin female. In a 10 min courtship assay, male dfmr1 mutant flies failed to activate advanced stages of the courtship behavior and spent less time engaged in active courtship than WT males.56 Courtship behavior was improved following inclusion of either 5 or 50 mM lithium chloride in the diet during development and the 4 days prior to testing.36 In an extension of this assay, training capability and short-term memory were tested in a conditioned courtship paradigm. A male WT fly exposed to a previously mated (unreceptive) female will soon learn to decrease his courtship activity (learning during training). When subsequently paired with a receptive virgin female, the male will display reduced courtship activity indicating memory of the training. Mutant dfmr1 flies showed learning during training but impaired memory of the training. The memory impairment was reversed by chronic treatment with either 5 or 50 mM lithium in the diet.36 In another study, dfmr1 mutants were treated with lithium only during development, and learning and memory were tested in aged flies (20 days). Results showed that treatment with lithium during development prevents both learning and memory impairments that occur in aged flies.62 It is possible that treatment during development might induce some lasting changes in neuronal signaling at synapses that continue to be beneficial to the behavioral performances during aging.
Lithium Treatment in the Fmr1 KO Mouse Model
The positive effects of lithium treatment in flies stimulated the study of this treatment and its effects on numerous behavioral, physiological, and cellular phenotypes in Fmr1 KO mice. The first report of efficacy of lithium treatment on behavior in Fmr1 KO mice was a study of audiogenic seizure incidence.63 Young (29–30 day), male WT and Fmr1 KO mice were administered a single i.p. injection of lithium citrate (120, 200, or 300 mg/kg) and tested 30 min later for seizure activity induced by a high intensity sound. Almost 80% of vehicle-injected Fmr1 KO mice died following status epilepticus, whereas none of the WT mice were so affected. Following acute lithium treatment, the incidence of status epilepticus in Fmr1 KO mice was reduced to 33, 30, and 7% at the three doses of lithium tested, respectively.63
In a comprehensive study of the effects of chronic lithium treatment,64Fmr1 KO male and WT control littermates were fed either lithium carbonate supplemented (0.3% w/w) or control diets commencing on the day of weaning and maintained throughout. An important consideration in these studies is the dose and the length of treatment. To achieve long-term consistent plasma concentrations, animals were fed diet containing lithium carbonate, and blood levels were measured to confirm that they were not toxic and approximate the therapeutic range. Lithium has a narrow therapeutic index, so the dose of lithium in the diet was carefully selected to achieve and maintain a concentration of lithium at the low end of the therapeutic range. To counteract potential hyponatremia caused by lithium-induced increased sodium excretion, animals’ drinking water was supplemented with sodium chloride. After 5 weeks of treatment, mice were subjected to a battery of behavioral tests each administered 1 week apart. Behavioral assessment began with open field testing, followed by social interaction, elevated plus and zero mazes, and passive avoidance tests. These tests assessed four robust phenotypes in Fmr1 KO mice: hyperactivity, deficits on social interaction, reduced general anxiety, and deficits on fear learning and memory.
Chronic lithium treatment corrected all four phenotypes and had little or no effect on WT mice. In the open field, Fmr1 KO mice consistently show hyperactivity and a higher percent activity in the center of the field, indicating lower levels of general anxiety. Both of these behaviors were reversed by chronic lithium treatment.64 On other tests of general anxiety, that is, elevated plus and elevated zero mazes, Fmr1 KO mice also behaved as if they were less anxious about being in the open as indicated by higher number of entrances and more time spent in open arms or quadrants of the mazes; these behaviors were also partially reversed by chronic lithium treatment.64 A form of anxiety that is increased in patients with FXS is social anxiety, characterized by avoidance in social situations. Social behavior in Fmr1 KO mice tested in an automated three-chambered apparatus indicates normal social approach behavior; that is, they showed a normal preference for a single novel mouse over a novel object. In the social novelty phase of the test, that is, the choice between a familiar mouse and a novel mouse, Fmr1 KO mice “shied away” from both mice and spent an increased amount of time in the center chamber. Chronic lithium treatment reversed this “social anxiety” phenotype. It is interesting to note that untreated Fmr1 KO mice exhibited increased “social anxiety” and decreased general anxiety phenotypes, dissociating these two forms of anxiety.8,64 This dissociation has also been observed clinically and in other animal disease models.65,66 Chronic lithium treatment appears to normalize both hypo-general anxiety and hyper-social anxiety.64 Aversive learning and memory tests such as the passive avoidance test yield strong evidence of impairment.9,12 Following a single trial in a two-chambered apparatus, in which the test mouse receives a mild foot shock upon entering the dark chamber, the mouse is returned to his home cage and tested 24 h later for latency to enter the dark chamber. Fmr1 KO mice exhibited a decreased latency to enter the dark chamber.9,12,67,64,68 This behavior is thought to demonstrate an impairment of learning/memory. Chronic lithium treatment reversed this deficit in Fmr1 KO mice but had no measurable effect in WT mice.64
In addition to the positive effects on behavior, chronic lithium treatment also reversed the dendritic spine phenotype in medial prefrontal cortex in Fmr1 KO mice. That the spine phenotype occurs in medial prefrontal cortex is of particular interest in light of the involvement of this cortical area in mood regulation and social cognition.69 Medial prefrontal cortex is also involved in executive function and control of impulsivity.70,71 Performance of Fmr1 KO mice on the passive avoidance test and possibly also tests of anxiety may be interpreted as a reflection of hyperactivity and impulsivity, known attributes of these mice.13 Correction of the spine phenotype in medial prefrontal cortex and performance on both passive avoidance and social interaction tests is consistent with decreased social anxiety, reduced locomotor activity, and improved inhibitory control following lithium treatment.
Lithium Treatment and Glycogen Synthase Kinase-3
A principal target of lithium is glycogen synthase kinase-3 (GSK-3), a serine/threonine kinase first identified as an enzyme involved in glycogen synthesis. It is now known that GSK-3 plays a role in numerous pathways regulating fundamental processes, including development, cell architecture, microtubule dynamics, gene expression, and apoptosis.72 GSK-3 has two isoforms, GSK-3α and GSK-3β,73 which share many functions. Activity of GSK-3 is negatively regulated by phosphorylation (GSK-3α at Ser21, GSK-3β at Ser9). Dysregulation of GSK-3 is thought to contribute to a number of diseases such as mood disorders, schizophrenia, Alzheimer’s disease, and Huntington’s disease.72
Accumulating evidence suggests that GSK-3 plays a pivotal role in expression of Fmr1 KO phenotypes. GSK3β mRNA is a target of FMRP,19 indicating that FMRP binds to GSK3β mRNA and can regulate its translation. Moreover, current data suggest that GSK-3 activity is upregulated in Fmr1 KO mice with the phosphorylated forms of GSK-3α and GSK-3β decreased relative to total enzyme in homogenates of striatum, cortex, hippocampus, and cerebellum63 and whole brain.64 Acute treatment of animals with the Gp1 mGluR negative allosteric modulator, MPEP, resulted in an increase in the relative levels of p-GSK-3α and p-GSK-3β, suggesting that this treatment decreases GSK-3 activity.67 Both acute and chronic lithium treatment had similar effects.67 In support of the idea that dysregulation of GSK-3 is important in FXS is the finding that treatment with selective inhibitors of GSK-3 reverses many phenotypes in Fmr1 KO mice. Acute i.p injection of the selective ATP competitive inhibitor of GSK-3, SB-216763, reduced the incidence of audiogenic seizures and normalized open field behavior.63 A study of hippocampal-dependent cognitive behaviors tested the efficacy of selective GSK-3 inhibitors, TDZD-8 (5 mg/kg, i.p.) or VP0.7 (5 mg/kg, i.p.), to improve performance of Fmr1 KO mice on novel object recognition, coordinate and categorical spatial processing and temporal ordering for visual objects.74 In all cases, treatment with either drug 1 h prior to testing reversed the deficits seen on these tasks in Fmr1 KO mice. Taken together, these results indicate the involvement of overactive GSK-3 in the hyperactivity, impulsivity, vulnerability to audiogenic seizures, social interaction deficits, hippocampal-dependent cognitive deficits, and dendritic spine abnormalities so characteristic of Fmr1 KO mice.
Lithium Treatment and Protein Synthesis
The ability of both lithium and GSK-3 inhibitors to reverse behavioral, physiological, and structural phenotypes in the FXS mouse model coupled with the finding that lithium treatment decreases activity of GSK-3 as indicated by the relative increase in the phosphorylated form of the enzyme suggest that overactive GSK-3 activity is an ideal target for treatment of FXS. Moreover, and in accord with the idea that the dysregulation of protein synthesis may be at the heart of the defect in FXS, chronic lithium treatment reversed increased rCPS measured in vivo in Fmr1 KO mice; effects of the treatment on WT mice were minor75 (Figure 1). The process of protein synthesis is primarily regulated by translation factors that transiently associate with ribosomes. A number of components involved in the initiation and elongation stages of translation are regulated by mammalian target of rapamycin (mTOR), a protein kinase that phosphorylates eIF4E-binding proteins (4E-BP) and the ribosomal protein p70 S6 kinase 1 (p70 S6K1). The PI3K/Akt signaling pathway is an important upstream regulator of mTOR, and PI3K/Akt signaling may be dysregulated in FXS. Basal PI3K activity in cortical synaptoneurosomes from developing Fmr1 KO mice was found to be excessive likely due to increased synthesis and synaptic localization of its catalytic subunit, p110β, both of which are normally regulated by FMRP.32 In hippocampal slices from Fmr1 KO mice, mTOR activity and downstream targets S6K1, 4E-BP, and eIF4E were also found to be elevated possibly due to an increase in PI3K enhancer (PIKE), which is also normally regulated by FMRP.76 It has been suggested that these effects may lead to exaggerated protein synthesis in FXS. Another study in hippocampal slices, however, failed to observe increased levels of mTOR pathway components in Fmr1 KO mice, but indicated that ERK1/2 is hypersensitive to mGluR stimulation leading to excessive protein synthesis in Fmr1 KO mice.23 The effects of lithium treatment on the above two pathways were tested in Fmr1 KO mice. Relative levels of p-Akt and p-mTOR were higher in hippocampal homogenates of Fmr1 KO mice compared to WT,64 although the differences were not as strong as previously reported in hippocampal slices.76 Chronic lithium treatment resulted in substantial decreases in the levels of p-Akt and p-mTOR in Fmr1 KO mice, but effects on p-p70 S6K1 and p-ERK1/2 were weak and did not reach statistical significance.75 These findings fail to support either PI3K/Akt or MAPK/ERK1/2 pathways alone as the means by which chronic lithium treatment normalizes protein synthesis in brain. The mechanisms underlying the upregulation of protein synthesis and the effects of lithium treatment in fragile X are likely more complicated than originally thought. Multiple physiological targets may exist for lithium.
Figure 1.
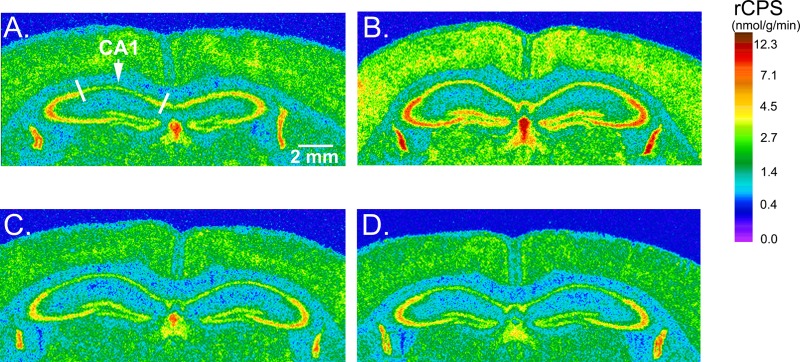
Effects of chronic lithium treatment on rates of protein synthesis measured in vivo in Fmr1 KO mice.75 (A–D) Digitized autoradiographic images color-coded for regional rates of cerebral protein synthesis (rCPS) at the level of dorsal hippocampus from each experimental group are as follows: (A) WT-control, (B) Fmr1 KO-control, (C) WT-lithium-treated, and (D) KO-lithium-treated. Color bar and the scale bar in (A) (2 mm) apply to all four images.
Lithium Treatment of FXS Patients
The findings demonstrating that lithium treatment can normalize an array of phenotypes in animal models of FXS suggest that it may be a promising treatment in patients. Currently, lithium is used in FXS for treatment of aggression and for mood stabilization in adolescents and adults.2 A pilot add-on trial has been conducted to evaluate the safety and efficacy of lithium in human subjects with FXS.77 Fifteen subjects with FXS, ages 6–23, received lithium titrated to levels of 0.8–1.2 mequiv/L. After 2 months of treatment, significant improvements were found in hyperactivity, inappropriate speech, aggression, abnormal vocalizations, self-abuse, work refusal, outbursts, overemotionality, anxiety, mood swings, tantrums, perseveration, crying, and maladaptive behavior. Scores on the Clinical Global Improvement Scale were significantly enhanced. One cognitive measure, the RBANS List Learning, also showed significant improvement over baseline performance after lithium treatment. Patients also showed trends of improvements in lethargy and stereotypy scores following lithium treatment. These results suggest that lithium has positive effects on behavioral and adaptive skills for individuals with FXS. Positive responses were distributed across the age range of the study cohort, suggesting that both children and young adults with FXS can be benefit from lithium treatment. As for the side effects, except polyuria and polydipsia, lithium was well-tolerated.77
Summary
Increased understanding of the function of FMRP and the consequences of its absence has led to a number of promising potential therapeutic approaches for FXS. Presently, there are six compounds in clinical trials for the treatment of FXS; others are on the horizon. Some of the drugs are expected to redress an excitatory/inhibitory imbalance, one is an anti-inflammatory agent. Lithium treatment has proven benefit for many of the abnormal phenotypes in Drosophila and mouse models of FXS. The precise mechanism through which lithium effects these changes is not totally understood, but it is noteworthy that increased GSK-3 activity is normalized by lithium treatment and that inhibitors of GSK-3 had similar effects on animal behavior. Most importantly, lithium treatment reversed upregulated protein synthesis in Fmr1 KO mice. Because the exaggerated protein synthesis as a consequence of the absence of FMRP is thought to be central to the pathogenesis of FXS, the reversal effect of lithium on protein synthesis may account for the improvements in multiple phenotypes. Moreover, results of a pilot clinical trial reported in 2008 support the possibility that it is a promising treatment for patients with FXS.77 With all of the evidence that lithium can reverse fragile X phenotypes in animal models and in the pilot human study (Table 1), and in view of the extensive clinical experience managing patients on this drug, it seems the time has come for a placebo-controlled trial of lithium in individuals with FXS.
Table 1. Fragile X Phenotypes: Effects of Lithium Treatment.
| phenotype | effects of lithium treatment |
|---|---|
| studies indfmr1mutantDrosophila | |
| courtship behavior56 | reversed37 |
| courtship behavior memory56 | reversed37 |
| studies inFmr1KO mice | |
| macroorchidism4 | small effect64 |
| audiogenic seizures7 | reversed63 |
| behavior | |
| hyperactivity4 | partially reversed64 |
| social interaction deficit8 | reversed64 |
| passive avoidance test9 | reversed64 |
| cellular | |
| dendritic spine abnormalities14 | reversed64 |
| cerebral protein synthesis22 | reversed75 |
| signaling | |
| p-GSK-363 | reversed67 |
| p-Akt76 | reversed75 |
| p-mTOR76 | reversed75 |
| p-p70 S6K176 | no effect75 |
| studies in adults with fragile X syndrome | |
| visual analogue scale for behavior | improvement77 |
| Clinical Global Improvement Scale | improvement77 |
| Aberrant behavior Checklist-Community Edition | improvement77 |
| Vineland Maladaptive Behavior Scale | improvement77 |
| RBans List Learning Test | improvement77 |
Acknowledgments
We thank Ms. Geeta Strange for editorial help with the manuscript.
Author Contributions
Z.H.L. and C.B.S. prepared the manuscript.
This work was supported by the Intramural Research Program of the National Institute of Mental Health, National Institutes of Health, Department of Health and Human Services.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Turner G.; Webb T.; Wake S.; Robinson H. (1996) Prevalence of fragile X syndrome. Am. J. Med. Gen. 64, 196–197. [DOI] [PubMed] [Google Scholar]
- Hagerman R. J. (2006) Lessons from fragile X regarding neurobiology, autism, and neurodegeneration. J. Dev. Behav. Pediatr. 27, 63–74. [DOI] [PubMed] [Google Scholar]
- Hagerman R. J. (2002) The physical and behavioral phenotype. In Fragile X Syndrome: Diagnosis, Treatment, and Research (Hagerman R. J., and Hagerman P. J., Eds.), 3rd ed., pp 3–109, Johns Hopkins University Press, Baltimore. [Google Scholar]
- Bakker C. E.; (1994) Fmr1 knockout mice: a model to study fragile X mental retardation. Cell 78, 23–33. [PubMed] [Google Scholar]
- Wan L.; Dockendorff T. C.; Jongens T. A.; Dreyfuss G. (2000) Characterization of dFMR1, a Drosophila melanogaster homolog of the fragile X mental retardation protein. Mol. Cell. Biol. 20, 8536–8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Den Broeder M. J.; van der Linde H.; Brouwer J. R.; Oostra B. A.; Willemsen R.; Ketting R. F. (2009) Generation and characterization of Fmr1 knockout zebrafish. PLoS One 4, e7910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musumeci S. A.; Bosco P.; Calabrese G.; Bakker C.; De Sarro G. B.; Elia M.; Ferri R.; Oostra B. A. (2000) Audiogenic seizures susceptibility in transgenic mice with fragile X syndrome. Epilepsia 41, 19–23. [DOI] [PubMed] [Google Scholar]
- Liu Z. H.; Smith C. B. (2009) Dissociation of social and nonsocial anxiety in a mouse model of fragile X syndrome. Neurosci. Lett. 454, 62–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin M.; Kang J.; Smith C. B. (2002) Increased rates of cerebral glucose metabolism in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U.S.A. 99, 15758–15763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M. G.; Toyoda H.; Ko S. W.; Ding H. K.; Wu L. J.; Zhuo M. (2005) Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J. Neurosci. 25, 7385–7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan F. X.; Albeck D. S.; Paylor R. (2006) Fmr1 knockout mice are impaired in a leverpress escape/avoidance task. Genes, Brain Behav. 5, 467–471. [DOI] [PubMed] [Google Scholar]
- Qin M.; Kang J.; Smith C. B. (2005b) A null mutation for Fmr1 in female mice: effects on regional cerebral metabolic rate for glucose and relationship to behavior. Neuroscience 135, 999–1009. [DOI] [PubMed] [Google Scholar]
- Moon J.; Beaudin A. E.; Verosky S.; Driscoll L. L.; et al. (2006) Attentional dysfunction, impulsivity, and resistance to change in a mouse model of fragile X syndrome. Behav. Neurosci. 120, 1367–1379. [DOI] [PubMed] [Google Scholar]
- Comery T. A.; Harris J. B.; Willems P. J.; Oostra B. A.; Oostra B. A.; Irwin S. A.; Weiler I. J.; Greenough W. T. (1997) Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc. Natl. Acad. Sci. U.S.A. 94, 5401–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölen G.; Osterweil E.; Rao B. S.; Smith G. B.; Auerbach B. D.; Chattarji S.; Bear M. F. (2007) Correction of fragile X syndrome in mice. Neuron. 56, 955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow E. G.; Till S. M.; Russell T. A.; Wijetunge L. S.; Kind P.; Contractor A. (2010) Critical period plasticity is disrupted in the barrel cortex of Fmr1 knockout mice. Neuron 65, 385–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laggerbauer B.; Ostareck D.; Keidel E. M.; Ostareck-Lederer A.; Fischer U. (2001) Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum. Mol. Genet. 10, 329–338. [DOI] [PubMed] [Google Scholar]
- Li Z.; Zhang Y.; Ku L.; Wilkinson K. D.; Warren S. T.; Feng Y. (2001) The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 29, 2276–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell J. C.; Van Driesche S. J.; Zhang C.; Hung K. Y. S.; Mele A.; Fraser C. E.; Stone E. F.; Chen C.; Fak J. J.; Chi S. W.; Licatalosi D. D.; Richter J. D.; Darnell R. B. (2011) FMRP stalls ribosomal translocation on mRNAs linkied to synaptic function and autism. Cell 146, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli I.; Mercaldo V.; Boyl P. P.; Eleuteri B.; Zalfa F.; De Rubeis S.; De Marino D.; Mohr E.; Massimi M.; Falconi M.; Witke W.; Costa-Mattioli M.; Sonnenberg N.; Achsel T.; Nagni C. (2008) The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 134, 1042–1054. [DOI] [PubMed] [Google Scholar]
- Caudy A. A.; Myers M.; Hannon G. J.; Hammond S. M. (2002) Fragile X-related protein and VIG associate with the RNA interference machinery. Genes Dev. 16, 2491–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin M.; Kang J.; Burlin T.; Jiang C. H.; Smith C. B. (2005a) Post-adolescent changes in regional cerebral protein synthesis: An in vivo study in the Fmr1 null mouse. J. Neurosci. 25, 5087–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterweil E. K.; Krueger D. D.; Reinhold K.; Bear M. F. (2010) Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J. Neurosci. 30, 15616–15627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson C.; Wijetunge L.; Kinoshita M. N.; Shumway M.; Hammond R. S.; Postma F. R.; Brynczka C.; Rush R.; Thomas A.; Paylor R.; Warren S. T.; Vanderklish P. W.; Kind P. C.; Carpenter R. L.; Bear M.; Healy A. M. (2012) Sci. Transl. Med. 4, 152ra128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalon A.; Sidorov M.; Ballard T. M.; Ozmen L.; Spooren W.; Wettstein J. G.; Jaeschke G.; Bear M.; Lindemann L. (2012) Chronic pharmacological mGluR 5 inhibition corrects fragile X in adult mice. Neuron 74, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber K. M.; Gallagher S. M.; Warren S. T.; Bear M. F. (2002) Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U.S.A. 99, 7746–7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler I. J.; Irwin S. A.; Klintsova A. Y.; et al. (1997) Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc. Natl., Acad. Sci. U.S.A. 94, 5395–5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear M. F.; Huber K. M.; Warren S. T. (2004) The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377. [DOI] [PubMed] [Google Scholar]
- Huber K. M.; Kayser M. S.; Bear M. F. (2000) Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288, 1254–1257. [DOI] [PubMed] [Google Scholar]
- Nosyreva E. D.; Huber K. M. (2006) Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J. Neurophysiol. 95, 3291–3295. [DOI] [PubMed] [Google Scholar]
- Zalfa F.; Giorgi M.; Primerano B.; Moro A.; Di Penta A.; Reis S.; Oostra B.; Bagni C. (2003) The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell 112, 317–327. [DOI] [PubMed] [Google Scholar]
- Gross C.; Nakamoto M.; Yao X.; Chan C. B.; Yim S. Y.; Ye K.; Warren S. T.; Bassell G. J. (2010) Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J. Neurosci. 30, 10624–10638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan U.; Nalavadi V.; Nakamoto M.; Pallas D. C.; Ceman S.; Bassell G. J.; Warren S. T. (2007) FMRP phosphorylation reveals an immediate-early signaling pathway triggered by Group 1 mGluR and mediated by PP2A. J. Neurosci. 27, 14349–14357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan U.; Nalavadi V.; Nakamoto M.; Thomas G.; Ceman S.; Bassell G. J.; Warren S. T. (2008) S6K1 phosphorylates and regulates fragile X mental retardation protein (FMRP) with the neuronal protein synthesis-dependent mammalian target of rapamycine (mTOR) signaling cascade. J. Biol. Chem. 283, 18478–18482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakar A. L.; Dolen G.; Bear M. F. (2012) The pathophysiology of fragile X and what it teaches us about synapses. Annu. Rev. Neurosci. 34, 417–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride S. M.; Choi C. H.; Wang Y.; Liebelt D.; Braunstein E.; Ferreiro D.; Sehgal A.; Siwicki K. K.; Dockendorff T. C.; Nguyen H. T.; McDonald T. V.; Jongens T. A. (2005) Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron. 45, 753–764. [DOI] [PubMed] [Google Scholar]
- Pan L.; Woodruff E.; Liang P.; Broadie K. (2008) Mechanistic relationships between Drosophila fragile X mental retardation protein and metabotropic glutamate receptor A signaling. Mol. Cell. Neurosci. 37, 747–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis E. M.; Hessl D.; Coffey S.; Hervey C.; Schneider A.; Yuhas J.; Hutchinson J.; Snape M.; Tranfaglia M.; Nguyen D. V.; Hagerman R. (2009) A pilot open-label single-dose trial of fenobam in adults with fragile X syndrome. J. Med. Genet. 46, 266–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S.; Curie A.; des Portes V.; Torrioli M. G.; Berry-Kravis E. M.; Hagerman R. J.; Ramos F. J.; Cornish K.; He Y.; Paulding C.; Neri G.; Chen F.; Hadjikhani N.; Martinet D.; Meyer J.; Beckman J. S.; Delange K.; Brun A.; Bussy G.; Gasparini F.; Hilse T.; Floesser A.; Branson J.; Bilbe G.; Johns D.; Gomez-Mancilla B. (2011) Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci. Transl. Med. 3, 64ra1. [DOI] [PubMed] [Google Scholar]
- Mitchell P. B.; Hadzi-Pavlovic D. (2000) Lithium treatment for bipolar disorder. Bull. W. H. O. 78, 515–520. [PMC free article] [PubMed] [Google Scholar]
- Prasad V.; Sheard M. (1982) Effect of lithium upon desipramine enhanced shock-elicited fighting in rats. Pharmacol., Biochem. Behav. 17, 377–378. [DOI] [PubMed] [Google Scholar]
- Bersudsky Y.; Shaldubina A.; Belmaker R. H. (2007) Lithium’s effect in forced-swim test is blood level dependent but not dependent on weight loss. Behav. Pharmacol. 18, 77–80. [DOI] [PubMed] [Google Scholar]
- Gould T. D.; Einat H.; O’Donnell K. C.; Picchini A. M.; et al. (2007) Beta-catenin overexpression in the mouse brain phenocopies lithium-sensitive behaviors. Neuropsychopharmacology 32, 2173–2183. [DOI] [PubMed] [Google Scholar]
- Borison R.; Sabelli H.; Maple P.; Havdala H.; et al. (1978) Lithium prevention of amphetamine-induced ‘manic’ excitement and of reserpine-induced ’depression’ in mice: possible role of 2-phenylethylamine. Psychopharmacology (Berlin, Ger.) 59, 259–262. [DOI] [PubMed] [Google Scholar]
- Nocjar C.; Hammonds M. D.; Shim S. S. (2007) Chronic lithium treatment magnifies learning in rats. Neuroscience 150, 774–788. [DOI] [PubMed] [Google Scholar]
- Tsaltas E.; Kontis D.; Boulougouris V.; Papakosta V. M.; et al. (2007) Enhancing effects of chronic lithium on memory in the rat. Behav. Brain Res. 177, 51–60. [DOI] [PubMed] [Google Scholar]
- Vasconcellos A. P.; Tabajara A. S.; Ferrari C.; Rocha E.; et al. (2003) Effect of chronic stress on spatial memory in rats is attenuated by lithium treatment. Physiol. Behav. 79, 143–149. [DOI] [PubMed] [Google Scholar]
- Shim S. S.; Hammonds M. D.; Ganocy S. J.; Calabrese J. R. (2007) Effects of sub-chronic lithium treatment on synaptic plasticity in the dentate gyrus of rat hippocampal slices. Prog. Neuropsychopharmacol. Biol. Psychiatry 31, 343–347. [DOI] [PubMed] [Google Scholar]
- Lim K. Y.; Yang J. J.; Lee D. S.; Noh J. S.; et al. (2005) Lithium attenuates stress-induced impairment of long-term potentiation induction. NeuroReport 16, 1605–1608. [DOI] [PubMed] [Google Scholar]
- Rowe M. K.; Chuang D. M. (2004) Lithium neuroprotection: molecular mechanisms and clinical implications. Expert Rev. Mol. Med. 6, 1–18. [DOI] [PubMed] [Google Scholar]
- Berridge M. J.; Downes C. P.; Hanley M. R. (1989) Neural and developmental actions of lithium: a unifying hypothesis. Cell 59, 411–419. [DOI] [PubMed] [Google Scholar]
- Basselin M.; Chang L.; Bell J. M.; Rapoport S. I. (2006) Chronic lithium chloride administration attenuates brain NMDA receptor-initiated signaling via arachidonic acid in unanesthetized rats. Neuropsychopharmacology 31, 1659–1674. [DOI] [PubMed] [Google Scholar]
- Jope R. S.; Roh M. S. (2006) Glycogen synthase kinase-3 (GSK3) in psychiatric diseases and therapeutic interventions. Curr. Drug Targets 7, 1421–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein P. S.; Melton D. A. (1996) A molecular mechanism for the effects of lithium on development. Proc. Natl. Acad. Sci. U.S.A. 93, 8455–8459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockendorff T. C.; Su H. S.; McBride S. M.; Yang Z.; Choi C. H.; Siwicki K. K.; Sehgal A.; Jongens T. A. (2002) Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron 34, 973–984. [DOI] [PubMed] [Google Scholar]
- Morales J.; Hiesinger P. R.; Schroeder A. J.; Kume K.; Verstreken P.; Jackson F. R.; Nelson D. L.; Hassan B. A. (2002) Drosophila fragile X protein, DFXR, regulates neuronal morphology and function in the brain. Neuron 34, 961–972. [DOI] [PubMed] [Google Scholar]
- Bolduc F. V.; Bell K.; Cox H.; Braodie K. S.; Tully T. (2008) Excess protein synthesis in Drosophila fragile X mutants impairs long-term memory. Nat. Neurosci. 11, 1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc F. V.; Bell K.; Rosenfelt C. (2010) Fragile X mental retardation 1 and filamin A interact genetically in Drosophila long-term memory. Front. Neural Circuits 3, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A.; Li W.; Xu K.; Bogert B. A.; Su K.; Gao F. B. (2003) Control of dendritic development by the Drosophila fragile X-related gene involves the small GTPase Rac1. Development 130, 5543–5552. [DOI] [PubMed] [Google Scholar]
- Michel C. I.; Kraft R.; Restifo L. L. (2004) Defective neuronal development in the mushroom bodies of Drosophila fragile X mental retardation 1 mutants. J. Neurosci. 24, 5798–57809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S.; Bray S. M.; Li Z.; Zarnescu D. C.; He C.; Jin P.; Warren S. T. (2008) Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat. Chem. Biol. 4, 256–63. [DOI] [PubMed] [Google Scholar]
- Choi C. H.; Schoenfeld B. P.; Bell A. J.; Hinchey P.; Kollaros M.; Gertner M. J.; Woo N. H.; Tranfaglia M. R.; Bear M. F.; Zukin R. S.; McDonald T. V.; Jongens T. A.; McBride S. M. (2011) Pharmacological reversal of synaptic plasticity deficits in the mouse model of fragile X syndrome by group II mGluR antagonist or lithium treatment. Brain Res. 1380, 106–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min W. W.; Yuskaitis C. J.; Yan Q.; Sikorski C.; Chen S.; Jope R. S.; Bauchwitz R. P. (2009) Elevated glycogen synthase kinase-3 activity in Fragile X mice: Key metabolic regulator with evidence for treatment potential. Neuropharmacology 56, 463–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z. H.; Chuang D. M.; Smith C. B. (2011) Lithium ameliorates phenotypic deficits in a mouse model of fragile X syndrome. Int. J. Neuropsychopharmacol. 14, 618–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielsky I. F.; Hu S. B.; Szegda K. L.; Westphal H.; Young L. J. (2004) Profound impairment in social recognition and reduction in anxiety-like behavior in vasopressin V1a receptor knockout mice. Neuropsychopharmacology 29, 483–493. [DOI] [PubMed] [Google Scholar]
- Martens M. A.; Wilson S. J.; Reutens D. C. (2008) Research review: Williams syndrome: a critical review of the cognitive, behavioral, and neuroanatomical phenotype. J. Child Psychol. Psychiatry 49, 576–608. [DOI] [PubMed] [Google Scholar]
- Yuskaitis C. J.; Mines M. A.; King M. K.; Sweatt J. D.; Miller C. A.; Jope R. S. (2010) Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of fragile X syndrome. Biochem. Pharmacol. 79, 632–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeraragavan S.; Bui N.; Perkins J. R.; Yuva-Paylor L. A.; Paylor R. (2011) The modulation of fragile X behaviors by the muscarinic M4 antagonist, tropicamide. Behav. Neurosci. 125, 783–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachev P. (2006) Cognition and medial frontal cortex in health and disease. Curr. Opin. Neurol. 19, 586–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrell J. M.; Brown V. J. (2000) Medial frontal cortex mediates perceptual attentional set shifting in the rat. J. Neurosci. 20, 4320–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir J. L.; Everitt B. J.; Robbins T. W. (1996) The cerebral cortex of the rat and visual attentional function: dissociable effects of mediofrontal, cingulate, anterior dorsolateral, and parietal cortex lesions on a five-choice serial reaction time task. Cereb. Cortex 6, 470–481. [DOI] [PubMed] [Google Scholar]
- Grimes C. A.; Jope R. S. (2001) The multifaceted roles of glycogen synthase kinase 3β in cellular signaling. Prog. Neurobiol. 65, 391–426. [DOI] [PubMed] [Google Scholar]
- Woodgett J. R. (1990) Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 9, 2431–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin A. V.; King M. K.; Palomo V.; Martinez A.; McMahon L. L.; Jope R. S. (2014) Glycogen synthase kinase-3 inhibitors reverse deficits in long-term potentiation and cognition in fragile X mice. Biol. Psychiatry 75, 198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z. H.; Huang T.; Smith C. B. (2012) Lithium reverses increased rates of cerebral protein synthesis in a mouse model of fragile X syndrome. Neurobiol. Dis. 45, 1145–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A.; Hoeffer C. A.; Takayasu Y.; Miyawaki T.; McBride S. M.; Klann E.; Zukin R. S. (2010) Dysregulation of mTOR signaling in fragile X syndrome. J. Neurosci. 30, 694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis E. M.; Sumis A.; Hervey C.; Nelson M.; Porges S. W.; Weng N.; Weiler I. J.; Greenough W. T. (2008) Open-label treatment trial of lithium to target the underlying defect in fragile X syndrome. J. Dev. Behav. Pediatr. 29, 293–302. [DOI] [PubMed] [Google Scholar]