

Abstract
Traumatic brain injury (TBI) is a leading cause of disability and death from trauma to central nervous system (CNS) tissues. For patients who survive the initial injury, TBI can lead to neurodegeneration as well as cognitive and motor deficits, and is even a risk factor for the future development of neurodegenerative disorders such as Alzheimer’s disease. Preclinical studies of multiple neuropathological and neurodegenerative disorders have shown that lithium, which is primarily used to treat bipolar disorder, has considerable neuroprotective effects. Indeed, emerging evidence now suggests that lithium can also mitigate neurological deficits incurred from TBI. Lithium exerts neuroprotective effects and stimulates neurogenesis via multiple signaling pathways; it inhibits glycogen synthase kinase-3 (GSK-3), upregulates neurotrophins and growth factors (e.g., brain-derived neurotrophic factor (BDNF)), modulates inflammatory molecules, upregulates neuroprotective factors (e.g., B-cell lymphoma-2 (Bcl-2), heat shock protein 70 (HSP-70)), and concomitantly downregulates pro-apoptotic factors. In various experimental TBI paradigms, lithium has been shown to reduce neuronal death, microglial activation, cyclooxygenase-2 induction, amyloid-β (Aβ), and hyperphosphorylated tau levels, to preserve blood-brain barrier integrity, to mitigate neurological deficits and psychiatric disturbance, and to improve learning and memory outcome. Given that lithium exerts multiple therapeutic effects across an array of CNS disorders, including promising results in preclinical models of TBI, additional clinical research is clearly warranted to determine its therapeutic attributes for combating TBI. Here, we review lithium’s exciting potential in ameliorating physiological as well as cognitive deficits induced by TBI.
Keywords: Anti-inflammation, behavioral deficits and cognitive improvements, combined therapy treatment, controlled cortical impact, functional recovery, GSK-3 (glycogen synthase kinase-3) inhibitor, lithium, mood stabilizer, neuroprotection, neuroregeneration, preclinical model, TBI (traumatic brain injury)
Traumatic brain injury (TBI), defined as damage to brain tissue by mechanical force or blunt force trauma, is the primary cause of morbidity and death in young adults in industrialized countries.1 TBI is also recognized as the “signature wound” of soldiers engaged in the Iraq and Afghanistan wars; since 2001, over 200 000 United States military personnel have sustained TBI. In addition, TBI afflicts an average of 1.7 million people annually in the United States, underscoring its importance as a significant public health concern with long-term personal and economic consequences.2
Of continuing concern, no FDA-approved drug is presently available for intervention in TBI despite intensive research efforts over the past decades. This dearth of treatment options is at least partly due to the complex pathophysiology of TBI. Specifically, TBI comprises an initial primary injury that mechanically damages neurons, glia, and vascular structures, then is often followed by a cascade of secondary injuries whose onset occurs hours or days after the initial trauma.3−6 These secondary brain injuries, which are often reversible, may include oxidative stress via free radical formation and lipid peroxidation; excitotoxicity via excess glutamate release, increased N-methyl-d-aspartate (NMDA) receptor activation that may result in increased calcium ion influx; neuroinflammation via pro-inflammatory cytokines, nitric oxide, or prostaglandins; mitochondrial disruption involving increased poly[ADP-ribose] polymerase 1 (PARP-1) activation, decreased NAD+/ATP levels, increased calpain activation, and permeabilization of mitochondrial permeability transition pore (mPTP); failure of the blood-brain barrier involving cerebral edema, hypoxia, and ischemia; and cellular death via necrosis, caspase-dependent (caspase-3) apoptosis, and caspase-independent (apoptosis inducing factor) apoptosis.3,4,7 Sequelae such as neuropsychiatric disturbances (e.g., depression, anxiety, and posttraumatic stress disorder) and behavioral and cognitive deficits typically accompany secondary injuries (for a review, see ref (8)). This complex pathophysiology may therefore require pharmacological agents that act either alone or in concert on multiple cell survival and cell death pathways.9 Notably, over the past decade and a half, research has clearly shown that lithium, and perhaps valproate, may be able to act in this manner.
The mood stabilizers lithium and valproate have historically been used to treat bipolar disorder. Given that these mood stabilizers act across multiple pathways, it is not surprising that recent research has highlighted their putative use as neuroprotective agents in a variety of neurological and neurodegenerative disorders, including cerebral injury. Here, we present a brief overview of some of lithium’s known mechanisms of action, and then review its potential use as a therapeutic agent for the neurological deficits and damage caused by TBI.
Lithium: Mechanisms of Action
Evidence from Bipolar Disorder (BD)
For over 100 years lithium, has been used in the treatment of manic depression,10 and as the treatment of choice for BD for over 60 years. Gerrod described the use of lithium in 1859 for the treatment of rheumatic conditions and gout, based on the prevailing hypothesis of the time because of lithium’s ability to dissolve nitrogen-containing compounds such as uric acid which were believed to accumulate in tissues, thus resulting in illnesses. In the 1880s, Carl Lange and others used lithium in the treatment of BD, and by 1885 lithium carbonate and citrate had earned a place in the British Pharmacopoeia. The urea hypothesis was also used as rationale for the use of lithium in food stuffs and tonics, most notably with the introduction in 1929 of “Bib-Label Lithiated Lemon-Lime Soda”, later to be renamed “7 UP”. Due to the decline of the urea hypothesis and a lack of a credible therapeutic mechanism, the lithium was subsequently removed by 1950 (summarized in ref (11)).
Working with BD patients, John Cade rediscovered the use of lithium in 1949.12 Based on the hypothesis that BD was caused by toxins which were thought to be recoverable in urine, he identified urea as the most toxic compound, and reasoned that alkali metal injections could dissolve the toxin. He then found that lithium caused sedation and stupor in test animals, suggesting to him that it may have mood-control properties. Testing lithium on himself and then with BD, depressed, and schizophrenic patients, he found great improvements in the BD group (summarized in ref (11)).
In the treatment of BD, typically, lithium is effective in approximately one-third of patients, partially effective in another third, and not effective in the remainder; exciting new data are emerging suggesting that some forms of nonresponsiveness to lithium treatment may be due to variants in the promoter of the glutamate decarboxylase-like protein 1 (GADL1) gene.13 Lithium’s mechanisms of action in BD are numerous and complex, and it is beyond the scope of this article to review exhaustively. The interested reader is directed to several articles addressing this topic.14−21
One particularly intriguing, although debated, area of research in BD stems from the observation that gray matter increases are associated with lithium treatment.16 Magnetic resonance imaging of BD patients treated with lithium showed higher concentrations of N-acetyl-aspartate (NAA) (a marker of neuronal integrity) that is typically otherwise suppressed in BD.22,23 BD patients treated with lithium, however, displayed an increased number of neurons (greater volume with higher density of gray matter) relative to patients not treated with lithium and healthy controls.16 A voxel-based morphometric comparative study found that BD-I patients receiving lithium had significantly lower gray matter volumes in the left middle temporal gyrus and larger gray matter volumes in the left parahippocampal gyrus.24 Earlier studies comparing juvenile BD patients and healthy controls reported that untreated BD patients had lower amygdalar gray matter volumes compared to those treated with lithium or valproate.25 BD patients also exhibited decreased gray matter in amygdala and prefrontal cortex and hyper-intensity of white matter.26 Other studies of BD patients found that lithium treatment significantly increased cortical gray matter density, most notably in the cingulate and paralimbic cortices, regions necessary for attention, motivation, and emotional modulation. Gray matter density increase was also noted in the right anterior cingulate.27 Finally, a longitudinal brain imaging study noted that lithium-induced increases in gray matter volume correlated with treatment response in BD subjects.28 Despite these multiple, intriguing findings, a recent report suggested that lithium-induced gray matter increase could be artifactual and actually stem from changes in MRI signal characteristics.29 Other approaches such as quantification of synaptic density and related neuropil may help resolve this question.
Glycogen Synthase Kinase-3 (GSK-3) Inhibition and Transcription Factor Activation
GSK-3 exists in two structurally similar isoforms, α and β, which share 85% sequence homology.30 Although GSK-3α and GSK-3β are highly homologous within their kinase domains and display similar biochemical and substrate properties, their functional roles are not always identical. For example, the transcription factors early growth response 1 and Smad3/4 are oppositely affected by GSK-3α and GSK-3β.31 In addition, GSK-3β plays a more important role than GSK-3α in mediating spontaneous neuronal death in extended cultures.32
Lithium directly inhibits GSK-3 by binding to the magnesium-sensitive site of the enzyme,33,34 and indirectly inhibits GSK-3 by enhancing phosphorylation to specific serine residues. The first evidence that lithium indirectly inhibits GSK-3 came from studies noting its ability to act on the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) pathway and elevate phosphorylation of GSK-3α at Ser21.35 Lithium can also indirectly inhibit the GSK-3β isoform by phosphorylation at Ser9.36 It was further shown to increase GSK-3 serine phosphorylation by disrupting β-arrestin 2/protein phosphatase 2A/Akt complex formation, which dephosphorylates and inactivates Akt.37 Phosphorylation of GSK-3 by activating upstream Akt and protein kinases A or C (PKA, PKC) then can result in effects on diverse biological processes (for a review, see refs (15−19, 36, and 38−43)). Notably, GSK-3 can phosphorylate various nuclear factors such as Wnt/β-catenin and nuclear factor of activated T cells (NFAT) (for a review, see refs (16 and 39)). In addition, lithium by inhibiting GSK-3 can also activate the transcription factors activator protein-1 (AP-1) and cyclic AMP-response element binding protein (CREB),44 as well as the mitogen-activated protein (MAP) kinase pathway;45 for a review, see ref (17). GSK-3 also has the ability to promote self-activation through enhancing phosphatase activity which removes its own N-terminal inhibitory phosphate groups,46 as well as through stabilization of the Akt/βArr2/PP2A signaling complex which results in Akt dephosphorylation, as mentioned above.37,47 Lithium can block both of these mechanisms as part of its role in GSK-3 inhibition.48
Thus, in light of its central role in signaling, GSK-3 may be considered as a major way station in mediating lithium’s actions with effects in major systems such as anti-inflammation by suppressing IL-6 production and astrocyte activation;49 neuroprotection by stimulating antiapoptotic factors including CREB, HSF-1, NF-κB, and β-catenin and eventually B-cell lymphoma 2 (Bcl-2) (for a review, see refs (39 and 50)); and the alleviation of depressive behaviors in experimental animals.41,51−55 Indeed, lithium has even been shown to attenuate amphetamine-induced hyperlocomotion behavior via Akt kinase activity in a mouse model.56
Neuroprotection
Lithium has been shown to be highly neuroprotective in numerous in vivo and in vitro models, and to prevent neuronal apoptosis via multiple mechanisms (for a review, see refs (14−16 and 18)). Pioneering studies found that chronic (6–7 days) lithium treatment at therapeutic concentrations provided nearly complete protection against glutamate-induced excitotoxicity in primary cerebellar, cerebral cortical, and hippocampal neuronal cultures; these neuroprotective effects occurred independently of lithium-mediated inositol monophosphatase activity blockade, but were associated with downregulation of NMDA receptor-mediated calcium influx57,58 due to lithium-induced Src kinase inactivation and resulting inhibition of the NR2B subunit tyrosine phosphorylation.59 It is currently unknown whether this aspect of lithium’s action requires GSK-3 inhibition. These neuroprotective effects also stimulated cell survival factors, such as BDNF/TrkB,60 antiapoptotic Bcl-2, and heat shock protein 70 (HSP70) in a GSK-3 dependent manner.18 Concomitantly, lithium was found to inhibit p38 kinase, AP-1 DNA binding activity, and glutamate-induced JNK activity, as well as induce the loss of phosphorylated CREB by reducing protein phosphatase 1 and increasing MEK activities.61 This suggests that CREB and CRE-responsive genes such as Bcl-2 may be a mechanism for lithium’s effects on neuronal survival. Notably, lithium also inhibited pro-apoptotic molecules such as p53 and Bax;62 pretreatment with lithium inhibited glutamate-induced mitochondrial cytochrome c release, activation of caspse-3, as well as cleavage of lamin B1, which is the nuclear substrate for caspase-3, a critical enzyme that mediates apoptosis.
Lithium can also increase the activity of the antiapoptotic serine-threonine Akt-1 kinase and its upstream PI3K in primary cerebellar neurons.35 Cerebellar neurons cultured with serum respond with higher levels of phosphorylated/active Akt-1. The PI3K inhibitors wortmannin and LY294002 blocked Akt-1 activity causing neuronal death, and this neuronal apoptosis was prevented by chronic lithium exposure. PI3K activity as well as Akt-1 phosphorylation and kinase activity were rapidly, but transiently, increased by lithium, thus reversing glutamate-induced loss of Akt-1 activity and cell viability. Acute lithium exposure also protected cortical neurons via a PI3K-mediated increase in intracellular calcium through the phospholipase Cγ pathway.63 Furthermore, in cultured human neuroblastoma SH-SY5Y cells, lithium treatment ameliorated HIV-gp120-mediated toxicity via the PI3K/Akt pathway.64
In addition, GSK-3 inhibitors—including lithium—have been shown to control inflammatory conditions in both the peripheral and central nervous systems. For example, an in vitro study found that lithium significantly increased both pro-inflammatory (IFN-γ, TNF-α, IL-8) and anti-inflammatory immunoregulatory cytokines/compounds (IL-10, IL-1 receptor antagonist).65 With regard to pro-inflammation, lithium can increase TNF-α in Raw264.7 macrophages and modulate differential signaling depending on Toll-like receptor 2/3 stimulation.66 With regard to anti-inflammation, lithium pretreatment in an experimental autoimmune encephalomyelitis (EAE) mouse spinal cord study abolished the onset of EAE and markedly decreased demyelination, microglial activation, and leukocyte infiltration.67 Also, in a rodent TBI model, lithium robustly suppressed injury-induced associated microglia activation (see below).
As the above evidence highlights, the effects of lithium on cellular systems are indeed far reaching. An earlier microarray analysis using neuroblastoma SH-SY5Y documented that lithium elicited over 900 statistically significant gene expression changes and, in particular, the Six1 gene mediated lithium protection against staurosporine-induced apoptosis by blockade of caspase-3 activation.68 A recent microarray analysis found that lithium differentially regulated the expression of over 50 genes including basic transcription factors, transcription activators, cell signaling proteins, cell adhesion proteins, oncogenes and tumor suppressors, intracellular transducers, survival and death genes, and cyclins.69 In another recent microarray study using rat cerebellar granule cells and SH-SY5Y cells, lithium and valproate combination was found to be protective against glutamate excitotoxicity and the neuroprotection was associated with downregulation of a prominent microRNA, miR-34a.70
Neurotrophins and Growth Factors
The effects of lithium on neurotrophin expression are numerous, and may depend on brain state, region, and species (for a review, see ref (16)) Depression has been reported to reduce brain levels of BDNF,71,72 and lithium is known to increase BDNF levels in the brain.72−75 Lithium and valproate both selectively activated BDNF promoter IV of primary neurons76 and elevated BDNF levels in rat hippocampal, frontal, and temporal cortices,77,78 but not the mouse thalamus.79 Acute and long-term lithium treatment were both found to increase the BDNF receptor TrkB in the mouse anterior cingulate but not in the hippocampus, while acute lithium significantly reduced CREB phosphorylation, an important intracellular target of TrkB-mediated signaling.80 This decrease in CREB phosphorylation is in contrast to an increased phospho-CREB level reported previously in an in vivo and in vitro study following lithium treatment,44 and the discrepancy may stem from differences in the experimental conditions such as the duration of drug treatment and brain regions examined. These above-mentioned findings support the suggestion that BDNF release and receptor activation can be increased by lithium. One study even found that certain BDNF polymorphisms can predict response to lithium in patients with bipolar depression,81 and a clinical trial in patients with Alzheimer’s disease found that lithium increased serum BDNF levels, and that this was accompanied by reductions in cognitive impairment.82 Interestingly, lowered levels of BDNF have been reported in BD patients,83 and lithium treatment has also been found to restore and even increase these levels.84−86 Additionally, in a study using peripheral blood mononuclear cells, hypermethylation (which block transcription factor binding ultimately leading to gene silencing) of the BDNF promoter region was found in BD II patients, and lithium significantly reduced this DNA methylation.87 However, these findings are not without debate, as other studies found lithium to lower BDNF levels in BD patients.88 Together, the data support that BDNF induction and secretion could be one mechanism by which lithium exerts its neuroprotective effects.
Lithium also has been reported to increase nerve growth factor (NGF) in the rat frontal cortex, hippocampus, amygdala, and limbic forebrain.73,74,89−91 However, no effects were noted on NGF levels in the striatum, hypothalamus, or midbrain.90 Neurotrophin-3 (NT-3) levels in the serum and hippocampus of rats were elevated by lithium after amphetamine-induced mania regardless of whether lithium treatment was initiated before, simultaneously, or after amphetamine treatment;92 in contrast, valproate only increased NT-3 when given prior to amphetamine.
Vascular endothelial growth factor (VEGF) is a secreted dimeric protein that exerts its effects by binding to four receptors (VEGFR1–4) and activates intracellular signaling cascades via intracellular tyrosine kinase domains,93,94 thereby stimulating angiogenesis and vasculogenesis (for a review, see ref (95)), and promoting development, cell survival, proliferation, migration, and permeability.96,97 In this respect, VEGF acts as a direct neurotrophic/neuroprotective factor both in vivo and in vitro.98−101 Multiple reports have confirmed that TBI increases the expression of VEGF and its receptor,102,103 supporting angiogenesis and astrogliosis.104,105 In a mouse TBI model, lateral ventricle infusion of VEGF increased the number of astrocytes, oligodendroglia, and some neurons. Furthermore, lesion size was decreased, and significant improvement of functional outcome was observed.106−108
Recent studies using an ischemic paradigm in rats found that the mood stabilizer valproate could enhance angiogenesis and promote functional recovery by upregulating VEGF through hypoxia-inducible factor-1α (HIF-1α).109 Earlier preclinical models of TBI had found that lithium was neuroprotective against induced stroke in both rats110−112 and mice.113−115 Expanding on previous investigations which showed that lithium increased VEGF expression,116−118 an in vitro study under serum-deprivation conditions using human brain microvascular endothelial cells and primary rat cortical astrocytes found that lithium promoted VEGF through a PI3K/GSK-3β-dependent pathway in brain endothelium, but through a PI3K/GSK-3β-independent pathway in astrocytes.119 Together, these data support the notion that lithium’s neuroprotective effects, including those observed in mouse models of TBI (see below), may be at least partly mediated by VEGF, similar to the neuroprotective effects of valproate.
Fibroblast growth factor-21 (FGF-21), a new member of the FGF superfamily, is a recently discovered metabolic regulator involved in glucose and lipid metabolism.120 It targets the liver, pancreatic islets, and adipose tissue,121 and is a possible therapeutic target for diabetes and obesity. FGF-21 transcripts have also been detected in the human prefrontal cortex122 (BrainCloud, www.braincloud.jhmi.edu). Earlier studies found that cotreatment with lithium and valproate could synergistically enhance GSK-3 inhibition over that of lithium alone,123 as well as prevent glutamate excitotoxicity in aging primary neurons. In a very recent rat model study, mRNA microarray strategy and quantitative real time PCR (q-PCR) were used to demonstrate for the first time that FGF-21 is expressed in primary brain neurons, and that lithium increases FGF-21 mRNA and protein expression levels via GSK-3 inhibition.124 That study also found that FGF-21 is selectively and synergistically elevated by cotreatment with lithium and valproate via enhanced lithium-induced GSK-3 inhibition and Akt-1 activation, as well as valproate-induced histone deacetylase (HDAC) inhibition.124 In this proposed complex pathway, the lithium-valproate combination induces Akt-1 to act as an upstream regulator of FGF-21 expression as well as a downstream mediator for FGF-21-induced neuroprotection.124 Considered together, these novel results indicate that FGF-21 is an important mediator of the protective effects of these mood stabilizers, and holds great promise as a new therapeutic target for CNS disorders, including TBI and others with an underlying excitotoxic component.
Neurogenesis
Numerous preclinical studies have found that lithium induces neurogenesis (for a review, see ref (16)). In a rodent quinolinic acid-induced striatal injury model, short-term lithium exposure activated neurogenesis; the proliferating cells showed astroglial and neuronal phenotypes, suggesting that the newly formed cells were neural stem cells.125 In other studies, chronic lithium treatment increased neurogenesis in the hippocampus of the rat dentate gyrus;126,127 however, in older rats, it enhanced long-term potentiation but not neurogenesis.128 Lithium also selectively increased differentiation of hippocampal neural progenitor cells both in vitro and in vivo.129
Insight into the potential mechanisms by which lithium can induce neurogenesis has advanced considerably. It was noted that lithium could prevent stress-induced reductions in VEGF levels and this, as previously mentioned, can stimulate both neurogenesis and angiogenesis.118 In rats, chronic mild stress led to decreased hippocampal cell proliferation; this type of stress could increase GSK-3β mRNA expression and total GSK-3β protein expression.130 Lithium could block the negative effects of chronic-mild stress, suggesting that GSK-3β inhibition is involved in preventing the negative effects of stress on behavior and cellular processes. However, another study using chronic stress found that GSK-3β levels did not increase in mouse hippocampus, but rather that phosphorylated levels of GSK-3β decreased, thus indicating enhanced GSK-3β activity (see supplementary material in ref (54)). Other studies found that lithium increased proliferation of adult hippocampal progenitor cells that involved Wnt/β-catenin activation due to GSK-3β inhibition.131
Lithium at therapeutically relevant concentrations was shown to stimulate the proliferation of nestin-positive progenitor cells in cultured brain neurons.132 Using an in vitro adult rat spinal cord model, lithium was also found to enhance proliferation and neuronal production of neural progenitor cells after transplantation. Specifically, lithium treatment increased the number of labeled neurons derived from transplanted cells, thus increasing neurogenesis while reducing microglia and macrophage activation.133 Acting through the PI3K/Akt pathway as a “gain control”, lithium upregulated the signaling of the developmental gene Notch in hippocampal neurons as well as in primary T cells or Chinese hamster oöcytes.134 In addition, an earlier report showed that lithium was found to disinhibit Notch by phosphorylating GSK-3β, which prevents GSK-3β activation and the resulting Notch inhibition.135
Because of its multiple mechanisms of action and noted neuroprotective properties, lithium has been used to investigate the molecular neuroprotective mechanisms underlying Alzheimer’s disease, Parkinson’s disease (for a review, see refs (15, 16, and 43)), Down syndrome, spinocerebellar ataxia type 1 (SCA1) and 3 (SCA3), TBI, ischemic stroke, postoperative cognitive dysfunction (for a review, see refs (15, 43, and 50)), and Huntington’s disease, among others (for a review, see refs (15 and 16)). It has also been used in early clinical studies of cerebral malaria, HIV encephalitis, diabetes, fragile X syndrome, and amyotrophic lateral sclerosis (ALS) (for a review, see refs (15, 16, and 43)).
Inroads to the Future: Lithium in TBI
The devastating effects of TBI on those afflicted are further amplified by the fact that there are as yet no specified treatment options. However, there is hope for the future treatment of both the primary and secondary injuries associated with TBI. Lithium is already known to ameliorate edema by repairing blood-brain-barrier disruption (see below); prevent inflammation by inhibiting microglia activation and COX-2 induction; protect neurons by preventing excess NMDA receptor activation and calcium influx; stabilize mitochondria via a Bcl-2 dependent mechanism;136 reduce mitochondrial release of cytochrome c and apoptosis-inducing factor as well as reduce calpain and caspase-3 activation.137 Furthermore, lithium can reduce cognitive impairments by decreasing APP overexpression and Aβ accumulation, as well as reduce BACE1-injury induced upregulation and tau hyperphosphorylation (see below).
In cases of mild TBI in mice, 30 min of pretreatment with lithium alleviated depressive behavior measured 24 h postinjury.138 Mild TBI stimulated hippocampal Akt phosphorylation (hence activation) as well as phosphorylation at Ser9 of GSK-3β (hence inhibition) and accumulation of downstream β-catenin. This suggests that one of lithium’s effects is to activate this protective pathway, and that GSK-3β inhibition may be a therapeutic target in TBI treatment. In addition, in a mouse model using controlled cortical impact to produce moderate TBI, 14-day pretreatment with lithium attenuated IL-1β expression, brain edema, hippocampal neurodegeneration, and loss of hemispheric tissues, as well as improved memory and spatial learning.139
Most notably, postinsult injections with therapeutic doses of lithium similarly exerted a marked neuroprotective effect when administered three to 6 h after controlled cortical impact-induced TBI. Specifically, lithium reduced lesion volume when measured 3 days and 3 weeks after injury induction in mice.113 TBI-induced neuroinflammation detected by microglial activation and cyclooxygenase-2 (COX-2) induction were also reduced, while blood-brain barrier integrity was maintained through inhibition of matrix metallopeptidase-9 (MMP-9) expression. Behaviorally, TBI-induced hyperlocomotor activity, anxiety-like behavior, and impairments in motor coordination were all normalized. In addition, GSK-3β Ser9 phosphorylation was robustly increased, again suggesting that GSK-3β inhibition is involved in mediating lithium’s therapeutic effects.113 Another controlled cortical impact study using lithium starting 30 min postinjury for 5 days also found increased GSK-3β Ser9 phosphorylation with subsequent β-catenin accumulation and reduced neuronal loss in the hippocampal CA3 region, as well as decreased hippocampal-dependent deficits in learning and memory in the period 14–28 days after initial injury.140 Furthermore, a selective GSK-3 inhibitor, SB-216763, partially mimicked the behavioral benefits of lithium, lending further support to the notion that lithium protects against TBI via GSK-3 inhibition.140 A very recent study using a rat diffuse severe brain injury model showed that combination treatment with lithium and etanercept, a TNFα inhibitor, decreased TNFα and glial fibrillary acidic protein (GFAP) levels, and alleviated neuronal degeneration, edema, and axonal swelling.141
Increasing evidence suggests that TBI is a major risk factor in the eventual development of Alzheimer’s disease (for a review, see ref (142)). Animal models as well as patient populations afflicted by TBI show increased memory impairment.143 Elevated levels of amyloid-β (Aβ) have also been found in the cerebrospinal fluid144,145 and post-mortem brain samples of TBI patients.146,147 Interestingly, GSK-3 hyperactivity has been implicated in the pathogenesis of Alzheimer’s disease by regulating amyloid precursor protein (APP) cleavage and thus increasing Aβ production.148,149 Lithium as a GSK-3 inhibitor can counteract increased Aβ production and can therefore produce beneficial effects in various Alzheimer’s disease models (for a review, see refs (15−17)). In addition, previous research found that lithium could protect cultured PC12 cells from Aβ42 toxicity, a more toxic form of Aβ comprising 42 amino acids, possibly by upregulating Bcl-2.150 Indeed, reducing Aβ by inhibiting β- or γ-secretase enzyme processing of APP, or by modulating ATP binding cassette protein A1 (ABCA1, which enhances Aβ clearance), were found to have beneficial effects including protection of hippocampal tissue.151,152 These results suggest that postinsult lithium treatment may be a rationale strategy to alleviate Alzheimer’s-disease-like syndromes associated with TBI.
In recent studies testing the above-mentioned hypothesis using a mouse controlled cortical impact TBI model, 1.5 mEq/kg lithium i.p. given 15 min post injury and then once daily thereafter robustly reduced TBI-induced APP increases, β-APP-cleaving enzyme-1 (BACE1) overexpression, Aβ load in the hippocampus and corpus callosum, tau hyperphosphorylation in the thalamus, and significantly preserved brain tissues.114 One group reported that a 27-kDa Aβ oligomer, which had previously been shown to be toxic,153 was robustly elevated by TBI, and this increase was attenuated by lithium.114 APP and Aβ42 levels were also elevated in the hippocampus by TBI, as well as in axonal bulbs of the corpus callosum; again, lithium significantly reduced these increases. Most notably, Y-maze and Morris water maze tests showed that lithium mitigated memory and spatial learning deficits induced by TBI, which were associated with suppression of TBI-induced loss of hippocampal volume.114 Moreover, lithium-induced downregulation of APP accumulation, Aβ generation, and tau hyperphosphorylation through GSK-3 inhibition has been reported in a transgenic mouse model of Alzheimer’s disease,154 and GSK-3 inhibitor-mediated inhibition of BACE1 activity was also reported.155 This evidence readily supports the notion that lithium, by reducing levels and processing of APP as well as levels of tau phosphorylation (most likely via GSK-3 inhibition) can reduce TBI-induced Aβ load and tau hyperphosphorylation and mitigate the resulting negative downstream effects, notably learning and memory deficits.113,114,140 Figure 1 illustrates the proposed pathophysiological events induced by lithium after TBI.
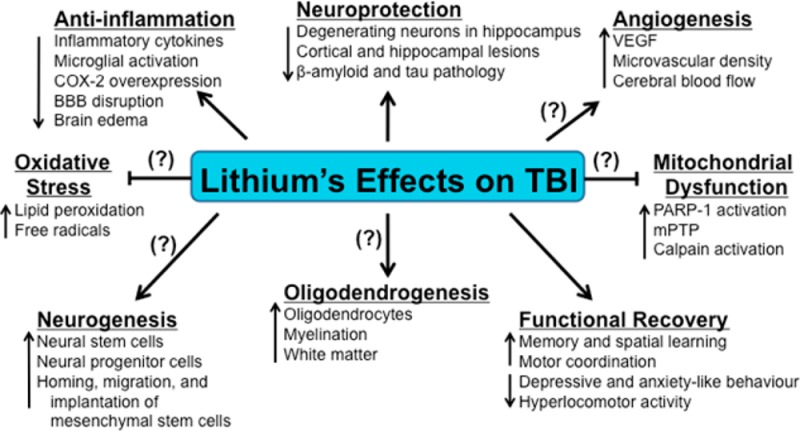
Figure 1.
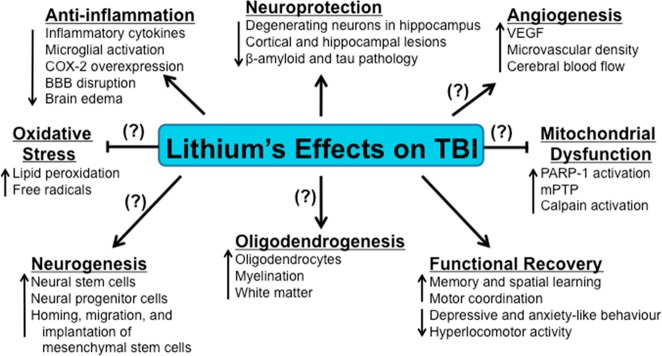
Lithium intervention in traumatic brain injury (TBI). Following TBI, a complex series of pathophysiological events are triggered that ultimately induce neurodegeneration and loss of neurological function. Preclinical studies on TBI have demonstrated that the mood stabilizing drug lithium exerts numerous beneficial effects on neuroinflammation, neuronal protection, and functional recovery. The neuroprotective effects of this novel pharmacological strategy on the modulation of TBI-induced oxidative stress, mitochondrial dysfunction, oligodendrogenesis, neurogenesis, as well as the ability to promote angiogenesis have not been fully determined. Arrows indicate increases or decreases in the respective effects due to lithium-induced actions on each pathophysiological category. Blocked ends indicate lithium-mediated prevention of these pathophysiological events. Question marks (?) indicate known effects of lithium, but not yet verified in TBI models.
In a very recent expansion of these experiments, subeffective doses of combined lithium and valproate were more useful in mitigating TBI injury than either agent used alone.115 Previous studies had found that cotreatment with lithium and valproate could synergistically enhance GSK-3 inhibition over that of lithium alone, and prevent glutamate excitotoxicity in aging primary neurons.123 Previous work also found that epigenetic regulation via HDAC inhibition, which restores histone acetylation levels and corrects transcriptional deficits, is neuroprotective in TBI and other brain disorder models, including maintenance of blood-brain barrier integrity, reduction of contusion volume, and improvement of spatial memory and motor function.156−161 Lithium (1 mEq/kg) and valproate (200 mg/kg) cotreatment was administered i.p. 15 min after TBI, and then once daily thereafter. At 3 days postinjury, cotreatment significantly reduced lesion volume, attenuated blood-brain barrier disruption, mitigated hippocampal neurodegeneration, and robustly increased histone H3 acetylation (an index of HDAC inhibition). Additionally, starting from seven to at least 21 days postinjury, motor coordination was improved as measured by the decrease in number of foot faults in a beam-walk test. As expected, treatment with either of these mood stabilizers alone at subeffective doses had no benefit against TBI.115 These findings are the first experimental evidence showing that cotreatment with subeffective doses of lithium and valproate can significantly reduce brain tissue damage and protect blood-brain barrier integrity from TBI. These benefits are associated with a synergistic increase in acetylated-histone H3 levels, suggesting a cross-talk between GSK-3 and HDAC inhibition. This pharmacological strategy may be a promising route for therapy in that lower doses can avoid undesirable side effects, particularly kidney and thyroid toxicity from lithium, and liver toxicity from valproate.15 In light of the possibility of inducing lithium toxicity, routine monitoring of blood levels should still be done to avoid unwanted side effects.
It should be noted that previous studies have also documented that combining lithium and valproate treatment provided more beneficial effects with regard to preventing excitotoxicity of neuronal cultures, and improved outcome in mouse models of amyotrophic lateral sclerosis (ALS) and Huntington’s disease than monotreatment with either drug.123,162,163 Moreover, it was found that in a model of rat brain ischemia, copriming mesenchymal stem cells (MSC) with these two mood stabilizers increased homing and migration of MSCs to the lesion site and improved functional outcome.164,165 Together, these preclinical studies suggest that combined therapy with lithium and valproate may be a rational strategy for ameliorating the neuropathology associated with CNS disorders including TBI.
Lithium’s Effects on Glia
Much of the research into the protective and regenerative properties of lithium has focused on neurons; in contrast, limited research has been conducted into lithium’s effects on white matter, which also plays an important part in TBI-related pathophysiology (for a review, see ref (166)). Very early post-mortem studies of patients afflicted by a demented state after experiencing TBI showed diffuse degeneration of cerebral white matter,167 and more modern studies using MRI showed white matter decrease after mild TBI.168 Recent studies using immunofluorescence staining have shown that increases in white matter microglia/macrophages correlate with the severity of TBI injury.169 Another study using diffusion tensor imaging in a rat model found that repetitive TBI significantly increased microstructural damage to white matter.170 Moreover, a review of imaging studies found that after the initial and secondary injury periods, pathological changes showed greater disruption to white matter neural connectivity.171 Studies that administered clinical doses of lithium in a rat spinal cord injury model found that it resulted in significant corticospinal and serotonergic axon sprouting and promoted locomoter functional recovery.172 Interestingly, patients suffering from BD may show white matter disruption, and a diffusion tensor imaging study of individuals with BD found that lithium improved white matter structure and functional integrity in multiple brain areas.173 In addition, a rat ischemic model using delayed (24 h postinjury) valproate treatment found increased oligodendrocyte survival as well as an increased number of newly generated oligodendrocytes.174 These model systems demonstrated the efficacy of using lithium and valproate to rescue, repair, and even regenerate white matter. Future investigations of lithium in white matter protection after TBI are clearly warranted.
Conclusion
In summary, recent preliminary data using lithium have demonstrated robust beneficial effects in experimental models of TBI. These include decreases in TBI-induced brain lesion, suppression of neuroinflammation, protection against blood-brain barrier disruption, normalization of behavioral deficits, and improvement of learning and memory, among others. These exciting findings raise the possibility that the mood stabilizers lithium and valproate, either alone or in combination, may be repurposed as drugs that can be used in clinical settings to treat multiple categories of neuropathological, neurodegenerative disorders including those with an excitotoxic component, such as acute ischemia and TBI. In addition, because of their long history of safe clinical use, and the recently discovered synergistic effects stemming from cotreatment, thus requiring lower doses that, in turn, would result in fewer side effects and improved tolerability, future investigation is clearly warranted both in the laboratory and in clinical settings.
Acknowledgments
The authors thank Ioline Henter (NIMH, NIH) for critical review and editorial assistance with the manuscript. We also wish to thank the many collaborators involved in the studies discussed in this report.
Glossary
Abbreviations
- BBB
blood-brain barrier
- COX-2
cyclooxygenase-2
- mPTP
mitochondrial permeability transition pore
- PARP-1
poly[ADP-ribose] polymerase 1
- VEGF
vascular endothelial growth factor
Author Contributions
D.-M.C. designed the review and directed its implementation. P.R.L. conducted literature search and drafting. G.R.L. drafted the figure. All authors read, edited, and approved the final manuscript.
This work was supported by the Intramural Research Program of the National Institute of Mental Health, National Institutes of Health, Department of Health and Human Services (IRP-NIMH-NIH-DHHS), and the Department of Defense in the Center for Neuroscience and Regenerative Medicine (CNRM).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Maas A. I.; Stocchetti N.; Bullock R. (2008) Moderate and severe traumatic brain injury in adults. Lancet Neurol. 7(8), 728–741. [DOI] [PubMed] [Google Scholar]
- Faul M., Xu L., Wald M. M., and Coronado V. G. (2010) Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006, Centers for Disease Control and Prevention, National Center for Injury Prevention and Control, Atlanta, GA. [Google Scholar]
- Loane D. J.; Faden A. I. (2010) Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol. Sci. 31(12), 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Algattas H.; Huang J. H. (2014) Traumatic brain injury pathophysiology and treatments: early, intermediate, and late phases post-injury. Int. J. Mol. Sci. 15(1), 309–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabadi S. V.; Faden A. I. (2014) Neuroprotective strategies for traumatic brain injury: improving clinical translation. Int. J. Mol. Sci. 15(1), 1216–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blennow K.; Hardy J.; Zetterberg H. (2012) The neuropathology and neurobiology of traumatic brain injury. Neuron 76(5), 886–899. [DOI] [PubMed] [Google Scholar]
- McIntosh T. K.; Smith D. H.; Meaney D. F.; Kotapka M. J.; Gennarelli T. A.; Graham D. I. (1996) Neuropathological sequelae of traumatic brain injury: relationship to neurochemical and biomechanical mechanisms. Lab. Invest. 74(2), 315–342. [PubMed] [Google Scholar]
- Ursano R. J.; Goldenberg M.; Zhang L.; Carlton J.; Fullerton C. S.; Li H.; Johnson L.; Benedek D. (2010) Posttraumatic stress disorder and traumatic stress: from bench to bedside, from war to disaster. Ann. N.Y. Acad. Sci. 1208, 72–81. [DOI] [PubMed] [Google Scholar]
- Margulies S.; Hicks R. (2009) Combination Therapies for Traumatic Brain Injury Workshop, L. Combination therapies for traumatic brain injury: prospective considerations. J. Neurotrauma 26(6), 925–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacheron-Trystram M. N.; Braitman A.; Cheref S.; Auffray L. (2004) Antipsychotics in bipolar disorders. L’Encephale 30(5), 417–424. [DOI] [PubMed] [Google Scholar]
- Williams R. S. B., and Harwood A. J. (2005) 3Li lithium metallotherapeutics. In Metallotherapeutic Drugs and Metal-Based Diagnostic Agents: The Use of Metals in Medicine (Gielen M., and Teikink E. R. T., Eds.), pp 1–18, John Wieley & Sons Ltd, Chichester, West Sussex, UK. [Google Scholar]
- Cade J. F. (1949) Lithium salts in the treatment of psychotic excitement. Med. J. Aust. 2(10), 349–352. [DOI] [PubMed] [Google Scholar]
- Chen C. H.; Lee C. S.; Lee M. T.; Ouyang W. C.; Chen C. C.; Chong M. Y.; Wu J. Y.; Tan H. K.; Lee Y. C.; Chuo L. J.; Chiu N. Y.; Tsang H. Y.; Chang T. J.; Lung F. W.; Chiu C. H.; Chang C. H.; Chen Y. S.; Hou Y. M.; Chen C. C.; Lai T. J.; Tung C. L.; Chen C. Y.; Lane H. Y.; Su T. P.; Feng J.; Lin J. J.; Chang C. J.; Teng P. R.; Liu C. Y.; Chen C. K.; Liu I. C.; Chen J. J.; Lu T.; Fan C. C.; Wu C. K.; Li C. F.; Wang K. H.; Wu L. S.; Peng H. L.; Chang C. P.; Lu L. S.; Chen Y. T.; Cheng A. T.; Taiwan Bipolar C. (2014) Variant GADL1 and response to lithium therapy in bipolar I disorder. New Engl. J. Med. 370(2), 119–128. [DOI] [PubMed] [Google Scholar]
- Chuang D. M. (2004) Neuroprotective and neurotrophic actions of the mood stabilizer lithium: can it be used to treat neurodegenerative diseases?. Crit. Rev. Neurobiol. 16(1–2), 83–90. [DOI] [PubMed] [Google Scholar]
- Chiu C. T.; Wang Z.; Hunsberger J. G.; Chuang D. M. (2013) Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder. Pharmacol. Rev. 65(1), 105–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young W. (2009) Review of lithium effects on brain and blood. Cell Transplant. 18(9), 951–975. [DOI] [PubMed] [Google Scholar]
- Chiu C. T.; Chuang D. M. (2010) Molecular actions and therapeutic potential of lithium in preclinical and clinical studies of CNS disorders. Pharmacol. Ther. 128(2), 281–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang D. M.; Wang Z.; Chiu C. T. (2011) GSK-3 as a Target for Lithium-Induced Neuroprotection Against Excitotoxicity in Neuronal Cultures and Animal Models of Ischemic Stroke. Front. Mol. Neurosci. 4, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jope R. S.; Roh M. S. (2006) Glycogen synthase kinase-3 (GSK3) in psychiatric diseases and therapeutic interventions. Curr. Drug Targets 7(11), 1421–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhi G. S.; Tanious M.; Das P.; Coulston C. M.; Berk M. (2013) Potential mechanisms of action of lithium in bipolar disorder. Current understanding. CNS Drugs 27(2), 135–153. [DOI] [PubMed] [Google Scholar]
- Dodd S.; Maes M.; Anderson G.; Dean O. M.; Moylan S.; Berk M. (2013) Putative neuroprotective agents in neuropsychiatric disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 42, 135–145. [DOI] [PubMed] [Google Scholar]
- Moore G. J.; Bebchuk J. M.; Hasanat K.; Chen G.; Seraji-Bozorgzad N.; Wilds I. B.; Faulk M. W.; Koch S.; Glitz D. A.; Jolkovsky L.; Manji H. K. (2000) Lithium increases N-acetyl-aspartate in the human brain: in vivo evidence in support of bcl-2′s neurotrophic effects?. Biol. Psychiatry 48(1), 1–8. [DOI] [PubMed] [Google Scholar]
- Silverstone P. H.; Wu R. H.; O’Donnell T.; Ulrich M.; Asghar S. J.; Hanstock C. C. (2003) Chronic treatment with lithium, but not sodium valproate, increases cortical N-acetyl-aspartate concentrations in euthymic bipolar patients. Int. Clin. Psychopharmacol. 18(2), 73–79. [DOI] [PubMed] [Google Scholar]
- Chen X.; Wen W.; Malhi G. S.; Ivanovski B.; Sachdev P. S. (2007) Regional gray matter changes in bipolar disorder: a voxel-based morphometric study. Aust. N. Z. J. Psychiatry 41(4), 327–336. [DOI] [PubMed] [Google Scholar]
- Chang K.; Karchemskiy A.; Barnea-Goraly N.; Garrett A.; Simeonova D. I.; Reiss A. (2005) Reduced amygdalar gray matter volume in familial pediatric bipolar disorder. J. Am. Acad. Child Adolesc. Psychiatry 44(6), 565–573. [DOI] [PubMed] [Google Scholar]
- Monkul E. S.; Malhi G. S.; Soares J. C. (2005) Anatomical MRI abnormalities in bipolar disorder: do they exist and do they progress?. Aust. N. Z. J. Psychiatry 39(4), 222–226. [DOI] [PubMed] [Google Scholar]
- Bearden C. E.; Thompson P. M.; Dalwani M.; Hayashi K. M.; Lee A. D.; Nicoletti M.; Trakhtenbroit M.; Glahn D. C.; Brambilla P.; Sassi R. B.; Mallinger A. G.; Frank E.; Kupfer D. J.; Soares J. C. (2007) Greater cortical gray matter density in lithium-treated patients with bipolar disorder. Biol. Psychiatry 62(1), 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyoo I. K.; Dager S. R.; Kim J. E.; Yoon S. J.; Friedman S. D.; Dunner D. L.; Renshaw P. F. (2010) Lithium-induced gray matter volume increase as a neural correlate of treatment response in bipolar disorder: a longitudinal brain imaging study. Neuropsychopharmacology 35(8), 1743–5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousins D. A.; Aribisala B.; Nicol Ferrier I.; Blamire A. M. (2013) Lithium, gray matter, and magnetic resonance imaging signal. Biol. Psychiatry 73(7), 652–657. [DOI] [PubMed] [Google Scholar]
- Woodgett J. R. (1990) Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 9(8), 2431–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang M. H.; Chuang D. M. (2006) Differential roles of glycogen synthase kinase-3 isoforms in the regulation of transcriptional activation. J. Biol. Chem. 281(41), 30479–30484. [DOI] [PubMed] [Google Scholar]
- Liang M. H.; Chuang D. M. (2007) Regulation and function of glycogen synthase kinase-3 isoforms in neuronal survival. J. Biol. Chem. 282(6), 3904–3917. [DOI] [PubMed] [Google Scholar]
- Klein P. S.; Melton D. A. (1996) A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. U.S.A. 93(16), 8455–8459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic V.; Ruel L.; Woodgett J. R. (1996) Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr. Biol. 6(12), 1664–1668. [DOI] [PubMed] [Google Scholar]
- Chalecka-Franaszek E.; Chuang D. M. (1999) Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc. Natl. Acad. Sci. U.S.A. 96(15), 8745–8750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jope R. S. (2003) Lithium and GSK-3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol. Sci. 24(9), 441–443. [DOI] [PubMed] [Google Scholar]
- Beaulieu J. M.; Marion S.; Rodriguiz R. M.; Medvedev I. O.; Sotnikova T. D.; Ghisi V.; Wetsel W. C.; Lefkowitz R. J.; Gainetdinov R. R.; Caron M. G. (2008) A beta-arrestin 2 signaling complex mediates lithium action on behavior. Cell 132(1), 125–316. [DOI] [PubMed] [Google Scholar]
- Meijer L.; Flajolet M.; Greengard P. (2004) Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol. Sci. 25(9), 471–480. [DOI] [PubMed] [Google Scholar]
- Rowe M. K.; Chuang D. M. (2004) Lithium neuroprotection: molecular mechanisms and clinical implications. Expert Rev. Mol. Med. 6(21), 1–18. [DOI] [PubMed] [Google Scholar]
- Huang H. C.; Klein P. S. (2006) Multiple roles for glycogen synthase kinase-3 as a drug target in Alzheimer’s disease. Current Drug Targets 7(11), 1389–1397. [DOI] [PubMed] [Google Scholar]
- Rowe M. K.; Wiest C.; Chuang D. M. (2007) GSK-3 is a viable potential target for therapeutic intervention in bipolar disorder. Neurosci. Biobehav. Rev. 31(6), 920–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Jope R. S. (2010) Is glycogen synthase kinase-3 a central modulator in mood regulation?. Neuropsychopharmacology 35(11), 2143–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King M. K.; Pardo M.; Cheng Y.; Downey K.; Jope R. S.; Beurel E. (2014) Glycogen synthase kinase-3 inhibitors: Rescuers of cognitive impairments. Pharmacol. Ther. 141(1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki N.; Chuang D. M. (1997) Lithium increases transcription factor binding to AP-1 and cyclic AMP-responsive element in cultured neurons and rat brain. J. Neurochem. 69(6), 2336–2344. [DOI] [PubMed] [Google Scholar]
- Chen R. W.; Qin Z. H.; Ren M.; Kanai H.; Chalecka-Franaszek E.; Leeds P.; Chuang D. M. (2003) Regulation of c-Jun N-terminal kinase, p38 kinase and AP-1 DNA binding in cultured brain neurons: roles in glutamate excitotoxicity and lithium neuroprotection. J. Neurochem. 84(3), 566–575. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Phiel C. J.; Spece L.; Gurvich N.; Klein P. S. (2003) Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J. Biol. Chem. 278(35), 33067–33077. [DOI] [PubMed] [Google Scholar]
- O’Brien W. T.; Huang J.; Buccafusca R.; Garskof J.; Valvezan A. J.; Berry G. T.; Klein P. S. (2011) Glycogen synthase kinase-3 is essential for beta-arrestin-2 complex formation and lithium-sensitive behaviors in mice. J. Clin. Invest. 121(9), 3756–3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freland L.; Beaulieu J. M. (2012) Inhibition of GSK3 by lithium, from single molecules to signaling networks. Front. Mol. Neurosci. 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurel E.; Jope R. S. (2009) Lipopolysaccharide-induced interleukin-6 production is controlled by glycogen synthase kinase-3 and STAT3 in the brain. J. Neuroinflammation 6, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. F.; Fessler E. B.; Chuang D. M. (2011) Beneficial effects of mood stabilizers lithium, valproate and lamotrigine in experimental stroke models. Acta Pharmacol. Sinica 32(12), 1433–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu J. M.; Zhang X.; Rodriguiz R. M.; Sotnikova T. D.; Cools M. J.; Wetsel W. C.; Gainetdinov R. R.; Caron M. G. (2008) Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc. Natl. Acad. Sci. U.S.A. 105(4), 1333–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould T. D.; Chen G.; Manji H. K. (2004) In vivo evidence in the brain for lithium inhibition of glycogen synthase kinase-3. Neuropsychopharmacolog 29(1), 32–38. [DOI] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O.; Milman A.; Weizman A.; Pick C. G.; Eldar-Finkelman H. (2004) Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on beta-catenin in mouse hippocampus. Biol. Psychiatry 55(8), 781–784. [DOI] [PubMed] [Google Scholar]
- Omata N.; Chiu C. T.; Moya P. R.; Leng Y.; Wang Z.; Hunsberger J. G.; Leeds P.; Chuang D. M. (2011) Lentivirally mediated GSK-3beta silencing in the hippocampal dentate gyrus induces antidepressant-like effects in stressed mice. Int. J. Neuropsychopharmacol. 14(5), 711–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa A. O.; Kaster M. P.; Binfare R. W.; Morales S.; Martin-Aparicio E.; Navarro-Rico M. L.; Martinez A.; Medina M.; Garcia A. G.; Lopez M. G.; Rodrigues A. L. (2008) Antidepressant-like effect of the novel thiadiazolidinone NP031115 in mice. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 32(6), 1549–1556. [DOI] [PubMed] [Google Scholar]
- Pan J. Q.; Lewis M. C.; Ketterman J. K.; Clore E. L.; Riley M.; Richards K. R.; Berry-Scott E.; Liu X.; Wagner F. F.; Holson E. B.; Neve R. L.; Biechele T. L.; Moon R. T.; Scolnick E. M.; Petryshen T. L.; Haggarty S. J. (2011) AKT kinase activity is required for lithium to modulate mood-related behaviors in mice. Neuropsychopharmacology 36(7), 1397–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka S.; Hough C. J.; Chuang D. M. (1998) Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-D-aspartate receptor-mediated calcium influx. Proc. Natl. Acad. Sci. U.S.A. 95(5), 2642–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto R.; Hough C.; Nakazawa T.; Yamamoto T.; Chuang D. M. (2002) Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J. Neurochem. 80(4), 589–597. [DOI] [PubMed] [Google Scholar]
- Hashimoto R.; Fujimaki K.; Jeong M. R.; Christ L.; Chuang D. M. (2003) Lithium-induced inhibition of Src tyrosine kinase in rat cerebral cortical neurons: a role in neuroprotection against N-methyl-d-aspartate receptor-mediated excitotoxicity. FEBS Lett. 538(1–3), 145–148. [DOI] [PubMed] [Google Scholar]
- Hashimoto R.; Takei N.; Shimazu K.; Christ L.; Lu B.; Chuang D. M. (2002) Lithium induces brain-derived neurotrophic factor and activates TrkB in rodent cortical neurons: an essential step for neuroprotection against glutamate excitotoxicity. Neuropharmacology 43(7), 1173–1179. [DOI] [PubMed] [Google Scholar]
- Kopnisky K. L.; Chalecka-Franaszek E.; Gonzalez-Zulueta M.; Chuang D. M. (2003) Chronic lithium treatment antagonizes glutamate-induced decrease of phosphorylated CREB in neurons via reducing protein phosphatase 1 and increasing MEK activities. Neuroscience 116(2), 425–435. [DOI] [PubMed] [Google Scholar]
- Chen R. W.; Chuang D. M. (1999) Long term lithium treatment suppresses p53 and Bax expression but increases Bcl-2 expression. A prominent role in neuroprotection against excitotoxicity. J. Biol. Chem. 274(10), 6039–6042. [DOI] [PubMed] [Google Scholar]
- Kang H. J.; Noh J. S.; Bae Y. S.; Gwag B. J. (2003) Calcium-dependent prevention of neuronal apoptosis by lithium ion: essential role of phosphoinositide 3-kinase and phospholipase Cgamma. Mol. Pharmacol. 64(2), 228–234. [DOI] [PubMed] [Google Scholar]
- Everall I. P.; Bell C.; Mallory M.; Langford D.; Adame A.; Rockestein E.; Masliah E. (2002) Lithium ameliorates HIV-gp120-mediated neurotoxicity. Mol. Cell. Neurosci. 21(3), 493–501. [DOI] [PubMed] [Google Scholar]
- Maes M.; Song C.; Lin A. H.; Pioli R.; Kenis G.; Kubera M.; Bosmans E. (1999) In vitro immunoregulatory effects of lithium in healthy volunteers. Psychopharmacology 143(4), 401–407. [DOI] [PubMed] [Google Scholar]
- Hull M.; Lee E.; Lee T.; Anand N.; LaLone V.; Parameswaran N. (2014) Lithium chloride induces TNFalpha in mouse macrophages via MEK-ERK-dependent pathway. J. Cell. Biochem. 115(1), 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sarno P.; Axtell R. C.; Raman C.; Roth K. A.; Alessi D. R.; Jope R. S. (2008) Lithium prevents and ameliorates experimental autoimmune encephalomyelitis. J. Immunol. 181(1), 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant K. E.; Anderson E.; Simecek N.; Brown R.; Forster S.; Spinks J.; Toms N.; Gibson G. G.; Lyon J.; Plant N. (2009) The neuroprotective action of the mood stabilizing drugs lithium chloride and sodium valproate is mediated through the up-regulation of the homeodomain protein Six1. Toxicol. Appl. Pharmacol. 235(1), 124–134. [DOI] [PubMed] [Google Scholar]
- Farah R.; Khamisy-Farah R.; Amit T.; Youdim M. B.; Arraf Z. (2013) Lithium’s gene expression profile, relevance to neuroprotection A cDNA microarray study. Cell. Mol. Neurobiol. 33(3), 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsberger J. G.; Fessler E. B.; Chibane F. L.; Leng Y.; Maric D.; Elkahloun A. G.; Chuang D. M. (2013) Mood stabilizer-regulated miRNAs in neuropsychiatric and neurodegenerative diseases: identifying associations and functions. Am. J. Transl. Res. 5(4), 450–464. [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K.; Shimizu E.; Iyo M. (2004) Critical role of brain-derived neurotrophic factor in mood disorders. Brain Res. Rev. 45(2), 104–114. [DOI] [PubMed] [Google Scholar]
- Post R. M. (2007) Role of BDNF in bipolar and unipolar disorder: clinical and theoretical implications. J. Psychiatr. Res. 41(12), 979–990. [DOI] [PubMed] [Google Scholar]
- Angelucci F.; Aloe L.; Jimenez-Vasquez P.; Mathe A. A. (2003) Lithium treatment alters brain concentrations of nerve growth factor, brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor in a rat model of depression. Int. J. Neuropsychopharmacol. 6(3), 225–231. [DOI] [PubMed] [Google Scholar]
- Angelucci F.; Mathe A. A.; Aloe L. (2004) Neurotrophic factors and CNS disorders: findings in rodent models of depression and schizophrenia. Prog. Brain Res. 146, 151–165. [DOI] [PubMed] [Google Scholar]
- Gama C. S.; Andreazza A. C.; Kunz M.; Berk M.; Belmonte-de-Abreu P. S.; Kapczinski F. (2007) Serum levels of brain-derived neurotrophic factor in patients with schizophrenia and bipolar disorder. Neurosci. Lett. 420(1), 45–48. [DOI] [PubMed] [Google Scholar]
- Yasuda S.; Liang M. H.; Marinova Z.; Yahyavi A.; Chuang D. M. (2009) The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol. Psychiatry 14(1), 51–59. [DOI] [PubMed] [Google Scholar]
- Frey B. N.; Andreazza A. C.; Cereser K. M.; Martins M. R.; Valvassori S. S.; Reus G. Z.; Quevedo J.; Kapczinski F. (2006) Effects of mood stabilizers on hippocampus BDNF levels in an animal model of mania. Life Sci. 79(3), 281–286. [DOI] [PubMed] [Google Scholar]
- Fukumoto T.; Morinobu S.; Okamoto Y.; Kagaya A.; Yamawaki S. (2001) Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brain. Psychopharmacology 158(1), 100–106. [DOI] [PubMed] [Google Scholar]
- Omata N.; Murata T.; Takamatsu S.; Maruoka N.; Mitsuya H.; Yonekura Y.; Fujibayashi Y.; Wada Y. (2008) Neuroprotective effect of chronic lithium treatment against hypoxia in specific brain regions with upregulation of cAMP response element binding protein and brain-derived neurotrophic factor but not nerve growth factor: comparison with acute lithium treatment. Bipolar Disord. 10(3), 360–368. [DOI] [PubMed] [Google Scholar]
- Rantamaki T.; Knuuttila J. E.; Hokkanen M. E.; Castren E. (2006) The effects of acute and long-term lithium treatments on trkB neurotrophin receptor activation in the mouse hippocampus and anterior cingulate cortex. Neuropharmacology 50(4), 421–417. [DOI] [PubMed] [Google Scholar]
- Wu R.; Fan J.; Zhao J.; Calabrese J. R.; Gao K. (2014) The relationship between neurotrophins and bipolar disorder. Expert Rev. Neurother. 14(1), 51–65. [DOI] [PubMed] [Google Scholar]
- Leyhe T.; Eschweiler G. W.; Stransky E.; Gasser T.; Annas P.; Basun H.; Laske C. (2009) Increase of BDNF serum concentration in lithium treated patients with early Alzheimer’s disease. J. Alzheimer’s Dis. 16(3), 649–656. [DOI] [PubMed] [Google Scholar]
- Machado-Vieira R.; Dietrich M. O.; Leke R.; Cereser V. H.; Zanatto V.; Kapczinski F.; Souza D. O.; Portela L. V.; Gentil V. (2007) Decreased plasma brain derived neurotrophic factor levels in unmedicated bipolar patients during manic episode. Biol. Psychiatry 61(2), 142–144. [DOI] [PubMed] [Google Scholar]
- Tramontina J. F.; Andreazza A. C.; Kauer-Sant’anna M.; Stertz L.; Goi J.; Chiarani F.; Kapczinski F. (2009) Brain-derived neurotrophic factor serum levels before and after treatment for acute mania. Neurosci. Lett. 452(2), 111–113. [DOI] [PubMed] [Google Scholar]
- Rybakowski J. K.; Suwalska A. (2010) Excellent lithium responders have normal cognitive functions and plasma BDNF levels. Int. J. Neuropsychopharmacol. 13(5), 617–622. [DOI] [PubMed] [Google Scholar]
- de Sousa R. T.; van de Bilt M. T.; Diniz B. S.; Ladeira R. B.; Portela L. V.; Souza D. O.; Forlenza O. V.; Gattaz W. F.; Machado-Vieira R. (2011) Lithium increases plasma brain-derived neurotrophic factor in acute bipolar mania: a preliminary 4-week study. Neurosci. Lett. 494(1), 54–56. [DOI] [PubMed] [Google Scholar]
- D’Addario C.; Dell’Osso B.; Palazzo M. C.; Benatti B.; Lietti L.; Cattaneo E.; Galimberti D.; Fenoglio C.; Cortini F.; Scarpini E.; Arosio B.; Di Francesco A.; Di Benedetto M.; Romualdi P.; Candeletti S.; Mari D.; Bergamaschini L.; Bresolin N.; Maccarrone M.; Altamura A. C. (2012) Selective DNA methylation of BDNF promoter in bipolar disorder: differences among patients with BDI and BDII. Neuropsychopharmacology 37(7), 1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias V. V.; Brissos S.; Frey B. N.; Andreazza A. C.; Cardoso C.; Kapczinski F. (2009) Cognitive function and serum levels of brain-derived neurotrophic factor in patients with bipolar disorder. Bipolar Disord. 11(6), 663–671. [DOI] [PubMed] [Google Scholar]
- Frey B. N.; Andreazza A. C.; Rosa A. R.; Martins M. R.; Valvassori S. S.; Reus G. Z.; Hatch J. P.; Quevedo J.; Kapczinski F. (2006) Lithium increases nerve growth factor levels in the rat hippocampus in an animal model of mania. Behav. Pharmacol. 17(4), 311–318. [DOI] [PubMed] [Google Scholar]
- Hellweg R.; Lang U. E.; Nagel M.; Baumgartner A. (2002) Subchronic treatment with lithium increases nerve growth factor content in distinct brain regions of adult rats. Mol. Psychiatry 7(6), 604–608. [DOI] [PubMed] [Google Scholar]
- Mudo G.; Jiang X. H.; Timmusk T.; Bindoni M.; Belluardo N. (1996) Change in neurotrophins and their receptor mRNAs in the rat forebrain after status epilepticus induced by pilocarpine. Epilepsia 37(2), 198–207. [DOI] [PubMed] [Google Scholar]
- Walz J. C.; Frey B. N.; Andreazza A. C.; Cereser K. M.; Cacilhas A. A.; Valvassori S. S.; Quevedo J.; Kapczinski F. (2008) Effects of lithium and valproate on serum and hippocampal neurotrophin-3 levels in an animal model of mania. J. Psychiatr. Res. 42(5), 416–421. [DOI] [PubMed] [Google Scholar]
- Matsumoto T.; Claesson-Welsh L. (2001) VEGF receptor signal transduction. Sci. Signaling 2001(112), re21. [DOI] [PubMed] [Google Scholar]
- Ferrara N.; Gerber H. P.; LeCouter J. (2003) The biology of VEGF and its receptors. Nat. Med. 9(6), 669–676. [DOI] [PubMed] [Google Scholar]
- Patan S. (2004) Vasculogenesis and angiogenesis. Cancer Treat. Res. 117, 3–32. [DOI] [PubMed] [Google Scholar]
- Carmeliet P.; Ferreira V.; Breier G.; Pollefeyt S.; Kieckens L.; Gertsenstein M.; Fahrig M.; Vandenhoeck A.; Harpal K.; Eberhardt C.; Declercq C.; Pawling J.; Moons L.; Collen D.; Risau W.; Nagy A. (1996) Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380(6573), 435–439. [DOI] [PubMed] [Google Scholar]
- Ferrara N.; Carver-Moore K.; Chen H.; Dowd M.; Lu L.; O’Shea K. S.; Powell-Braxton L.; Hillan K. J.; Moore M. W. (1996) Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 380(6573), 439–442. [DOI] [PubMed] [Google Scholar]
- Rosenstein J. M.; Mani N.; Khaibullina A.; Krum J. M. (2003) Neurotrophic effects of vascular endothelial growth factor on organotypic cortical explants and primary cortical neurons. J. Neurosci. 23(35), 11036–11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K. L.; Mao X. O.; Greenberg D. A. (2000) Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Proc. Natl. Acad. Sci. U.S.A. 97(18), 10242–10247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosthuyse B.; Moons L.; Storkebaum E.; Beck H.; Nuyens D.; Brusselmans K.; Van Dorpe J.; Hellings P.; Gorselink M.; Heymans S.; Theilmeier G.; Dewerchin M.; Laudenbach V.; Vermylen P.; Raat H.; Acker T.; Vleminckx V.; Van Den Bosch L.; Cashman N.; Fujisawa H.; Drost M. R.; Sciot R.; Bruyninckx F.; Hicklin D. J.; Ince C.; Gressens P.; Lupu F.; Plate K. H.; Robberecht W.; Herbert J. M.; Collen D.; Carmeliet P. (2001) Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat. Genet. 28(2), 131–138. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Jin K.; Xie L.; Childs J.; Mao X. O.; Logvinova A.; Greenberg D. A. (2003) VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J. Clin. Invest. 111(12), 1843–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodobski A.; Chung I.; Kozniewska E.; Ivanenko T.; Chang W.; Harrington J. F.; Duncan J. A.; Szmydynger-Chodobska J. (2003) Early neutrophilic expression of vascular endothelial growth factor after traumatic brain injury. Neuroscience 122(4), 853–867. [DOI] [PubMed] [Google Scholar]
- Mellergard P.; Sjogren F.; Hillman J. (2010) Release of VEGF and FGF in the extracellular space following severe subarachnoidal haemorrhage or traumatic head injury in humans. Br. J. Neurosurg. 24(3), 261–267. [DOI] [PubMed] [Google Scholar]
- Rosenstein J. M.; Mani N.; Silverman W. F.; Krum J. M. (1998) Patterns of brain angiogenesis after vascular endothelial growth factor administration in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 95(12), 7086–7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krum J. M.; Khaibullina A. (2003) Inhibition of endogenous VEGF impedes revascularization and astroglial proliferation: roles for VEGF in brain repair. Exp. Neurol. 181(2), 241–257. [DOI] [PubMed] [Google Scholar]
- Thau-Zuchman O.; Shohami E.; Alexandrovich A. G.; Leker R. R. (2010) Vascular endothelial growth factor increases neurogenesis after traumatic brain injury. J. Cereb. Blood Flow Metab. 30(5), 1008–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thau-Zuchman O.; Shohami E.; Alexandrovich A. G.; Leker R. R. (2012) Combination of vascular endothelial and fibroblast growth factor 2 for induction of neurogenesis and angiogenesis after traumatic brain injury. J. Mol. Neurosci. 47(1), 166–172. [DOI] [PubMed] [Google Scholar]
- Thau-Zuchman O.; Shohami E.; Alexandrovich A. G.; Leker R. R. (2012) Subacute treatment with vascular endothelial growth factor after traumatic brain injury increases angiogenesis and gliogenesis. Neuroscience 202, 334–341. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Tsai L. K.; Munasinghe J.; Leng Y.; Fessler E. B.; Chibane F.; Leeds P.; Chuang D. M. (2012) Chronic valproate treatment enhances postischemic angiogenesis and promotes functional recovery in a rat model of ischemic stroke. Stroke 43(9), 2430–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren M.; Senatorov V. V.; Chen R. W.; Chuang D. M. (2003) Postinsult treatment with lithium reduces brain damage and facilitates neurological recovery in a rat ischemia/reperfusion model. Proc. Natl. Acad. Sci. U.S.A. 100(10), 6210–6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian Q.; Shi T.; Chuang D. M.; Qian Y. (2007) Lithium reduces ischemia-induced hippocampal CA1 damage and behavioral deficits in gerbils. Brain Res. 1184, 270–276. [DOI] [PubMed] [Google Scholar]
- Kim Y. R.; van Meer M. P.; Tejima E.; Murata Y.; Mandeville J. B.; Dai G.; Chuang D. M.; Rosen B. R.; Lo E. H. (2008) Functional MRI of delayed chronic lithium treatment in rat focal cerebral ischemia. Stroke 39(2), 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F.; Wang Z.; Tchantchou F.; Chiu C. T.; Zhang Y.; Chuang D. M. (2012) Lithium ameliorates neurodegeneration, suppresses neuroinflammation, and improves behavioral performance in a mouse model of traumatic brain injury. J. Neurotrauma 29(2), 362–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F.; Zhang Y.; Chuang D. M. (2012) Lithium reduces BACE1 overexpression, beta amyloid accumulation, and spatial learning deficits in mice with traumatic brain injury. J. Neurotrauma 29(13), 2342–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F.; Wang Z.; Tanaka M.; Chiu C. T.; Leeds P.; Zhang Y.; Chuang D. M. (2013) Posttrauma cotreatment with lithium and valproate: reduction of lesion volume, attenuation of blood-brain barrier disruption, and improvement in motor coordination in mice with traumatic brain injury. J. Neurosurg. 119(3), 766–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W. J.; Li J. K.; Wang Q. Y.; Hou J. B.; Yu B. (2009) Lithium chloride preconditioning optimizes skeletal myoblast functions for cellular cardiomyoplasty in vitro via glycogen synthase kinase-3beta/beta-catenin signaling. Cells Tissues Organs 190(1), 11–19. [DOI] [PubMed] [Google Scholar]
- Kaga S.; Zhan L.; Altaf E.; Maulik N. (2006) Glycogen synthase kinase-3beta/beta-catenin promotes angiogenic and anti-apoptotic signaling through the induction of VEGF, Bcl-2 and survivin expression in rat ischemic preconditioned myocardium. J. Mol. Cell. Cardiol. 40(1), 138–147. [DOI] [PubMed] [Google Scholar]
- Silva R.; Martins L.; Longatto-Filho A.; Almeida O. F.; Sousa N. (2007) Lithium prevents stress-induced reduction of vascular endothelium growth factor levels. Neurosci. Lett. 429(1), 33–38. [DOI] [PubMed] [Google Scholar]
- Guo S.; Arai K.; Stins M. F.; Chuang D. M.; Lo E. H. (2009) Lithium upregulates vascular endothelial growth factor in brain endothelial cells and astrocytes. Stroke 40(2), 652–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki T.; Dutchak P.; Zhao G.; Ding X.; Gautron L.; Parameswara V.; Li Y.; Goetz R.; Mohammadi M.; Esser V.; Elmquist J. K.; Gerard R. D.; Burgess S. C.; Hammer R. E.; Mangelsdorf D. J.; Kliewer S. A. (2007) Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 5(6), 415–425. [DOI] [PubMed] [Google Scholar]
- Kharitonenkov A.; Larsen P. (2011) FGF21 reloaded: challenges of a rapidly growing field. Trends Endocrinol. Metab. 22(3), 81–86. [DOI] [PubMed] [Google Scholar]
- Colantuoni C.; Lipska B. K.; Ye T.; Hyde T. M.; Tao R.; Leek J. T.; Colantuoni E. A.; Elkahloun A. G.; Herman M. M.; Weinberger D. R.; Kleinman J. E. (2011) Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 478(7370), 519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng Y.; Liang M. H.; Ren M.; Marinova Z.; Leeds P.; Chuang D. M. (2008) Synergistic neuroprotective effects of lithium and valproic acid or other histone deacetylase inhibitors in neurons: roles of glycogen synthase kinase-3 inhibition. J. Neurosci. 28(10), 2576–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng Y.; Wang Z.; Tsai L. K.; Leeds P.; Fessler E. B.; Wang J.; Chuang D. M. (2014) FGF-21, a novel metabolic regulator, has a robust neuroprotective role and is markedly elevated in neurons by mood stabilizers. Mol. Psychiatry 10.1038/mp.2013.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senatorov V. V.; Ren M.; Kanai H.; Wei H.; Chuang D. M. (2004) Short-term lithium treatment promotes neuronal survival and proliferation in rat striatum infused with quinolinic acid, an excitotoxic model of Huntington’s disease. Mol. Psychiatry 9(4), 371–385. [DOI] [PubMed] [Google Scholar]
- Chen G.; Rajkowska G.; Du F.; Seraji-Bozorgzad N.; Manji H. K. (2000) Enhancement of hippocampal neurogenesis by lithium. J. Neurochem. 75(4), 1729–1734. [DOI] [PubMed] [Google Scholar]
- Son H.; Yu I. T.; Hwang S. J.; Kim J. S.; Lee S. H.; Lee Y. S.; Kaang B. K. (2003) Lithium enhances long-term potentiation independently of hippocampal neurogenesis in the rat dentate gyrus. J. Neurochem. 85(4), 872–881. [DOI] [PubMed] [Google Scholar]
- Yu I. T.; Kim J. S.; Lee S. H.; Lee Y. S.; Son H. (2003) Chronic lithium enhances hippocampal long-term potentiation, but not neurogenesis, in the aged rat dentate gyrus. Biochem. Biophys. Res. Commun. 303(4), 1193–1198. [DOI] [PubMed] [Google Scholar]
- Kim J. S.; Chang M. Y.; Yu I. T.; Kim J. H.; Lee S. H.; Lee Y. S.; Son H. (2004) Lithium selectively increases neuronal differentiation of hippocampal neural progenitor cells both in vitro and in vivo. J. Neurochem. 89(2), 324–336. [DOI] [PubMed] [Google Scholar]
- Silva R.; Mesquita A. R.; Bessa J.; Sousa J. C.; Sotiropoulos I.; Leao P.; Almeida O. F.; Sousa N. (2008) Lithium blocks stress-induced changes in depressive-like behavior and hippocampal cell fate: the role of glycogen-synthase-kinase-3beta. Neuroscience 152(3), 656–669. [DOI] [PubMed] [Google Scholar]
- Wexler E. M.; Geschwind D. H.; Palmer T. D. (2008) Lithium regulates adult hippocampal progenitor development through canonical Wnt pathway activation. Mol. Psychiatry 13(3), 285–292. [DOI] [PubMed] [Google Scholar]
- Hashimoto R.; Senatorov V.; Kanai H.; Leeds P.; Chuang D. M. (2003) Lithium stimulates progenitor proliferation in cultured brain neurons. Neuroscience 117(1), 55–61. [DOI] [PubMed] [Google Scholar]
- Su H.; Chu T. H.; Wu W. (2007) Lithium enhances proliferation and neuronal differentiation of neural progenitor cells in vitro and after transplantation into the adult rat spinal cord. Exp. Neurol. 206(2), 296–307. [DOI] [PubMed] [Google Scholar]
- McKenzie G.; Ward G.; Stallwood Y.; Briend E.; Papadia S.; Lennard A.; Turner M.; Champion B.; Hardingham G. E. (2006) Cellular Notch responsiveness is defined by phosphoinositide 3-kinase-dependent signals. BMC Cell Biol. 7, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa L.; Ingles-Esteve J.; Aguilera C.; Bigas A. (2003) Phosphorylation by glycogen synthase kinase-3 beta down-regulates Notch activity, a link for Notch and Wnt pathways. J. Biol. Chem. 278(34), 32227–32235. [DOI] [PubMed] [Google Scholar]
- Bachmann R. F.; Wang Y.; Yuan P.; Zhou R.; Li X.; Alesci S.; Du J.; Manji H. K. (2009) Common effects of lithium and valproate on mitochondrial functions: protection against methamphetamine-induced mitochondrial damage. Int. J. Neuropsychopharmacol. 12(6), 805–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.; Li H.; Roughton K.; Wang X.; Kroemer G.; Blomgren K.; Zhu C. (2010) Lithium reduces apoptosis and autophagy after neonatal hypoxia-ischemia. Cell Death Dis. 1, e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira M.; Licht A.; Milman A.; Pick C. G.; Shohami E.; Eldar-Finkelman H. (2007) Role of glycogen synthase kinase-3beta in early depressive behavior induced by mild traumatic brain injury. Mol. Cell. Neurosci. 34(4), 571–577. [DOI] [PubMed] [Google Scholar]
- Zhu Z. F.; Wang Q. G.; Han B. J.; William C. P. (2010) Neuroprotective effect and cognitive outcome of chronic lithium on traumatic brain injury in mice. Brain Res. Bull. 83(5), 272–277. [DOI] [PubMed] [Google Scholar]
- Dash P. K.; Johnson D.; Clark J.; Orsi S. A.; Zhang M.; Zhao J.; Grill R. J.; Moore A. N.; Pati S. (2011) Involvement of the glycogen synthase kinase-3 signaling pathway in TBI pathology and neurocognitive outcome. PLoS One 6(9), e24648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekici M. A.; Uysal O.; Cikriklar H. I.; Ozbek Z.; Turgut Cosan D.; Baydemir C.; Kazanci B.; Hafizoglu D. (2014) Effect of etanercept and lithium chloride on preventing secondary tissue damage in rats with experimental diffuse severe brain injury. Eur. Rev. Med. Pharmacol. Sci. 18(1), 10–27. [PubMed] [Google Scholar]
- Sivanandam T. M.; Thakur M. K. (2012) Traumatic brain injury: a risk factor for Alzheimer’s disease. Neurosci. Biobehav. Rev. 36(5), 1376–1381. [DOI] [PubMed] [Google Scholar]
- Spikman J. M.; Timmerman M. E.; Milders M. V.; Veenstra W. S.; van der Naalt J. (2012) Social cognition impairments in relation to general cognitive deficits, injury severity, and prefrontal lesions in traumatic brain injury patients. J. Neurotrauma 29(1), 101–111. [DOI] [PubMed] [Google Scholar]
- Olsson A.; Csajbok L.; Ost M.; Hoglund K.; Nylen K.; Rosengren L.; Nellgard B.; Blennow K. (2004) Marked increase of beta-amyloid(1–42) and amyloid precursor protein in ventricular cerebrospinal fluid after severe traumatic brain injury. J. Neurol. 251(7), 870–876. [DOI] [PubMed] [Google Scholar]
- Raby C. A.; Morganti-Kossmann M. C.; Kossmann T.; Stahel P. F.; Watson M. D.; Evans L. M.; Mehta P. D.; Spiegel K.; Kuo Y. M.; Roher A. E.; Emmerling M. R. (1998) Traumatic brain injury increases beta-amyloid peptide 1–42 in cerebrospinal fluid. J. Neurochem. 71(6), 2505–2509. [DOI] [PubMed] [Google Scholar]
- Uryu K.; Chen X. H.; Martinez D.; Browne K. D.; Johnson V. E.; Graham D. I.; Lee V. M.; Trojanowski J. Q.; Smith D. H. (2007) Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp. Neurol. 208(2), 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts G. W.; Gentleman S. M.; Lynch A.; Graham D. I. (1991) beta A4 amyloid protein deposition in brain after head trauma. Lancet 338(8780), 1422–1423. [DOI] [PubMed] [Google Scholar]
- Hooper C.; Killick R.; Lovestone S. (2008) The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 104(6), 1433–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phiel C. J.; Wilson C. A.; Lee V. M.; Klein P. S. (2003) GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 423(6938), 435–439. [DOI] [PubMed] [Google Scholar]
- Wei H.; Leeds P. R.; Qian Y.; Wei W.; Chen R.; Chuang D. (2000) beta-amyloid peptide-induced death of PC 12 cells and cerebellar granule cell neurons is inhibited by long-term lithium treatment. Eur. J. Pharmacol. 392(3), 117–123. [DOI] [PubMed] [Google Scholar]
- Loane D. J.; Pocivavsek A.; Moussa C. E.; Thompson R.; Matsuoka Y.; Faden A. I.; Rebeck G. W.; Burns M. P. (2009) Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat. Med. 15(4), 377–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane D. J.; Washington P. M.; Vardanian L.; Pocivavsek A.; Hoe H. S.; Duff K. E.; Cernak I.; Rebeck G. W.; Faden A. I.; Burns M. P. (2011) Modulation of ABCA1 by an LXR agonist reduces beta-amyloid levels and improves outcome after traumatic brain injury. J. Neurotrauma 28(2), 225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert M. P.; Barlow A. K.; Chromy B. A.; Edwards C.; Freed R.; Liosatos M.; Morgan T. E.; Rozovsky I.; Trommer B.; Viola K. L.; Wals P.; Zhang C.; Finch C. E.; Krafft G. A.; Klein W. L. (1998) Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95(11), 6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E.; Torrance M.; Adame A.; Mante M.; Bar-on P.; Rose J. B.; Crews L.; Masliah E. (2007) Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer’s disease are associated with reduced amyloid precursor protein phosphorylation. J. Neurosci. 27(8), 1981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly P. T.; Wu Y.; Zou H.; Wang R.; Zhou W.; Kinoshita A.; Zhang M.; Yang Y.; Cai F.; Woodgett J.; Song W. (2013) Inhibition of GSK3beta-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Invest. 123(1), 224–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang D. M.; Leng Y.; Marinova Z.; Kim H. J.; Chiu C. T. (2009) Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 32(11), 591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash P. K.; Orsi S. A.; Moore A. N. (2009) Histone deactylase inhibition combined with behavioral therapy enhances learning and memory following traumatic brain injury. Neuroscience 163(1), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash P. K.; Orsi S. A.; Zhang M.; Grill R. J.; Pati S.; Zhao J.; Moore A. N. (2010) Valproate administered after traumatic brain injury provides neuroprotection and improves cognitive function in rats. PLoS One 5(6), e11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shein N. A.; Grigoriadis N.; Alexandrovich A. G.; Simeonidou C.; Lourbopoulos A.; Polyzoidou E.; Trembovler V.; Mascagni P.; Dinarello C. A.; Shohami E. (2009) Histone deacetylase inhibitor ITF2357 is neuroprotective, improves functional recovery, and induces glial apoptosis following experimental traumatic brain injury. FASEB J. 23(12), 4266–4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Leng Y.; Tsai L. K.; Leeds P.; Chuang D. M. (2011) Valproic acid attenuates blood-brain barrier disruption in a rat model of transient focal cerebral ischemia: the roles of HDAC and MMP-9 inhibition. J. Cereb. Blood Flow Metab. 31(1), 52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; West E. J.; Van K. C.; Gurkoff G. G.; Zhou J.; Zhang X. M.; Kozikowski A. P.; Lyeth B. G. (2008) HDAC inhibitor increases histone H3 acetylation and reduces microglia inflammatory response following traumatic brain injury in rats. Brain Res. 1226, 181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu C. T.; Liu G.; Leeds P.; Chuang D. M. (2011) Combined treatment with the mood stabilizers lithium and valproate produces multiple beneficial effects in transgenic mouse models of Huntington’s disease. Neuropsychopharmacology 36(12), 2406–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H. L.; Leng Y.; Ma C. H.; Zhang J.; Ren M.; Chuang D. M. (2008) Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience 155(3), 567–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai L. K.; Leng Y.; Wang Z.; Leeds P.; Chuang D. M. (2010) The mood stabilizers valproic acid and lithium enhance mesenchymal stem cell migration via distinct mechanisms. Neuropsychopharmacology 35(11), 2225–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai L. K.; Wang Z.; Munasinghe J.; Leng Y.; Leeds P.; Chuang D. M. (2011) Mesenchymal stem cells primed with valproate and lithium robustly migrate to infarcted regions and facilitate recovery in a stroke model. Stroke 42(10), 2932–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson V. E.; Stewart W.; Smith D. H. (2013) Axonal pathology in traumatic brain injury. Exp. Neurol. 246, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strich S. J. (1956) Diffuse degeneration of the cerebral white matter in severe dementia following head injury. J. Neurol Neurosurg. Psychiatry 19(3), 163–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton M. L.; Gellella E.; Lo C.; Gold T.; Ardekani B. A.; Shifteh K.; Bello J. A.; Branch C. A. (2008) Multifocal white matter ultrastructural abnormalities in mild traumatic brain injury with cognitive disability: a voxel-wise analysis of diffusion tensor imaging. J. Neurotrauma 25(11), 1335–1342. [DOI] [PubMed] [Google Scholar]
- Wang G.; Zhang J.; Hu X.; Zhang L.; Mao L.; Jiang X.; Liou A. K.; Leak R. K.; Gao Y.; Chen J. (2013) Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J. Cereb. Blood Flow Metab. 33(12), 1864–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese E.; Du F.; Garman R. H.; Johnson G. A.; Riccio C.; Tong L. C.; Long J. B. (2014) Diffusion tensor imaging reveals white matter injury in a rat model of repetitive blast-induced traumatic brain injury. J. Neurotrauma 10.1089/neu.2013.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigler E. D. (2013) Traumatic brain injury, neuroimaging, and neurodegeneration. Front. Hum. Neurosci. 7, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill J.; Wang H.; Zhou F.; Li S. (2008) Inactivation of glycogen synthase kinase 3 promotes axonal growth and recovery in the CNS. J. Neurosci. 28(36), 8914–8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti F.; Bollettini I.; Barberi I.; Radaelli D.; Poletti S.; Locatelli C.; Pirovano A.; Lorenzi C.; Falini A.; Colombo C.; Smeraldi E. (2013) Lithium and GSK3-beta promoter gene variants influence white matter microstructure in bipolar disorder. Neuropsychopharmacology 38(2), 313–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. S.; Chopp M.; Kassis H.; Jia L. F.; Hozeska-Solgot A.; Zhang R. L.; Chen C.; Cui Y. S.; Zhang Z. G. (2012) Valproic acid increases white matter repair and neurogenesis after stroke. Neuroscience 220, 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]