

Abstract
The vascular endothelium expresses endothelial nitric oxide synthase (eNOS) that generates nitric oxide (NO) to help maintain vascular integrity due to its anti-inflammatory, antiproliferative, and antithrombogenic effects. Pharmacological blockade of NO production has been shown to exacerbate renal injury in chronic renal disease and induces endothelial cell loss. However, pharmacological inhibition of NO nonspecifically blocks other types of NOS and therefore does not define the specific role of eNOS in kidney disease. We hypothesized that a lack of endothelial eNOS can induce a loss of glomerular and peritubular capillary endothelium and exacerbate renal injury in progressive renal disease. We tested out this hypothesis using remnant kidney (RK) in eNOS knockout (eNOS KO) mice. Systolic blood pressure was significantly higher, and renal function was worse in RK-eNOS KO mice compared with those in RK-C57BL6 mice. eNOS deficiency resulted in more severe glomerulosclerosis, mesangiolysis, and tubular damage. Glomerular and tubular macrophage infiltration and collagen deposition were also greater in RK-eNOS KO mice. Renal injuries in the RK-eNOS KO mice were accompanied by a greater loss of endothelial cells that was shown to be due to both a decrease in endothelial cell proliferation and an increase in apoptosis. A lack of eNOS accelerates both glomerular and tubulointerstitial injury with a loss of glomerular capillaries and peritubular capillaries. Impaired endothelial function is likely a direct risk factor for renal disease.
Keywords: endothelial nitric oxide synthase, glomerular endothelial cell, mesangiolysis
nitric oxide (NO), a free radical in the form of a highly diffusible gas, regulates renal hemodynamics (2). The production of NO is regulated by three different forms of NO synthase (NOS), including neuronal NOS, inducible NOS, and endothelial NOS (eNOS). Expression of these three isoforms is cell specific and subject to distinct control mechanisms (16). Neuronal NOS is predominantly expressed in tubular epithelial cells of the macula densa and inner medullary collecting duct (1), whereas eNOS is restricted to endothelial cells in the kidney, consistent with eNOS having an important role in the maintenance of endothelial function (10). eNOS knockout mice (eNOS KO) have been reported to develop focal congenital renal abnormalities, including glomerular hypoplasia and tubular cell death, and atubular glomeruli (7). In addition, Heeringa et al. (11) demonstrated that a lack of eNOS aggravates anti-glomerular basement membrane glomerulonephritis in association with more severe inflammation. The renal injuries in these mice were accompanied by severe neutrophil infiltration and thrombus formation not observed in controls (11). These data suggest that eNOS may play an important role in both renal development as well as acute glomerulonephritis.
We and others examined the role of NO in the rat RK model by using nitro-l-arginine methyl ester (l-NAME), which is an inhibitor of all NOS enzymes. We (13) found that the inhibition of NO synthesis resulted in more severe renal injury, suggesting a protective role of NO. Recently, the role of eNOS in the progressive renal disease was examined by using eNOS KO mice. Mendoza et al. (17) demonstrated that eNOS deficiency accelerates renal injury and that the injury could be ameliorated by l-arginine and antioxidants. The other study (25) reported that lack of eNOS contributed to the development of glomerular hypertrophy in the RK. However, these studies did not focus on the effect of eNOS deficiency on the response of the endothelium, which is critically modulated by NO. Thus we provided more detailed histological characterization, especially related to the changes in the glomerular and peritubular capillaries in this model. This study should provide insight into the specific role of endothelial function on progressive renal disease.
MATERIALS AND METHODS
Experimental animals.
All experiments were approved by the Animal Care and Use Committee of the University of Florida. Eight-week-old C57BL/6J male mice (n = 10; RK-C57BL6) or C57BL/6J-Nos3tm1Unc male mice (n = 10; RK-eNOS KO; Jackson Laboratory, Bar Harbor, ME) weighing 20–25 g underwent a right subcapsular nephrectomy and surgical resection of the poles of the left kidney. C57BL/6J male mice (n = 8; Cont-C57BL6) or C57BL/6J-Nos3tm1Unc male mice (n = 8; Cont-eNOS KO) underwent a sham operation. Eight mice in RK-C57BL6 and five mice in RK-eNOS KO mice survived the operation, whereas no sham-operated mice died in this study. Systolic blood pressure was measured by a tail-cuff sphygmomanometer (Visitech BP2000; Visitech Systems, Apex, NC) every month, and it was determined as the average value of 5–10 consecutive measurements. After 2 mo of observation, mice were killed to obtain kidney, blood, and urine. Serum urea nitrogen and creatinine were measured by blood urea nitrogen assay (Diagnostic Chemicals, Oxford, CT) and creatinine assay (Bio Quant, San Diego, CA), respectively. Urinary albumin was quantified by Albuwell M (Exocell, Philadelphia, PA), and the albumin-to-creatinine ratio was calculated.
Renal histology.
Kidneys were fixed in Fekete's fixative and embedded in paraffin (20). Two-micrometer sections were used for the periodic acid-Schiff reagent (PAS) and immunohistochemistry with the following antibodies; endothelial cells with rabbit anti-mouse thrombomodulin antibody (20) or rat anti-mouse CD34 antibody (BD Pharmingen, San Jose, CA), type III collagen deposition with a rabbit anti-mouse type III collagen antibody (Southern Biotech, Birmingham, AL), macrophage infiltration with rat anti-mouse F4/80 antigen (Raleigh, Serotec, NC), type IV collagen deposition with a polyclonal rabbit anti-mouse type IV collagen antibody (Chemicon International, Temecula, CA), and VEGF with goat anti-rat VEGF antigen (R&D Systems, Minneapolis, MN). To detect endothelial cell proliferation, we performed double immunostaining with rat anti-mouse Ki67 antibody (Dako, Glostrup, Denmark) and rat anti-mouse CD34 antibody. As a secondary antibody, Match2 goat anti-rabbit IgG-HRP-polymer (Biocare, Concord, CA) was used for thrombodomulin and collagen IV; nonbiotinylated rabbit anti-rat IgG (Vector Labs, Burlingame, CA) was used for CD34, Ki67, and F4/80; and biotinylated horse anti-goat IgG for collagen III was used. To detect apoptosis, we performed terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL) staining with ApopTag in situ apoptosis detection kit, which labels DNA fragments. Color was developed using 3,3-diaminobenzidine (Vector Labs) as a chromogen. In double staining, Fast red (BioCare Medical, Concord, CA) was used for thrombomodulin/CD34.
Quantification of morphology.
All quantifications were performed in a blinded manner by two observers. With the use of coronal sections of the kidney, all glomeruli (>50 glomeruli) were evaluated, whereas only the cortical tubulointestitial area was examined. For glomerular mesangial expansion, the extent of the mesangial matrix (defined as mesangial area) was determined by assessing the PAS-positive and nuclei-free area in the mesangium. The glomerular tuft area was measured by tracing along the outline of the capillary loop using the AxioVision image analyzer (Carl Zeiss). To evaluate the degree of mesangiolysis, the number of glomeruli containing mesangiolysis was divided by that of total glomeruli. Glomerulosclerosis was defined as obstruction of capillary lumen caused by mesangial matrix expansion or collapsed capillaries (3). Each biopsy gave a score of glomerular lesion based on the ratio of injured glomeruli to total glomeruli. Tubular damage was scored based on the ratio of injured tubules defined as tubular cast formation, sloughing of tubular epithelial cells, tubular dilatation, tubular atrophy, and vacuolization of the epithelium per 100 tubules. Glomerular type IV collagen deposition, F4/80-positive macrophage infiltration or CD34-positive endothelial cells were evaluated as percent positive area in glomeruli by using the AxioVision image analyzer (Carl Zeiss). Tubulointerstitial macrophage infiltration (with F4/80), interstitial collagen III deposition, or peritubular capillary endothelial cell density (with CD34 or thrombomodulin) was evaluated as the percent positive area in cortex of whole kidney. The area of peritubular capillary staining by CD34 was measured as the percent positive area per 100 tubules as described previously (15). To examine proliferating endothelial cells, double staining was performed with anti-CD34 antibody and anti-bromodeoxyuridine antibody. On the other hand, TUNEL staining was used to detect apoptotic cells. VEGF expression was measured as the percent positive area in the tubulointerstitium.
Statistical analyses.
All data are shown as means ± SD. The unpaired t-test was used to test the means of two groups of unpaired measurements. One-way ANOVA followed by the Tukey's test was used for three or more group comparisons. The Pearson correlation coefficient was calculated to measure the extent of the association. Statistical significance was defined as P < 0.05.
RESULTS
General parameters.
As shown in Table 1, 2 mo after surgery, blood pressure was significantly elevated in C57BL6 mice. In contrast, blood pressure was high in sham-operated eNOS KO mice but was not further increased by the RK procedure. Reduced renal function was evident in both RK-C57BL6 mice and RK-eNOS KO mice, as evidenced by a significant increase in serum creatinine and blood urea nitrogen. Compared with RK-C57BL6 mice, renal function was significantly worse in RK-eNOS KO mice. Similarly, urinary albumin excretion tended to be increased in both RK groups but was higher in RK-eNOS KO mice.
Table 1.
General characteristics of control and eNOS KO mice at 2 mo after operation
| C57BL6 |
eNOS KO |
|||||
|---|---|---|---|---|---|---|
| Cont | RK | Cont | RK | |||
| Body wt. g | 25.8±2 | 26.2±2 | 22.5±1.1 | 24.4±1.8 | ||
| SBP, mmHg | 114±4 | 135±6* | 149±9*† | 148±5*† | ||
| Kidney wt/body wt, ×10−3 | 5.8±0.3 | 4.3±0.5* | 5.1±0.7 | 3.6±0.5‡ | ||
| Creatinine, ×10−1mg/dl | 0.2±0.3 | 2.0±0.2*‡ | 0.4±0.4 | 2.9±0.1*†‡ | ||
| Urea nitrogen, mg/dl | 25.2±5.6 | 40.7±2.8* | 31.3±4.9 | 63.1±4.2*†‡ | ||
| Urine albumin, μg/ml | 18.0±8.1 | 30.7±47.6 | 99.5±65.8 | 140.4±110.4 | ||
| Urine albumin/creatinine | 0.3±0.1 | 3.7±2.2 | 0.9±1.3 | 5.4±4.5 | ||
eNOS KO, endothelial nitric oxide knockout; Cont, control group; RK, remnant kidney group, SBP, systolic blood pressure.
P < 0.01 vs. C57BL6 Cont.
P < 0.01 vs. C57BL6 RK.
P < 0.01 vs. eNOS KO Cont.
Glomerular and tubulointerstitial damage in RK-eNOS KO mice.
RK-C57BL6 mice did not developed glomerulosclerosis and mesangial expansion, while these changes were significantly induced in RK-eNOS KO mice as assessed by PAS staining and quantification (Fig. 1, A and B). Similarly, while glomerular hypertrophy was induced in RK-C57BL6 mice (40% increase) (c) compared with sham-operated mice (a), it was significantly greater in eNOS KO mice (d) than that in RK-C57BL6 mice (c). Interestingly, mesangiolysis, microaneurysms, and intraluminal thrombi developed in RK-eNOS KO mice (e–h). In contrast, RK-C57BL6 had minimal evidence for these lesions (Fig. 1).
Fig. 1.
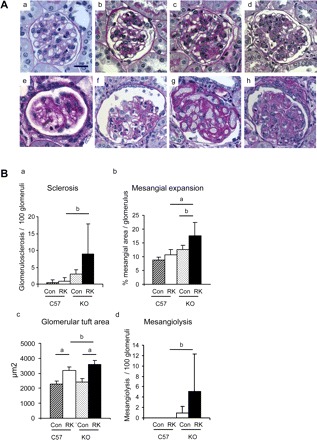
Glomerular injury in the remnant kidney model (RK). A: periodic acid-Schiff reagent staining of glomerulus in the C57BL6 mice and the eNOS KO mice. Normal glomerulus can be observed in C57BL6 mice (a) and eNOS KO mice (b). Mild mesangial proliferation occurs in both RK-C57BL6 mice (c) and RK-eNOS KO mice (d). RK-eNOS KO mice develop mesangiolysis (e–h). Some glomeruli had minimal mesangial cells (e and f), while others exhibited dilatation of glomerular capillaries with loss of mesangial cell (g). There are also some glomeruli, which has mesangiolysis and increased matrix deposition (h). Bar = 20 μm. B: quantification of glomerular lesions in the C57BL6 and the eNOS KO is shown for sclerosis (a), mesangial expansion (b), glomerular tuft area (c), and mesangiolysis (d). Con, control; RK, remnant kidney; C57, C57BL6 mice; KO, eNOS KO mice. Data are means ± SD. aP < 0.01; bP < 0.05.
Similarly, both RK-C57BL6 and RK-eNOS KO mice developed tubulointerstitial disease, but eNOS deficiency caused more severe injury (44% increase) compared with C57BL6 mice, especially in the renal cortex. Tubulointerstitial injury included tubular cast formation, sloughing of tubular epithelial cells, tubular dilatation, tubular atrophy, and vacuolization of the epithelium (Fig. 2). There was also nuclear exfoliation in tubular epithelial cells (Fig. 2B).
Fig. 2.
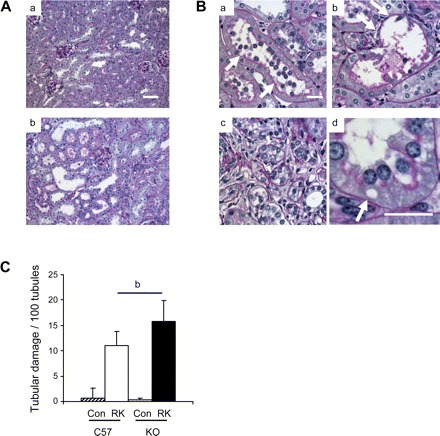
Tubular damages in the RK model. A: low power views demonstrated severe tubulointerstitial damages in RK-eNOS KO mice (a) compared with those in RK-C57BL6 mice (b). Bar = 50 μm. B: damaged tubules display nuclear exfoliation in tubular epithelial cell (a), sloughing of tubular epithelial cell (b), tubular dilatation and atrophy (c), and vacuolization (arrow in d) in epithelial cell. Bar = 20 μm. C: quantification of tubular lesions in cortical tubules and outer strip. Data are means ± SD. bP < 0.05.
To evaluate glomerular matrix expansion and tubulointerstitial fibrosis, we examined collagen IV and III deposition, respectively. As shown in Fig. 3, glomerular collagen IV deposition was significantly increased (89%) in the mesangial area of RK-C57BKL6 mice compared with sham-operated C57BL6 mice, and this was further enhanced in RK-eNOS KO mice (46% increase) compared with RK-C57BL6 (Fig. 3, A and C). Similarly, in the interstitial area, collagen III deposition was significantly increased in RK-C57BL6 (3.5-fold increase) and this deposition was more severe in the renal cortex in RK-eNOS KO mice (38% increase) compared with RK-C57BL6 (Fig. 3, B and C).
Fig. 3.
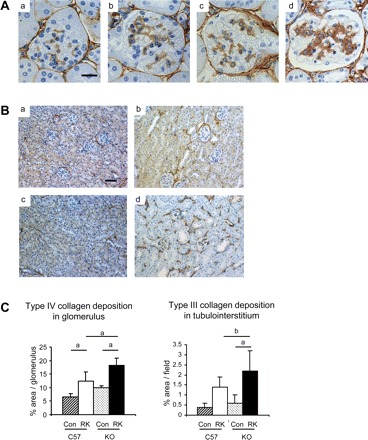
Collagen depositions in the RK model. A: immunohistochemistry demonstrates glomerular collagen IV deposition in RK-C57BL6 mice (b) that is further increased in RK-eNOS KO mice (d) compared with control C57BL6 (a) or eNOS KO mice (c) (bar = 20 μm). B: deposition of collagen III is increased in RK of both C57BL6 (b) and eNOS KO (d) compared with control C57BL6 (a) and eNOS KO mice (c) (bar = 50 μm). C: quantification of type IV collagen deposition in glomeruli and tubulointerstitium. Data are means ± SD. aP < 0.01; bP < 0.05.
Macrophage and T-cell infiltration in RK-eNOS KO mice.
Since NO can inhibit inflammatory reactions, we examined if eNOS deficiency could enhance macrophage and T-cell infiltration in this renal disease by using F4/80 as a marker of macrophages and CD3 for T cells. In sham-operated control C57BL6 mice, no macrophage was observed in glomerulus, while some were present in tubulointerstitium. RK-eNOS KO mice had a significant increase in macrophage infiltration compared with RK-C57BL6 mice in the glomeruli (15.4-fold increase), as well as in the tubulointerstitium (2.7-fold increase; Fig. 4). On the other hand, there were some residential CD3+ T cells in sham-operated kidney. The RK resulted in an increase in the T-cell infiltration in glomerulus (62% increase) but not in the tubulointerstitium. Interestingly, eNOS deficiency was associated with similar T-cell infiltration in the RK as control mice with RKs (Fig. 4C).
Fig. 4.
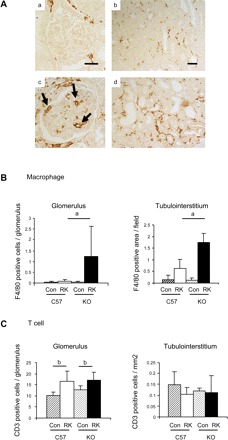
Macrophage infiltration in the RK model. A: macrophage (F4/80) infiltration (arrows) is prominent in glomeruli in RK-eNOS KO mice (c), whereas it is barely observed in control RK-C57BL6 mice (a). Bar = 20 μm in a and c. Similarly, F4/80-positive macrophage infiltration is increased in the tubulointerstitium of RK-eNOS KO mice (d) compared with RK-C57BL6 mice (b). Bar = 50 μm in b and d. Quantification of F4/80-positive macrophage infiltration (B) and CD3-positive T cell (C) is shown. Data are means ± SD. aP < 0.01; bP < 0.05.
Endothelial cell loss in RK-eNOS KO mice.
Next, we examined the integrity of the vascular endothelium in this model using antibodies for CD34 and thrombomodulin as markers of endothelial cells. As shown in Fig. 5, endothelial cell density was slightly reduced in glomeruli as well as in the cortical tubuloiterestitium in RK C57BL6 mice, whereas a significantly greater reduction was observed in RK-eNOS KO mice (50% reduction) compared with RK-C57BL6 (Fig. 5, A and B). Interestingly, the endothelial cell loss was more prominent in the glomeruli and renal cortex, as illustrated by reduced thrombomodulin-positive cells in these areas (Fig. 5B). CD34-positive cells were also reduced in RK-eNOS KO mice (17.4 ± 1.3 vs. 21.7 ± 1.9% in glomerulus of RK-C57BL6, P < 0.01; 6.5 ± 2.4 vs. 10.3 ± 1.2% in tubulointerstitium, P < 0.01). As shown in Fig. 5C, the loss of the endothelium was associated with both greater glomerulosclerosis and tubular damage.
Fig. 5.
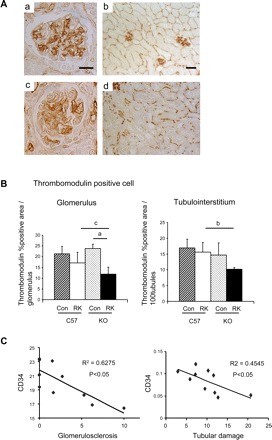
Changes in the renal microvasculature in the RK model. A: glomerular and peritubular capillary endothelial (CD34) staining. Severe endothelial cell loss in glomerular (c) and peritubular capillaries (d) in RK-eNOS KO mice were observed compared with those in C57BL6 mice (a and b). Bar = 20 μm in a and c. Bar = 50 μm in b and d. B: quantification of thrombomodulin-positive cells indicates the endothelial cell loss in glomerular and peritubular capillary of cortex. Data are means ± SD. aP < 0.01; bP < 0.05; cP = 0.051. C: negative correlation between CD34-positive endothelial cell and glomerular injury or tubulointerstitial damage in RK model of C57BL6 and eNOS KO mice.
Endothelial cell proliferation and apoptosis in RK-eNOS KO mice.
We further examined the mechanism by which endothelial cells were lost in RK-eNOS KO mice. Since the endothelial cell number results from the balance between proliferation and apoptosis, we first examined endothelial cell proliferation evidenced by Ki67-positive endothelial cells (Fig. 6A). Double staining for Ki67 and CD34 demonstrated that proliferating endothelial cells were significantly reduced in cortical peritubular capillaries in RK-eNOS KO mice (36% reduction) compared with RK-C57BL6 mice. Similarly, glomerular capillary endothelial cells tended to be lower in RK-eNOS KO mice, although this did not reach significance (Fig. 6B). In contrast, total Ki67-positive cells in RK-eNOS KO mice are almost identical to that in RK-C57BL6 mice (Fig. 6C).
Fig. 6.
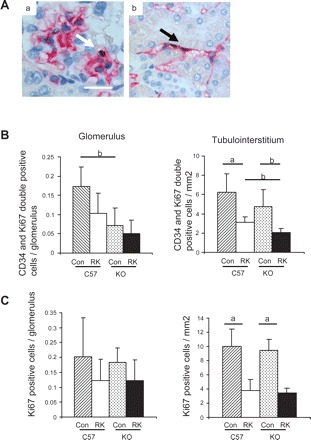
Endothelial cell proliferation in the remnant kidney model. A: double staining of CD34 (red) and bromodeoxyuridine (black) reveals proliferating endothelial cells in glomeruli (a) and peritubular capillaries (b). Bar = 20 μm. B: quantification of proliferating endothelial cells in the C57BL6 and the eNOS KO mice demonstrates less number of endothelial proliferation in RK-eNOS KO compared with that in RK-C57BL6 mice. Total Ki67-positive cells were quantified in C. Data are means ± SD. aP < 0.01; bP < 0.05.
Next, we examined apoptotic cell in this model using TUNEL staining. Using serial sections, immunohistochemistry demonstrated that there are many cells undergoing apoptosis. These include glomerular endothelial cells (Fig. 7A, a and b) and peritubular capillary endothelial cells (Fig. 7A, c and d). By quantification of these cells, it was found that apoptosis was significantly increased in peritubular endothelial cells in RK-eNOS KO mice (4.6-fold increase) compared with RK-C57BL6 (Fig. 7B). Similarly, endothelial apoptotic cells were also slightly increased in glomeruli in RK-eNOS KO mice.
Fig. 7.
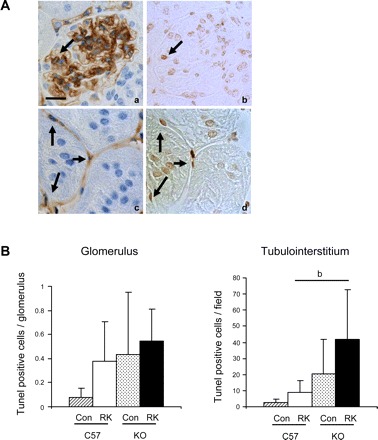
Apoptotic endothelial cells in the RK model. A: serial sections were stained for CD34 (a and b) and TUNEL-positive apoptotic cells (b and d) and demonstrate that some endothelial cells are undergoing apoptosis (arrow) in glomeruli (a and b). Similarly, there are several apoptotic glomerular (arrow) and peritubular capillary endothelial cells (c and d). B: quantification shows that there is significantly high number of apoptotic endothelial cells in cortex while it tends to be higher in glomeruli compared with RK-C57BL6 mice. bP < 0.05.
VEGF expression in RK-eNOS KO mice.
As shown in Fig. 8, a significant increase in cortical VEGF expression was observed in both C57BL6 (2.3-fold increase) and eNOS KO mice (3.1-fold increase) with RKs. On the other hand, glomerular VEGF expression was not altered in these groups.
Fig. 8.
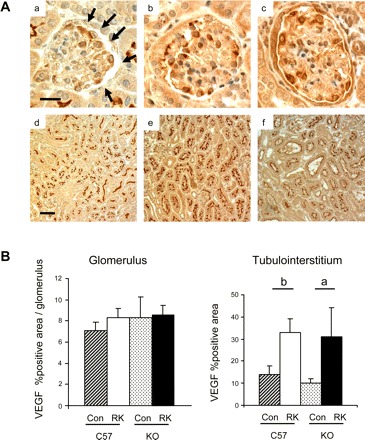
Renal VEGF expression in eNOS KO mice A: glomerular and tubulointerstitial VEGF in sham-operated (a and d, respectively), RK-C57BL6 (b and e), or RK-eNOS KO mice (c and f). B: quantitative analysis.
DISCUSSION
To determine the role of endothelial-derived NO in chronic renal disease, we studied RKs using eNOS KO mice. Since endothelial NO is expected to serve a protective role, we hypothesized that the RK model would result in more severe renal injury in eNOS KO mice compared with that in wild-type mice. As expected, the absence of functional eNOS resulted in worse renal function, greater urinary albumin excretion, and a more severe histological injury in both glomeruli and tubulointerstitium. In addition to worse glomerulosclerosis and tubulointerstitial fibrosis, RK-eNOS KO mice also developed occasional intraluminal thrombi in glomeruli with mesangiolysis and microaneurysm formation. Since thrombotic lesions do not usually develop in RK model of wild-type mice, it is likely the consequence of more severe endothelial injury. Consistent with this finding, eNOS deficiency resulted in greater endothelial cell loss and more macrophage infiltration. The endothelial loss was likely a consequence of both less proliferation and more apoptosis. Finally, a protective role of eNOS in progressive renal disease was suggested by the finding that endothelial cell density was negatively associated with renal injury.
Compatible with our study, it has been recently shown that eNOS KO mice exhibit an impaired angiogenic response in skeletal muscle. Murohara et al. (19) studied the hindlimb ischemia model in eNOS KO mice and found that tissue ischemia failed to induce angiogenesis. These studies indicate that endothelial NO is an indispensable factor for angiogenesis. However, the role of NO on endothelial cell is likely complicated. Gooch et al. (9) demonstrated that endothelial cell proliferation is positively and negatively regulated by NO and that this diverse effect depends on the amount of NO. For instance, a high amount of NO has been shown to negatively regulate endothelial proliferation, whereas endothelial proliferation can be positively stimulated by low amount of NO. On the other hand, NO may also serve as a survival factor to counteract apoptotic process in endothelial cell. Although the role of NO in regulation of apoptosis is still controversial, it has been demonstrated that NO donors at physiologically relevant concentrations blocked endothelial cell apoptosis induced by various stimuli, including the proinflammatory stimuli LPS or TNF-α and reactive oxygen species (4, 5). Given the complexity in regulation, eNOS could be a critical factor to regulate endothelial integrity in various pathological conditions.
In our study, RK-eNOS KO mice developed features consistent with a mild thrombotic microangiopathy (TMA), as noted by the presence of mesangiolysis, intraluminal thrombus formation, and occasional microaneurysms. Since these glomerular lesions did not develop in wild-type mice, they are presumably caused by severe endothelial dysfunction or eNOS deficiency. Compatible with this notion, it has been recently demonstrated (21) that selective damage on glomerular endothelial cell using administration of anti-glomerular endothelial cell antibody resulted in the formation of TMA in rat. Importantly, in this latter model, the blockade of NO resulted in even more severe injury, whereas use of a specific inducible NOS inhibitor had no effect, suggesting that NO derived from eNOS may be a critical factor in the development of TMA (22).
Hypertension is a major risk factor for renal progression. However, it seems unlikely for hypertension per se to fully explain the renal injury observed in this model, since RK-eNOS KO mice developed more severe renal injury despite showing no difference in blood pressure compared with sham-operated eNOS KO mice. Therefore, there might be another factor involved in the development of renal injury. As discussed above, endothelial dysfunction could be an important factor in development of severe glomerular injury. Although sham-operated eNOS KO mice had endothelial dysfunction due to eNOS deficiency, endothelial dysfunction was more severe in RK-eNOS KO mice, as evidenced by greater loss of endothelial cells. Thus, while there is no doubt that hypertension is a risk factor for renal injury, there may be a synergistic effect when hypertension and endothelial dysfunction are combined with nephron loss.
Endothelial cell loss is commonly associated with renal injury in progressive renal diseases (15, 23). In this study, we documented a reduction in endothelial staining, as noted by staining for CD34 and thrombomodulin, respectively, which strongly suggests a reduction in capillary density in the RK-eNOS KO mice. Nevertheless, we did not note significant differences in endothelial cell proliferation or apoptosis, although there was a trend for less endothelial cell proliferation and more apoptosis in the RK-eNOS KO mice. While it is possible that no difference in cell proliferation or apoptosis at this time point indeed exists, the sensitivity of this test is also limited due to the relative rarity and transient nature of these processes and because identification of proliferating or apoptotic endothelial cells requires inclusion of the cell nucleus in the tissue section examined. Given that we could show reduced endothelial staining with antibodies directed to two different antigens, it is likely that there is a true reduction in glomerular endothelial cells that is greater in the eNOS KO compared with the wild-type mouse with RKs.
The protection of endothelial function could be a therapeutic strategy to prevent renal disease. In several studies, VEGF supplementation has been used to examine if endothelial protection can prevent renal injury. Recently, our group (14) has shown that endothelial cell loss was associated with the progression of renal disease in the rat RK model, while the administration of VEGF slowed progression of renal disease and preserved endothelial cell number. The protective role of VEGF could be due to its ability to stimulate NO in endothelial cells. In fact, we and others (8, 13) have found that blocking NO production with l-NAME resulted in worse renal disease in the RK model. For instance, l-NAME administration caused higher blood pressure, greater proteinuria, worse renal function, and more severe glomerulosclerosis. This could be a consequence of increased glomerular hypertension and activation of renin-angiotensin system (8). We also documented worse interstitial fibrosis with greater tubular osteopontin expression, macrophage infiltration, and type III collagen deposition in l-NAME-treated RK rats. Similar to the current study the injury was accompanied by greater endothelial cell loss in both glomerular and peritubular capillaries (13). The increase in VEGF observed in the tubulointerstitium in both control and eNOS KO mice could represent a compensatory response to the renal injury. Nevertheless, if the benefit of VEGF is mediated via stimulation of endothelial NO, then this could potentially provide another explanation for why renal injury was worse in the eNOS KO mice (as this compensation would no longer be effective in this group).
While the loss of the endothelium observed in eNOS KO mice could be a direct effect of NO deficiency, it is also possible that systemic humoral factors could contribute to renal endothelial loss. For example, TNF-α can induce endothelial apoptosis while this pathway is negatively regulated by NO (12). Since serum TNF-α level is elevated in the rat RK model (6), a lack of eNOS could increase a susceptibility of renal endothelial cells to TNF-α. Given this fact, the involvement of systemic factors could provide another mechanism for endothelial loss in this model (24).
Interestingly, recent studies (18) suggest that the low levels of inflammation that occur in the RK model may have a role in renal disease progression. However, we did not observe any differences in T-cell infiltration in the eNOS KO mice compared with controls with RKs. Hence, it does not appear that endothelial NO has a significant role in mediating T-cell infiltration, at least in this model of renal disease.
In conclusion, renal injury in chronic renal disease is aggravated in mice lacking eNOS. Endothelial function was confirmed to be an important factor to determine the fate of renal disease. These studies emphasize that endothelial dysfunction may be a key risk factor for progressive renal disease and suggest that treatments targeting the improvement of endothelial function may provide new treatment strategies for this important condition.
Acknowledgments
This study was supported by a Program Project from the Juvenile Diabetes Research Foundation and by generous funds from Gatorade. T. Nakayama and R. J. Johnson also are supported by National Institutes of Health Grants DK-52121 and HL-68607.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Bachmann S, Bosse HM, Mundel P. Topography of nitric oxide synthesis by localizing constitutive NO synthases in mammalian kidney. Am J Physiol Renal Fluid Electrolyte Physiol 268: F885–F898, 1995 [DOI] [PubMed] [Google Scholar]
- 2.Bachmann S, Mundel P. Nitric oxide in the kidney: synthesis, localization, and function. Am J Kidney Dis 24: 112–129, 1994 [DOI] [PubMed] [Google Scholar]
- 3.D'Agati VD, Fogo AB, Bruijn JA, Jennette JC. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis 43: 368–382, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Dimmeler S, Haendeler J, Nehls M, Zeiher AM. Suppression of apoptosis by nitric oxide via inhibition of interleukin-1beta-converting enzyme (ICE)-like and cysteine protease protein (CPP)-32-like proteases. J Exp Med 185: 601–607, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dimmeler S, Hermann C, Galle J, Zeiher AM. Upregulation of superoxide dismutase and nitric oxide synthase mediates the apoptosis-suppressive effects of shear stress on endothelial cells. Arterioscler Thromb Vasc Biol 19: 656–664, 1999 [DOI] [PubMed] [Google Scholar]
- 6.Fedulov AV, Ses TP, Gavrisheva NA, Rybakova MG, Vassilyeva JG, Tkachenko SB, Kallner A, MacMillan JC. Serum TGF-beta 1 and TNF-alpha levels and cardiac fibrosis in experimental chronic renal failure. Immunol Invest 34: 143–152, 2005 [PubMed] [Google Scholar]
- 7.Forbes MS, Thornhill BA, Park MH, Chevalier RL. Lack of endothelial nitric-oxide synthase leads to progressive focal renal injury. Am J Pathol 170: 87–99, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fujihara CK, De Nucci G, Zatz R. Chronic nitric oxide synthase inhibition aggravates glomerular injury in rats with subtotal nephrectomy. J Am Soc Nephrol 5: 1498–1507, 1995 [DOI] [PubMed] [Google Scholar]
- 9.Gooch KJ, Dangler CA, Frangos JA. Exogenous, basal, and flow-induced nitric oxide production and endothelial cell proliferation. J Cell Physiol 171: 252–258, 1997 [DOI] [PubMed] [Google Scholar]
- 10.Han KH, Lim JM, Kim WY, Kim H, Madsen KM, Kim J. Expression of endothelial nitric oxide synthase in developing rat kidney. Am J Physiol Renal Physiol 288: F694–F702, 2005 [DOI] [PubMed] [Google Scholar]
- 11.Heeringa P, van Goor H, Itoh-Lindstrom Y, Maeda N, Falk RJ, Assmann KJ, Kallenberg CG, Jennette JC. Lack of endothelial nitric oxide synthase aggravates murine accelerated anti-glomerular basement membrane glomerulonephritis. Am J Pathol 156: 879–888, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang-Decker N, Cao S, Chatterjee S, Yao J, Egan LJ, Semela D, Mukhopadhyay D, Shah V. Nitric oxide promotes endothelial cell survival signaling through S-nitrosylation and activation of dynamin-2. J Cell Sci 120: 492–501, 2007 [DOI] [PubMed] [Google Scholar]
- 13.Kang D, Nakagawa T, Feng L, Johnson RJ. Nitric oxide modulates vascular disease in the remnant kidney model. Am J Pathol 161: 239–248, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang DH, Hughes J, Mazzali M, Schreiner GF, Johnson RJ. Impaired angiogenesis in the remnant kidney model: II. Vascular endothelial growth factor administration reduces renal fibrosis and stabilizes renal function. J Am Soc Nephrol 12: 1448–1457, 2001 [DOI] [PubMed] [Google Scholar]
- 15.Kang DH, Joly AH, Oh SW, Hugo C, Kerjaschki D, Gordon KL, Mazzali M, Jefferson JA, Hughes J, Madsen KM, Schreiner GF, Johnson RJ. Impaired angiogenesis in the remnant kidney model: I. Potential role of vascular endothelial growth factor and thrombospondin-1. J Am Soc Nephrol 12: 1434–1447., 2001 [DOI] [PubMed] [Google Scholar]
- 16.Kone BC. Localization and regulation of nitric oxide synthase isoforms in the kidney. Semin Nephrol 19: 230–241, 1999 [PubMed] [Google Scholar]
- 17.Mendoza MG, Castillo-Henkel C, Medina-Santillan R, Jarillo Luna RA, Robles HV, Romo E, Rios A, Escalante B. Kidney damage after renal ablation is worsened in endothelial nitric oxide synthase −/− mice and improved by combined administration of l-arginine and antioxidants. Nephrology (Carlton) 13: 218–227, 2008 [DOI] [PubMed] [Google Scholar]
- 18.Mu W, Ouyang X, Agarwal A, Zhang L, Long DA, Cruz PE, Roncal CA, Glushakova OY, Chiodo VA, Atkinson MA, Hauswirth WW, Flotte TR, Rodriguez-Iturbe B, Johnson RJ. IL-10 suppresses chemokines, inflammation, and fibrosis in a model of chronic renal disease. J Am Soc Nephrol 16: 3651–3660, 2005 [DOI] [PubMed] [Google Scholar]
- 19.Murohara T, Asahara T, Silver M, Bauters C, Masuda H, Kalka C, Kearney M, Chen D, Symes JF, Fishman MC, Huang PL, Isner JM. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest 101: 2567–2578, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakagawa T, Sato W, Glushakova O, Heinig M, Clarke T, Campbell-Thompson M, Yuzawa Y, Atkinson M, Johnson RJ, Croker B. Diabetic eNOS knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol 18: 539–550, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Nangaku M, Alpers CE, Pippin J, Shankland SJ, Adler S, Kurokawa K, Couser WG, Johnson RJ. A new model of renal microvascular endothelial injury. Kidney Int 52: 182–194, 1997 [DOI] [PubMed] [Google Scholar]
- 22.Shao J, Miyata T, Yamada K, Hanafusa N, Wada T, Gordon KL, Inagi R, Kurokawa K, Fujita T, Johnson RJ, Nangaku M. Protective role of nitric oxide in a model of thrombotic microangiopathy in rats. J Am Soc Nephrol 12: 2088–2097, 2001 [DOI] [PubMed] [Google Scholar]
- 23.Shimizu A, Masuda Y, Mori T, Kitamura H, Ishizaki M, Sugisaki Y, Fukuda Y. Vascular endothelial growth factor165 resolves glomerular inflammation and accelerates glomerular capillary repair in rat anti-glomerular basement membrane glomerulonephritis. J Am Soc Nephrol 15: 2655–2665, 2004 [DOI] [PubMed] [Google Scholar]
- 24.Stenvinkel P, Ketteler M, Johnson RJ, Lindholm B, Pecoits-Filho R, Riella M, Heimburger O, Cederholm T, Girndt M. IL-10, IL-6, and TNF-alpha: central factors in the altered cytokine network of uremia–the good, the bad, and the ugly. Kidney Int 67: 1216–1233, 2005 [DOI] [PubMed] [Google Scholar]
- 25.Yamashita C, Tazawa N, Ohkita M, Matsumura Y. Exaggerated renal pathology of partial ablation-induced chronic renal failure in eNOS deficient mice. Biol Pharm Bull 31: 1029–1031, 2008 [DOI] [PubMed] [Google Scholar]