

Abstract
Chitosan, the deacetylated derivative of chitin, can be found in the cell wall of some fungi and is utilized in translational applications. We have shown that highly purified preparations of chitosan, but not chitin, activate the NLRP3 inflammasome in primed mouse bone marrow-derived macrophages (BMMΦ), inducing a robust IL-1β response. Here, we further define specific cell types that are activated and delineate mechanisms of activation. BMMΦ differentiated to promote a classically activated (M1) phenotype released more IL-1β in response to chitosan than intermediate or alternatively activated macrophages (M2). Chitosan but not chitin induced a robust IL-1β response in mouse DCs, peritoneal macrophages, and human PBMCs. Three mechanisms for NLRP3 inflammasome activation may contribute: K+ efflux, reactive oxygen species (ROS), and lysosomal destabilization. The contributions of these mechanisms were tested using a K+ efflux inhibitor, high extracellular potassium, a mitochondrial ROS inhibitor, lysosomal acidification inhibitors, and a cathepsin B inhibitor. These studies revealed that each of these pathways participated in optimal NLRP3 inflammasome activation by chitosan. Finally, neither chitosan nor chitin stimulated significant release from unprimed BMMΦ of any of 22 cytokines and chemokines assayed. In conclusion, 1) chitosan, but not chitin, stimulates IL-1β release from multiple murine and human cell types; 2) multiple non-redundant mechanisms appear to participate in inflammasome activation by chitosan; and 3) chitin and chitosan are relatively weak stimulators of inflammatory mediators from unprimed BMMΦ. These data have implications for understanding the nature of the immune response to microbes and biomaterials that contain chitin and chitosan.
Introduction
Chitosan, a β-(1,4)-linked polymer of glucosamine (GlcN), is the deacetylated derivative of chitin, a β-(1,4)-linked polymer of N-acetylglucosamine (GlcNAc). Chitosan is not as prevalent naturally as chitin, though chitin deacetylases, which catalyze conversion of chitin to chitosan, are present in some medically important fungi such as Cryptococcus neoformans and members of the Zygomycetes (1, 2). Chitin is an essential component of fungal cell walls as well as a major component in crustacean shells, insect exoskeletons, and some parasites, including helminths and protozoa (3–9). Human exposure to these polysaccharides, particularly chitosan, may occur not only during fungal infection but may arise as a result of their presence in pharmaceutical and commercial applications such as gene and drug delivery constructs, tissue scaffolds, and wound dressings (10–13).
We previously found that chitosan, but not chitin, activates the NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome of bone marrow-derived macrophages (BMMΦ) (14). The NLRP3 inflammasome is a cytosolic complex containing NLRP3, the adaptor molecule Apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and caspase-1. Activation is a two-step process with the first step priming the system and resulting in an upregulation of both pro-IL-1β and NLRP3 (15), and the second step inducing caspase-1 dependent cleavage of pro-IL-1β to the active form of IL-1β. The NLRP3 inflammasome has been shown to be activated by a wide variety of stimuli such as ATP, amyloid-β, alum, silica, and nigericin, as well as a variety of fungi, bacteria and viruses (16). Unlike other described inflammasomes with more specific stimuli, such as AIM2 with DNA (17), and IPAF with flagellin (18), the NLRP3 inflammasome is unlikely to be activated by direct interaction with each of its varied activators.
While BMMΦ have been the most often studied cell type by inflammasome researchers, other pro-inflammatory cell types have also been investigated. Macrophages are polarized between classically activated macrophage (M1) and alternatively activated macrophage (M2) phenotypes. M1 macrophages are generally considered pro-inflammatory while M2 macrophages are considered anti-inflammatory; however, there is reversible plasticity between the phenotypes and some macrophages exhibit intermediate polarities (19). M1 macrophages have been shown to have a strong inflammasome response, which diminishes as macrophages become polarized towards intermediate and M2 phenotypes (20). Similar to cultured cells, primary cells such as peritoneal macrophages have also been shown to have strong inflammasome responses (21). Activation of the inflammasome in murine dendritic cells (DC) may be an important intermediary between the innate immune response and the adaptive immune response. DC activation is crucial for vaccine adjuvants to stimulate protective adaptive immunity (22) and the IL-1β produced by DCs is required for the optimal priming of T cells (23). Many parallels exist between mouse and human cell inflammasome activation. However, one important difference is that human blood monocytes have constitutively active caspase-1 and can be stimulated by LPS alone to secrete IL-1β (24).
Three mechanisms for NLRP3 inflammasome activation have been proposed: K+ efflux, reactive oxygen species (ROS) generation, and lysosomal destabilization. K+ efflux has been shown to be required for NLRP3 inflammasome activation by many different stimuli. This model was first described for ATP, with ATP-mediated activation of the NLRP3 inflammasome being dependent upon activation of P2X7, the ATP-gated ion channel, which triggers rapid K+ efflux (25). This K+ efflux is then somehow sensed, thereby activating the NLRP3 inflammasome. The second proposed mechanism of activation involves NLRP3 functioning as a more general sensor of cellular stress by recognizing and being activated by ROS. ROS involvement in the activation of the inflammasome has been shown for all NLRP3 stimuli tested (16) and recent work has suggested that the important ROS source for NLRP3 inflammasome activation is mitochondrial in origin (26, 27). ROS generation is frequently accompanied by K+ efflux, and one may trigger the other, combining to activate the NLRP3 inflammasome (28). In the third model, particulate stimuli are phagocytosed leading to phagosomal maturation and lysosomal fusion, ultimately resulting in phagosomal destabilization and lysosomal rupture. With the destabilization of the lysosome, cathepsins including cathepsin B are released into the cytoplasm, resulting in activation of the NLRP3 inflammasome (29). Additional work on how these mechanisms work in relation to each other is still needed.
The immunostimulatory properties of chitin and chitosan remain poorly understood. These polysaccharides have been characterized as relatively inert, proinflammatory, and pro-allergenic in different reports (14, 30–34). Chitin has been shown to induce an allergic response consisting of an accumulation of eosinophils and basophils expressing IL-4 and alternatively activated macrophages (31). Conversely, in another report, chitin down-regulated the allergic response to ragweed in mice (35). Possible explanations for the disparate findings include the sources of chitin (e.g., shrimp, crab, fungal), the manufacturing processes (which in turn could affect the degree of deacetylation and tertiary structure), and the presence of contaminants (36–38). Another possible explanation for the varied immunological response is particle size. The importance of size has been suggested by studies demonstrating differential stimulation of TNFα and IL-10 by size-fractionated chitin. Particles of intermediate size (40–70 μm) induced just TNFα whereas smaller particles (<40 μm) induced both TNFα and IL-10 (39).
In this study we assayed chitosan-induced IL-1β release in mouse macrophages with an M1, intermediate, or M2 phenotype, dendritic cells, and peritoneal cells, as well as human PBMCs. For all cell types tested, chitosan, but not chitin, induced IL-1β release. We then analyzed the role of each of the proposed NLRP3 inflammasome activation mechanisms described above. We found evidence that all three mechanisms; K+ efflux, ROS, and lysosomal destabilization, participate in the NLRP3 inflammasome activation by chitosan. Finally, with use of a multiplex assay, we determined that purified chitosan and chitin are relatively weak inducers of cytokines and chemokines from unprimed BMMΦ.
Materials and Methods
Reagents and cell culture
All materials were obtained from Sigma-Aldrich unless otherwise stated. Ultrapure LPS (free of TLR2-stimulating lipopeptides) was treated with deoxycholate twice followed by phenol extraction and ethanol precipitation (40) to further purify the original Sigma-Aldrich stock (catalog # L2630). Chitosan was obtained from Primex (ChitoClear, high molecular weight shrimp chitosan, 76% deacetylated). Complete media is defined as RPMI 1640 media (Invitrogen Life Technologies) supplemented with 10% heat-inactivated FBS (Tissue Culture Biologicals), 2 mM L-glutamine (Invitrogen), 100 U/ml penicillin, and 100 μg/ml streptomycin. Cell culture was at 37°C in humidified air supplemented with 5% CO2. All experiments were performed under conditions designed to minimize endotoxin contamination.
Cell populations
BMMΦ were generated as described (41). Briefly, bone marrow was extracted from the femurs and tibiae of wild type C57BL/6 mice (The Jackson Laboratory) and NLRP3−/− mice (originally obtained from Millennium Pharmaceuticals and kindly provided to us by Dr. Kate Fitzgerald, UMass Medical School). Cells were cultured in complete media supplemented with 10 ng/ml recombinant M-CSF (eBiosciences) and fed on days 4 and 7 with fresh media containing M-CSF. On day 8, non-adherent cells were washed away and the adherent macrophages were treated with 0.05% trypsin-EDTA, harvested and washed once in complete media before use in experiments. For the experiments examining macrophage skewing shown in Figure 1, BMMΦ were cultured with recombinant GM-CSF (5 ng/ml, Miltenyi Biotec), IFNγ (150 U/ml, eBioscience), M-CSF (10 ng/ml, eBioscience), and/or IL-4 (20 U/ml, eBioscience) (42). The cytokines were also in the media during the subsequent priming and stimulation steps. Bone marrow-derived dendritic cells (BMDCs) were generated as described (43). Briefly, bone marrow was extracted and cultured as described for M1-like macrophages except on day 8 non-adherent cells were collected for use in experiments. Resident peritoneal cells were harvested by lavaging the peritoneal cavity of C57BL/6 mice or NLRP3−/− mice with 10 ml of PBS. Experimental protocols involving animals were approved by the University of Massachusetts Medical School Institutional Animal Care and Use Committee. Human PBMCs were isolated from the blood of adult, healthy donors under a protocol approved by the University of Massachusetts Medical School Institutional Review Board using Ficoll-Hypaque density centrifugation.
Figure 1. Effect of macrophage skewing on stimulated IL-1β release.

BMMΦ were cultured with M-CSF, M-CSF+IL-4, GM-CSF, and GM-CSF+IFNγ as described in Methods, and then plated at 1 x 105 cells/well. A and B. Cells were incubated for 9 hours with 100 ng/ml LPS and then assayed for nitrite (A) and urea (B) as measures of NO and arginase activity, respectively. For nitrite measurements; p<0.001 comparing either M-CSF-containing group with either GM-CSF-containing group, and p<0.001 comparing GM-CSF with GM-CSF+IFNγ. For urea measurements; p<0.0001 comparing M-CSF+IL-4 with any other group. C. Cells were primed for 3 h with 100 ng/ml LPS, and then left unstimulated (unstim) or stimulated for 6 h with silica, chitosan, or chitin (all at 0.1 mg/ml). Supernatants were collected and analyzed by ELISA for IL-1β. For all BMMΦ culture conditions; p < 0.05 comparing chitosan to either unstim or chitin, and p < 0.001 comparing chitosan to silica for BMMΦ cultured with MCSF+IL-4. Data are means ± SE of three independent experiments, each performed in triplicate. All comparisons were analyzed by 2-way ANOVA with Tukey’s multiple comparison test.
Assays for nitric oxide (NO) and arginase
NO generation was quantified by measuring nitrite concentration in media removed from wells of cultured cells (44). Each sample (70 μl) was mixed with 70 μl of water and 70 μl of Greiss Reagent in a 96-well plate, and then incubated at 22°C for 30 min followed by measurement of absorbance at 540 nm. Standard curves were made using serial dilutions of sodium nitrite. Following the removal of media, cells were lysed and arginase activity was measured (45). Briefly, cells that had been cultured in 24-well plates were lysed by addition of 100 μl 0.1% Triton X-100, 1x Protease Inhibitor Cocktail (Roche) and incubation at 22°C for 30 min. To each well, 100 μl 25 mM Tris-HCl, pH 7.4 and 35 μl 10 mM MnCl2 were added followed by incubation at 55°C for 10 min. Next, 12.5 μl of sample was mixed with 12.5 μl 0.5M L-arginine, pH 9.7 and incubated for 1 h at 37°C. The reaction was stopped with 200 μl H2SO4/H3PO4/H20 (1:3:7) followed by addition of 12.5 μl 9% (w/v) α-isonitrosopropiophenone (dissolved in 100% ethanol) and heating at 99°C for 30 min. Standards were serial dilutions of urea and absorbance readings were at 540 nm.
Chitosan digestion, purification, and conversion to chitin
Chitosan was cleaved by pepsin to reduce the polymer length, purified and converted to chitin as previously described (14). Briefly, chitosan (250 mg) was dissolved in 25 ml 0.1 M sodium acetate, pH 4.5. Pepsin (Sigma, P7000) was added (100 U/ml) for 18 h at 37°C to partially digest the chitosan (46). This was followed by extraction with chloroform:isoamyl alcohol (24:1) then mixing the recovered aqueous layer with an equal volume of 12% potassium hydroxide and heating at 80°C for 90 min. Precipitated chitosan was collected by centrifugation and washed 3x with water followed by PBS to neutralize. Half of the chitosan was converted to chitin by suspending in 20 ml 1.0 M sodium bicarbonate, followed by addition of 1 ml acetic anhydride (Acros) and incubation at 22°C for 20 min with periodic mixing. The acetylation reaction was repeated, and terminated with heating at 100°C for 10 min. Chitin was collected by centrifugation and washed 3x with PBS. The degree of acetylation for chitin was approximately 93% as previously reported (14). Chitin and chitosan suspensions were passed through a 100 μm filter (BD Falcon) to remove the largest particles then treated in 0.1 M sodium hydroxide at 22°C for 30 min, followed by washing with PBS and storage at 4°C.
Stimulation of IL-1β release
Murine cells were plated at 1 x 105 cells/well in a 96-well plate. PBMCs were plated at 5x106/well in 24 well plates and after 1 h, non-adherent cells were washed away. Mouse cells were primed with 100 ng/ml ultrapure LPS, while PBMCs were primed with 50 pg/ml ultrapure LPS for 3 h (control cells were left unprimed), followed by incubation with the stimuli for 6 h (18 h for PBMCs). Positive stimuli controls included silica (topsize 15 microns, US Silica, MIN-U-SIL-15, used as described (29)), synthetic double stranded DNA: poly(dA:dT), ATP, and streptolysin O (SLO) or SLO + flagellin (FLA-ST, InvivoGen). Supernatants were collected for cytokine measurement and assayed by ELISA for IL-1β (eBioscience) or IL-18 (MBL International). Western blotting was performed as described (14) to confirm that the IL-1β was processed. Only mature IL-1β was detected; no pro-IL-1β was identified (data not shown). For K+ efflux, ROS and lysosomal inhibition assays, the inhibitors were added 1 h prior to secondary stimuli addition. K+ efflux was inhibited with glibenclamide, mitochondrial ROS was inhibited with Mito-TEMPO (Enzo Life Sciences), total ROS inhibited with diphenyleneiodonium chloride (DPI), lysosomal acidification was inhibited with bafilomycin A1 or chloroquine, and cathepsin B was selectively inhibited with CA-074-Me (Enzo Life Sciences). All inhibitors were assayed for cytotoxicity using the cytotoxicity detection kit (LDH) from Roche and none of the inhibitors at the concentrations used induced significant cell death above background (data not shown).
Inhibition of K+ efflux with high extracellular potassium
BMMΦ were plated and primed as described above. Ten min after stimuli were added, cells given sSLO or SLO + flagellin were washed three times with fresh media. Supernatants were collected for all stimuli after 2 h and analyzed by ELISA. For the K+ and Na+ buffer experiments, after the priming step the media was replaced with either K+ buffer (150 mM KCl, 5 mM NaH2PO4, 10 mM HEPES, 1 mM MgCl2, 1 mM CaCl2, 1% BSA) or Na+ buffer (150 mM NaCl, 5 mM KH2PO4, 10 mM HEPES, 1 mM MgCl2, 1 mM CaCl2, 1% BSA). SLO and SLO + flagellin treated cells were again washed three times after 10 minutes incubation and fresh buffer added, then supernatants for all stimuli were collected after 2 h and analyzed by ELISA for IL-1β.
Multiplex Assay
BMMΦ were plated as described above and stimulated for 6 h with 0.1 mg/ml chitin or chitosan. Supernatants were collected and analyzed by Bio-Plex Pro Assays (Bio-Rad).
Statistical Analysis
Data were analyzed and figures prepared using GraphPad Prism. Significance was assessed by 2-way ANOVA with Bonferroni post-hoc test, 1-way ANOVA with Dunnett post-hoc test, or Kruskal-Wallis 1-way ANOVA by ranks with Dunn post-hoc tests, as indicated. p-values of <0.05 after correction for multiple comparisons were considered significant.
Results
Chitosan, but not chitin, induces an IL-1β response in all cell types tested
We previously demonstrated that chitosan elicited a robust NLRP3 inflammasome-dependent IL-1β response in BMMΦ, while little IL-1β was elicited by chitin (14). To characterize the spectrum of cells that release IL-1β in response to chitosan and chitin, we studied a variety of cultured and primary cell types from mice and humans. First, to skew macrophages along a spectrum ranging from classically activated (M1) to alternatively activated (M2) phenotypes, mouse bone marrow cells were cultured in M-CSF, M-CSF + IL-4, GM-CSF, or GM-CSF + IFNγ (42, 47). M1/M2 skewing was assessed by assaying for NO and arginase, respectively (Fig. 1A and 1B). Culture with M-CSF resulted in an intermediate phenotype with little NO or arginase activity observed. The addition of IL-4 to M-CSF strongly biased the cells towards an M2 phenotype. In contrast, GM-CSF induced an M1 type phenotype which was more pronounced with the addition of IFNγ. An IL-1β response was induced by chitosan in all four cell types (Fig. 1C). The response to chitosan was more pronounced in the GM-CSF-promoted M1 phenotype, which is consistent with the response to other stimuli (20). No significant differences were observed between the responses with M-CSF alone and M-CSF plus IL-4 following chitosan stimulation. Interestingly though, the addition of IL-4 to M-CSF did result in a diminished IL-1β response to silica stimulation. We also observed a robust IL-1β response to chitosan in the primary peritoneal macrophages (Fig. 2A) and BMDCs (Fig. 2B). Finally we tested if human PBMCs respond to chitosan similarly as the mouse cells tested, and once again we saw a strong IL-1β response (Fig. 2C). Significant amounts of IL-1β above background levels were not released in response to chitin for any of the cell types tested (Figs. 1C and 2).
Figure 2. Chitosan, but not chitin, induces IL-1β release from mouse peritoneal macrophages, BMDCs and human PBMCs.

Mouse peritoneal macrophages (A) and BMDCs (B) were plated, primed and stimulated as in Fig. 1C. For peritoneal cells, p < 0.05 comparing chitosan to unstimulated and silica to unstimulated as analyzed by 1-way ANOVA. For BMDCs, p <0.01 comparing chitosan to unstimulated as analyzed by 1-way ANOVA. C. Human PBMCs were plated at 5 x 106/well in a 24-well plate, primed with 50 pg/ml LPS, and then stimulated overnight (same stimuli concentrations as in Fig. 1C). Supernatants were collected and analyzed by ELISA for IL-1β. p < 0.001 comparing unstimulated to chitosan and silica as analyzed by Kruskal-Wallis test. Data are means ± SE of three independent experiments, each performed in triplicate.
The IL-1β response of murine cells to chitosan is NLRP3-dependent
Our previous study demonstrated the inflammasome response induced by chitosan in BMMΦ was NLRP3 dependent (14). NLRP3−/− mice were used to generate peritoneal macrophages and DCs, which were then challenged with chitosan to assess if inflammasome activation in these cell types was NLRP3-dependent. For both peritoneal macrophages and DCs, IL-1β induced by chitosan was abolished in the NLRP3−/− cells (Fig. 3).
Figure 3. IL-1β release from mouse peritoneal cells and BMDCs is NLRP3-dependent.
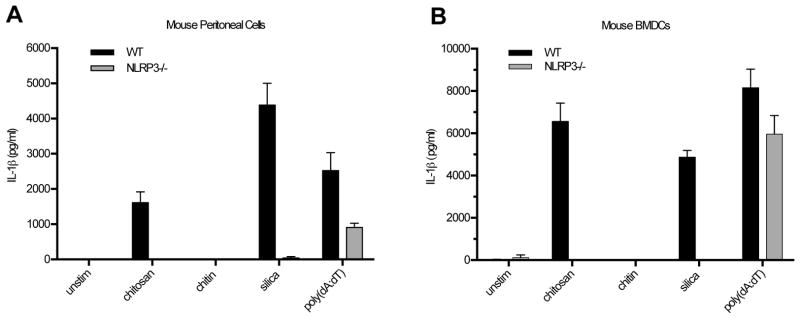
Peritoneal cells (A) and DCs (B) were cultured and stimulated as described in Figure 2, with the addition of the AIM2 inflammasome stimulus, poly(dA:dT) (2 μg/ml). A. p < 0.05 comparing WT to NLRP3−/− cells for chitosan and p < 0.0001 for silica. B. p < 0.0001 comparing WT to NLRP3−/− cells for chitosan and p < 0.0001 for silica. All comparisons were analyzed by 2-way ANOVA. Data are means ± SE of two (A) and three (B) independent experiments, each performed in triplicate.
Chitosan, but not chitin, induces IL-18
To further analyze the spectrum of the inflammasome response to chitosan, we assayed release of the inflammasome-related cytokine, IL-18, in BMMΦ. As was observed when measuring IL-1β release, IL-18 release was stimulated by chitosan and silica, but not chitin (Fig. 4).
Figure 4. Chitosan, but not chitin, induces IL-18 in BMMΦ.

BMMΦ (1 x 105/well) were primed for 3 h with 100 ng/ml LPS, and then left unstimulated or stimulated for 6 h with chitosan, chitin, or silica (all at 0.1 mg/ml). Supernatants were collected and analyzed by ELISA for IL-18. p < 0.001 comparing unstimulated to chitosan or silica and comparing chitin to chitosan or silica as analyzed by 1-way ANOVA. Data are means ± SE of three independent experiments, each performed in triplicate.
Importance of K+ efflux for chitosan-stimulated IL-1β release
To understand how chitosan stimulates the NLRP3 inflammasome, we analyzed each of the three proposed mechanisms mentioned above in murine BMMΦ. To examine the contribution of cellular K+ efflux, we blocked K+ efflux using the K+ ion channel inhibitor glibenclamide (Fig. 5A). The release of IL-1β in response to both chitosan and silica was significantly inhibited in a dose-dependent manner by glibenclamide. However, the IPAF inflammasome activator flagellin (delivered to the cytosol by SLO) still induced IL-1β release even at the highest dose of inhibitor tested. To further confirm a role for K+ efflux, we destroyed the gradient required for K+ efflux by replacing the extracellular media with a buffer containing 150 mM K+, which is approximately equal to the intracellular K+ concentration. As a control we used a buffer containing 150 mM Na+ which preserves the K+ gradient. While all three stimuli induced IL-1β in the Na+ buffer, neither chitosan nor silica induced significant quantities of IL-1β in the high K+ buffer (Fig. 5B). Once again, flagellin delivered with SLO induced IL-1β even when the K+ gradient was collapsed. Thus, K+ efflux appears to be required for chitosan inflammasome activation.
Figure 5. K+ efflux is required for NLRP3 inflammasome activation by chitosan.
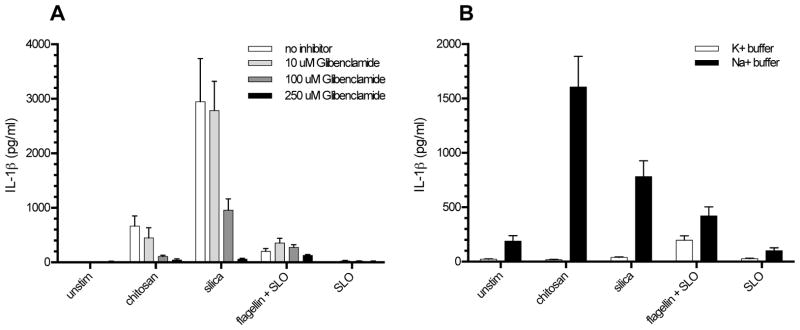
A, BMMΦ (1 x 105/well) were primed for 2 h with 100 ng/ml LPS. Glibenclamide (10 μM, 100 μM, or 250 μM) was then added to the wells receiving inhibitor 1 h prior to addition of the following stimuli: chitosan (0.1 mg/ml), silica (0.1 mg/ml), SLO (5 μg/ml), and flagellin (1 μg/ml) + SLO. Control wells were left unstimulated (unstim). After 6 h, supernatants were collected and analyzed for IL-1β by ELISA. p < 0.0001 comparing 100 μM and 250 μM to no inhibitor for both chitosan and silica, p < 0.05 comparing 10 μM to no inhibitor for chitosan as analyzed by 2-way ANOVA. B, BMMΦ were primed for 3 h with 100 ng/ml LPS. After priming, the media was replaced with K+ buffer or Na+ buffer followed by the addition of stimuli as above. p < 0.001 comparing chitosan and silica to unstim in Na+ buffer and comparing K+ buffer to Na+ buffer in the presence of chitosan and silica as analyzed by 2-way ANOVA. Data are means ± SE of three independent experiments, each performed in triplicate.
Effect of mitochondrial ROS on chitosan-induced IL-1β release
To analyze the role of mitochondrial ROS we used the mitochondrial ROS inhibitor, Mito-TEMPO. Using an inhibitor concentration of 100 μM, there was a significant reduction in IL-1β release induced by both chitosan and silica (Fig. 6A). To demonstrate specificity, we examined the effect of Mito-TEMPO on IL-1β release stimulated by the ROS-independent AIM2 inflammasome activator poly(dA:dT) (48). No significantly inhibition of poly(dA:dT)-stimulated IL-1β was observed in the presence of Mito-TEMPO (Fig. 6A). A less specific ROS inhibitor, DPI, blocked IL-1β release in response to both NLRP3 and AIM2 stimuli (Fig. 6B). These data suggest that mitochondrial ROS is required for optimal NLRP3 inflammasome stimulation by chitosan.
Figure 6. ROS is required for NLRP3 inflammasome activation by chitosan.
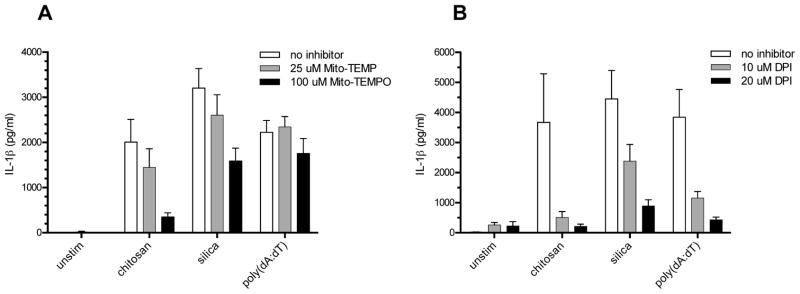
BMMΦ (1 x 105/well) were primed for 2 h with 100 ng/ml LPS. A, Mito-TEMPO (25 μM or 100 μM) or B, DPI (10 μM or 20 μM) was added to the indicated wells 1 h prior to addition of the following stimuli: chitosan (0.1 mg/ml), silica (0.1 mg/ml), and poly(dA:dT) (2 μg/ml). Control wells were left unstimulated (unstim). After 6 h, supernatants were collected and analyzed for IL-1β by ELISA. For Mito-TEMPO, p < 0.01 comparing 100 μM to no inhibitor for both chitosan and silica as analyzed by 2-way ANOVA. For DPI, p < 0.001 comparing 20 μM to no inhibitor for chitosan, silica and poly(dA:dT), p < 0.01 comparing 10 μM to no inhibitor for chitosan and poly(dA:dT), and p < 0.05 comparing 10 μM to no inhibitor for silica as analyzed by 2-way ANOVA. Data are means ± SE of three independent experiments, each performed in triplicate.
The role of lysosomal destabilization in chitosan-stimulated IL-1β release
Upon phagolysosomal fusion, acidification occurs which has been shown to be necessary for NLRP3 inflammasome activation through the lysosomal destabilization pathway (29). Lysosomal acidification was inhibited with either bafilomycin A1 (Fig. 7A) or chloroquine (Fig. 7B). IL-1β release from both chitosan and silica was significantly inhibited by bafilomycin A concentrations as low as 10 nM. Chloroquine concentrations as low as 1 μM significantly blocked IL-1β release induced by silica. A trend, albeit not statistically significant, towards a reduction in IL-1β release induced by chitosan in the presence of chloroquine was noted. To further explore the lysosomal destabilization pathway, we inhibited cathepsin B using CA-074-me (Fig. 7C). Upon lysosomal destabilization, cathepsin B is likely released into the cytosol where it activates the NLRP3 inflammasome (29). IL-1β release by the particulate stimuli chitosan and silica was significantly inhibited at CA-074-me concentrations of 10 μM. IL-1β release induced by the soluble inflammasome stimulator ATP was unaffected by these inhibitors, as expected for a phagocytosis-independent stimulus.
Figure 7. Lysosomal destabilization is required for NLRP3 inflammasome activation by chitosan.

BMMΦ (1 x 105/well) were primed for 2 h with 100 ng/ml LPS. A, Bafilomycin A1 (0.4 nM, 2 nM, 10 nM, 50 nM, or 250 nM) B, Chloroquine (1 μM, 10 μM, or 100 μM) and C, Ca-074-me (0.1 μM, 1 μM, or 10 μM) were added to the wells receiving inhibitor 1 h prior to addition of the following stimuli: chitosan (0.1 mg/ml), silica (0.1 mg/ml) and ATP (5 mM). Control wells were left unstimulated (unstim). After a 6 h stimulation period, supernatants were collected and analyzed for IL-1β by ELISA. For bafilomycin A1, p < 0.01 comparing 10 μM, 50 μM, and 250 μM to no inhibitor with chitosan stimulation, p < 0.05 comparing 10 μM to no inhibitor with silica stimulation, and p < 0.0001 comparing 50 μM and 250 μM to no inhibitor with silica stimulation. For chloroquine, p < 0.0001 comparing any inhibitor concentration to no inhibitor in the presence of silica. For CA-074-me, p < 0.001 comparing 10 μM to no inhibitor in the presence of chitosan or silica. Comparisons are by 2-way ANOVA. Data are means ± SE of three independent experiments, each performed in triplicate.
Spectrum of cytokines and chemokines elicited by chitosan and chitin
The above studies examined IL-1β release from primed BMMΦ. To examine the spectrum of cytokines and chemokines stimulated by chitosan and chitin, a multiplex assay was run on the supernatants from unprimed BMMΦ stimulated with these polysaccharides (Table I). None of the 22 cytokines and chemokines assayed was significantly induced by either chitosan or chitin. Trends of higher responses for chitosan compared to chitin were observed for some of the cytokines and chemokines, but these did not achieve statistical significance following corrections for multiple comparisons. The positive control, LPS, stimulated significant amounts of all the cytokines and chemokines tested except MCP-1 and RANTES.
Table I.
Induction of cytokines and chemokines by chitosan and chitin.
| unstim | chitin | chitosan | LPS | |
|---|---|---|---|---|
| IL-1α | 0.2 ± 0.1 | 1.6 ± 0.7 | 4.5 ± 1.2 | 60.5 ± 10.2* |
| IL-2 | 1.3 ± 0.6 | 3.9 ± 1.4 | 4.7 ± 1.6 | 19.3 ± 3.9* |
| IL-3 | 0.4 ± 0.2 | 1.3 ± 0.2 | 2.3 ± 0.5 | 19.2 ± 3.5* |
| IL-4 | 0.6 ± 0.2 | 1.6 ± 0.3 | 3.0 ± 0.6 | 26.9 ± 4.8* |
| IL-5 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 6.9 ± 1.0* |
| IL-6 | 0.9 ± 0.7 | 1.7 ± 1.1 | 6.5 ± 2.9 | 489.7 ± 85.9* |
| IL-9 | 9.6 ± 6.3 | 23.6 ± 9.1 | 52.2 ± 14.6 | 517.2 ± 89.8* |
| IL-10 | 2.9 ± 1.3 | 5.4 ± 1.4 | 10.3 ± 2.1 | 145.8 ± 22.6* |
| IL-12(p40) | 3.5 ± 1.3 | 5.3 ± 0.9 | 5.4 ± 1.2 | 1002.2 ± 322.7* |
| IL-12(p70) | 2.9 ± 2.0 | 11.4 ± 2.8 | 23.9 ± 4.2 | 221.9 ± 36.8* |
| IL-13 | 2.5 ± 1.2 | 23.3 ± 7.0 | 53.9 ± 13.5 | 774.6 ± 74.1* |
| IL-17 | 0 ± 0 | 0.2 ± 0.1 | 1.3 ± 0.3 | 17.7 ± 2.2* |
| Eotaxin | 6.8 ± 6.8 | 124.2 ± 49.2 | 251.9 ± 63.9 | 2359.3 ± 370.0* |
| G-CSF | 2.9 ± 1.6 | 5.4 ± 2.5 | 5.2 ± 1.5 | 316.7 ± 110.9* |
| GM-CSF | 0 ± 0 | 4.3 ± 2.8 | 8.3 ± 4.2 | 135.6 ±22.5* |
| IFN-γ | 0.9 ± 0.9 | 2.8 ± 1.1 | 10.8 ± 1.8 | 162.9 ±29.4* |
| KC | 55.8 ± 12.5 | 68.7 ± 13.6 | 80.3 ± 15.3 | 910.8 ± 147.6* |
| MCP-1 | 191.0 ± 50.8 | 263.4 ± 52.3 | 362.2 ± 68.9 | 1026.6 ± 193.1 |
| MIP-1α | 34.3 ± 10.6 | 106.9 ± 23.7 | 269.7 ± 57.7 | 22089.2 ± 5358.4* |
| MIP-1β | 280.4 ± 57.2 | 832.6 ± 137.9 | 1295.5 ± 139.7 | 30927.4 ± 6449.7* |
| RANTES | 1261.7 ± 254.5 | 1841.9 ± 367.2 | 2044.7 ± 329.7 | 10783.2 ± 1222.5 |
| TNFα | 0.2 ± 0.2 | 0.5 ± 0.3 | 1.2 ± 0.7 | 709.5 ± 340.6* |
Unprimed BMMΦ (1 x 105/well) were left unstimulated (unstim) or stimulated with chitosan (0.1 mg/ml), chitin (0.1 mg/ml) or LPS (100 ng/ml) for 6 h. Supernatants were analyzed by multiplex assay.
p < 0.01 by the Kruskal-Wallis test. Data are means (pg/ml) ± SE of two independent experiments, each with quadruplicate determinations. Shaded boxes indicate that the mean value was below the lower limit of detection for the assay.
Discussion
We previously demonstrated that phagocytosable particles of chitosan, but not chitin, elicited a strong NLRP3 inflammasome response in BMMΦ (14). Here we examined the capacity of these particulate glycans to stimulate responses in other cell populations, characterized the mechanisms responsible for inflammasome activation by chitosan, and more fully elucidated the profile of cytokines and chemokines stimulated by chitosan and chitin. Chitosan induced significant IL-18 and IL-1β responses, activating the inflammasome of a broad range of cell populations and across species, while chitin failed to induce a significant response from the cell types tested. While macrophages polarized towards M1, M2 and intermediate phenotypes were all stimulated by chitosan, the M1 response was greater, consistent with their more pro-inflammatory nature. The similar chitosan-stimulated IL-1β responses of intermediate phenotype and M2 macrophages suggest that the M1 phenotype is a primary driver of inflammasome activation. Indeed, the IL-1β response seen from the M2 macrophages may be partly explained by the plasticity macrophages and a switch towards a more M1 phenotype caused by the LPS priming step (49). The finding that chitosan stimulates IL-1β secretion in human cells has implications for the translational use of this polysaccharide. For example, the use of chitosan as a drug or vaccine delivery system could result in a potent, targeted inflammatory response (38).
In analyzing the mechanistic basis for NLRP3 inflammasome activation, we found evidence that K+ efflux, mitochondrial ROS, and lysosomal destabilization each contribute to chitosan activation of the NLRP3 inflammasome. K+ efflux from the cytosol has been shown to be required for NLRP3 inflammasome activation by many different stimuli. Some of the most potent NLRP3 inflammasome inducers are in fact K+ channels inducers, including nigericin, gramicidin, maitotoxin and α-toxin (50). Originally, these were thought to act by creating pores that allowed the NLRP3 stimuli access to the cytoplasm, whereby they could potentially interact directly with NRLP3. However, direct interaction with NLRP3 is unlikely given the variety of stimuli, therefore NLRP3 activation may be through sensing the K+ efflux by an unknown mechanism. Inhibition of chitosan-induced IL-1β release through blocking K+ efflux is consistent with previous published reports for other NLRP3 inflammasome stimuli (16, 50), showing that chitosan also requires K+ efflux to activate the NLRP3 inflammasome.
The second proposed model for NLRP3 inflammasome activation involves ROS induced activation of NRLP3. Some debate exists on what type of ROS is important for NLRP3 activation as ROS scavengers and inhibitors strongly impede NLRP3 activation (51, 52). In addition, it has been shown that knock-down of the p22phox subunit of the NOX 1–4 subfamily of NADPH oxidases also impairs NLRP3 activation (51). However, multiple studies have shown that cells from patients with chronic granulomatous disease (CGD), a disease with defects in NADPH oxidase, exhibit no NLRP3 deficiencies (53–55). Recent work has strongly suggested that the important ROS for NLRP3 inflammasome activation is mitochondrial in origin and that mitochondria are central to NLRP3 activation (26, 27). Oxidized mitochondrial DNA has also been proposed to act as an agonist of NLRP3 (56). Here we demonstrated that blocking mitochondrial ROS inhibited chitosan induced IL-1β release. The targeted mitochondrial ROS inhibitor had no significant effect on the AIM2 inflammasome activator poly(dA:dT), whereas the less specific ROS inhibitor, DPI blocked IL-1β for both NLRP3 and AIM2 stimuli. ROS may not only be involved in the NLRP3 activation but may also be important for the priming step, the upregulation of pro-IL-1β and NLRP3 (48). This could explain why DPI has more far reaching effects than the more targeted Mito-TEMPO.
Acidification-dependent lysosomal destabilization resulting in release of cathepsin B and subsequent activation of the NLRP3 inflammasome has been demonstrated following phagocytosis of particulate stimuli including silica and alum (29). We hypothesized that the polycationic properties of chitosan would enable its escape from lysosomes by sponging protons delivered by the vacuolar-ATPase. This in turn would lead to the retention of Cl− ions and water molecules resulting in lysosome swelling, leakage and eventual rupture (57, 58). Indeed, we found that inhibition of lysosomal acidification (which is required for the proton sponge effect), prevented chitosan from activating the inflammasome. The neutral charge of chitin due to acetylation of the polymer is a possible reason for its inability to activate the inflammasome. We were also able to block chitosan-induced NLRP3 inflammasome activation by blocking the downstream proposed activator of the lysosomal destabilization mechanism, cathepsin B, with the cathepsin B specific inhibitor Ca-074-me. Inhibition was incomplete which may be due to stimulation of the NLRP3 inflammasome by other lysosomal contents, such as cathepsin L or cathepsin D (59–61).
The preparations of chitosan and chitin that were used in our studies were extensively purified to remove potential contaminants including proteins, nucleic acids and lipopolysaccharides. The purification steps included solubilization, chloroform:isoamyl alcohol extraction, reprecipitation, hot-alkali treatment and extensive washing. Using the resulting ultrapure preparations, chitin and chitosan did not stimulate unprimed macrophages to release statistically significant quantities of any of the 22 cytokines and chemokines analyzed by multiplex assay. However, due to our use of stringent criteria to correct for the large number of comparisons, the possibility of a Type II error causing us to miss significant associations cannot be ruled out. Indeed some of the cytokines and chemokines trended higher, with chitosan being a more potent stimulus compared with chitin. It should also be noted that the concentrations of cytokines and chemokines stimulated by chitosan and chitin were relatively low compared to LPS.
Others have reported that chitin can elicit IL-10, TNFα, IL-17A, IL-12, or IL-18 (39, 62, 63). The explanation for discrepancies between these published data and ours is speculative but may be due to differences in chitin source, contaminants, tertiary structures and/or particle size (38). Less work has been done on the immunological activity of chitosan, although our inflammasome and multiplex assays suggest that chitosan is the more immunostimulatory of the two polymers. Defining the conditions under which chitin and chitosan trigger or fail to trigger immune responses has translational relevance given frequent natural exposure and the increasing use of these polymers in biomedical applications.
Acknowledgments
The work was supported by National Institutes of Health Grants, R21 AI093302, RO1 AI025780, RO1 AI072195, RO1 AI072195, RO1 HL112671 and T32 AI095213; and National Science Foundation grant 1022336.
The authors thank Dr. Kate Fitzgerald for helpful discussions and providing the NLRP3−/− mice, Dr. Vijay Rathinam for help with the western blots and Dr. Sumanth Gandra for statistical assistance.
References
- 1.Bartnicki-Garcia S. Cell wall chemistry, morphogenesis, and taxonomy of fungi. Annu Rev Microbiol. 1968;22:87–108. doi: 10.1146/annurev.mi.22.100168.000511. [DOI] [PubMed] [Google Scholar]
- 2.Banks IR, Specht CA, Donlin MJ, Gerik KJ, Levitz SM, Lodge JK. A chitin synthase and its regulator protein are critical for chitosan production and growth of the fungal pathogen Cryptococcus neoformans. Eukaryot Cell. 2005;4:1902–1912. doi: 10.1128/EC.4.11.1902-1912.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boot RG, Renkema GH, Verhoek M, Strijland A, Bliek J, de Meulemeester TMAMO, Mannens MMAM, Aerts JMFG. The Human Chitotriosidase Gene. Journal of Biological Chemistry. 1998;273:25680–25685. doi: 10.1074/jbc.273.40.25680. [DOI] [PubMed] [Google Scholar]
- 4.Neville AC, Parry DA, Woodhead-Galloway J. The chitin crystallite in arthropod cuticle. Journal of Cell Science. 1976;21:73–82. doi: 10.1242/jcs.21.1.73. [DOI] [PubMed] [Google Scholar]
- 5.Boot RG, Blommaart EFC, Swart E, Ghauharali-van der Vlugt K, Bijl N, Moe C, Place A, Aerts JMFG. Identification of a Novel Acidic Mammalian Chitinase Distinct from Chitotriosidase. Journal of Biological Chemistry. 2001;276:6770–6778. doi: 10.1074/jbc.M009886200. [DOI] [PubMed] [Google Scholar]
- 6.Fuhrman JA, Piessens WF. Chitin synthesis and sheath morphogenesis in Brugia malayi microfilariae. Molecular and Biochemical Parasitology. 1985;17:93–104. doi: 10.1016/0166-6851(85)90130-6. [DOI] [PubMed] [Google Scholar]
- 7.Shahabuddin M, Kaslow DC. Plasmodium: Parasite Chitinase and Its Role in Malaria Transmission. Experimental Parasitology. 1994;79:85–88. doi: 10.1006/expr.1994.1066. [DOI] [PubMed] [Google Scholar]
- 8.Araujo AC, Souto-Padrón T, de Souza W. Cytochemical localization of carbohydrate residues in microfilariae of Wuchereria bancrofti and Brugia malayi. Journal of Histochemistry & Cytochemistry. 1993;41:571–578. doi: 10.1177/41.4.8450196. [DOI] [PubMed] [Google Scholar]
- 9.Debono M, Gordee RS. Antibiotics that inhibit fungal cell wall development. Annu Rev Microbiol. 1994;48:471–497. doi: 10.1146/annurev.mi.48.100194.002351. [DOI] [PubMed] [Google Scholar]
- 10.Jayakumar R, Prabaharan M, Nair SV, Tamura H. Novel chitin and chitosan nanofibers in biomedical applications. Biotechnol Adv. 2010;28:142–150. doi: 10.1016/j.biotechadv.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 11.Morganti P, Morganti G. Chitin nanofibrils for advanced cosmeceuticals. Clin Dermatol. 2008;26:334–340. doi: 10.1016/j.clindermatol.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 12.Nakagawa Y, Murai T, Hasegawa C, Hirata M, Tsuchiya T, Yagami T, Haishima Y. Endotoxin contamination in wound dressings made of natural biomaterials. J Biomed Mater Res B Appl Biomater. 2003;66:347–355. doi: 10.1002/jbm.b.10020. [DOI] [PubMed] [Google Scholar]
- 13.Read RC, Naylor SC, Potter CW, Bond J, Jabbal-Gill I, Fisher A, Illum L, Jennings R. Effective nasal influenza vaccine delivery using chitosan. Vaccine. 2005;23:4367–4374. doi: 10.1016/j.vaccine.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 14.Bueter CL, Lee CK, Rathinam VA, Healy GJ, Taron CH, Specht CA, Levitz SM. Chitosan but not chitin activates the inflammasome by a mechanism dependent upon phagocytosis. J Biol Chem. 2011;286:35447–35455. doi: 10.1074/jbc.M111.274936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787–791. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 17.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, Inohara N, Vandenabeele P, Bertin J, Coyle AJ, Grant EP, Nunez G. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in Salmonella-infected macrophages. Nature Immunology. 2006;7:576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 19.Stout RD, Suttles J. Functional plasticity of macrophages: reversible adaptation to changing microenvironments. Journal of Leukocyte Biology. 2004;76:509–513. doi: 10.1189/jlb.0504272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pelegrin P, Surprenant A. Dynamics of macrophage polarization reveal new mechanism to inhibit IL-1beta release through pyrophosphates. Embo J. 2009;28:2114–2127. doi: 10.1038/emboj.2009.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 22.Kool M, Petrilli V, De Smedt T, Rolaz A, Hammad H, van Nimwegen M, Bergen IM, Castillo R, Lambrecht BN, Tschopp J. Cutting Edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J Immunol. 2008;181:3755–3759. doi: 10.4049/jimmunol.181.6.3755. [DOI] [PubMed] [Google Scholar]
- 23.Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, Perfettini JL, Schlemmer F, Tasdemir E, Uhl M, Genin P, Civas A, Ryffel B, Kanellopoulos J, Tschopp J, Andre F, Lidereau R, McLaughlin NM, Haynes NM, Smyth MJ, Kroemer G, Zitvogel L. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1[beta]-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 24.Netea MG, Nold-Petry CA, Nold MF, Joosten LA, Opitz B, van der Meer JH, van de Veerdonk FL, Ferwerda G, Heinhuis B, Devesa I, Funk CJ, Mason RJ, Kullberg BJ, Rubartelli A, van der Meer JW, Dinarello CA. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood. 2009;113:2324–2335. doi: 10.1182/blood-2008-03-146720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, Panther E, Di Virgilio F. The P2X7 Receptor: A Key Player in IL-1 Processing and Release. The Journal of Immunology. 2006;176:3877–3883. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- 26.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 27.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free Radical Biology and Medicine. 2009;47:333–343. doi: 10.1016/j.freeradbiomed.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 29.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee CG, Da Silva CA, Lee JY, Hartl D, Elias JA. Chitin regulation of immune responses: an old molecule with new roles. Curr Opin Immunol. 2008;20:684–689. doi: 10.1016/j.coi.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reese TA, Liang HE, Tager AM, Luster AD, Van Rooijen N, Voehringer D, Locksley RM. Chitin induces accumulation in tissue of innate immune cells associated with allergy. Nature. 2007;447:92–96. doi: 10.1038/nature05746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wagner CJ, Huber S, Wirth S, Voehringer D. Chitin induces upregulation of B7-H1 on macrophages and inhibits T-cell proliferation. Eur J Immunol. 2010;40:2882–2890. doi: 10.1002/eji.201040422. [DOI] [PubMed] [Google Scholar]
- 33.Lee CG. Chitin, chitinases and chitinase-like proteins in allergic inflammation and tissue remodeling. Yonsei Med J. 2009;50:22–30. doi: 10.3349/ymj.2009.50.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shibata Y, Foster LA, Metzger WJ, Myrvik QN. Alveolar macrophage priming by intravenous administration of chitin particles, polymers of N-acetyl-D-glucosamine, in mice. Infect Immun. 1997;65:1734–1741. doi: 10.1128/iai.65.5.1734-1741.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shibata Y, Foster LA, Bradfield JF, Myrvik QN. Oral administration of chitin down-regulates serum IgE levels and lung eosinophilia in the allergic mouse. J Immunol. 2000;164:1314–1321. doi: 10.4049/jimmunol.164.3.1314. [DOI] [PubMed] [Google Scholar]
- 36.Aranaz I, Mengibar M, Harris R, Panos I, Miralles B, Acosta N, Galed G, Heras A. Functional Characterization of Chitin and Chitosan. Current Chemical Biology. 2009;3:203–230. [Google Scholar]
- 37.Mora-Montes HM, Netea MG, Ferwerda G, Lenardon MD, Brown GD, Mistry AR, Kullberg BJ, O’Callaghan CA, Sheth CC, Odds FC, Brown AJP, Munro CA, Gow NAR. Recognition and Blocking of Innate Immunity Cells by Candida albicans Chitin. Infect Immun. 2011:IAI.01282–01210. doi: 10.1128/IAI.01282-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bueter CL, Specht CA, Levitz SM. Innate sensing of chitin and chitosan. PLoS Pathog. 2013;9:e1003080. doi: 10.1371/journal.ppat.1003080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Da Silva CA, Chalouni C, Williams A, Hartl D, Lee CG, Elias JA. Chitin is a size-dependent regulator of macrophage TNF and IL-10 production. J Immunol. 2009;182:3573–3582. doi: 10.4049/jimmunol.0802113. [DOI] [PubMed] [Google Scholar]
- 40.Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ. Cutting Edge: Repurification of Lipopolysaccharide Eliminates Signaling Through Both Human and Murine Toll-Like Receptor 2. The Journal of Immunology. 2000;165:618–622. doi: 10.4049/jimmunol.165.2.618. [DOI] [PubMed] [Google Scholar]
- 41.Johnson CR, Kitz D, Little JR. A method for the derivation and continuous propagation of cloned murine bone marrow macrophages. Journal of Immunological Methods. 1983;65:319–332. doi: 10.1016/0022-1759(83)90127-8. [DOI] [PubMed] [Google Scholar]
- 42.Fleetwood AJ, Dinh H, Cook AD, Hertzog PJ, Hamilton JA. GM-CSF- and M-CSF-dependent macrophage phenotypes display differential dependence on type I interferon signaling. J Leukoc Biol. 2009;86:411–421. doi: 10.1189/jlb.1108702. [DOI] [PubMed] [Google Scholar]
- 43.Huang H, Ostroff GR, Lee CK, Agarwal S, Ram S, Rice PA, Specht CA, Levitz SM. Relative Contributions of Dectin-1 and Complement to Immune Responses to Particulate β-Glucans. The Journal of Immunology. 2012;189:312–317. doi: 10.4049/jimmunol.1200603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miranda KM, Espey MG, Wink DA. A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite. Nitric Oxide. 2001;5:62–71. doi: 10.1006/niox.2000.0319. [DOI] [PubMed] [Google Scholar]
- 45.Corraliza IM, Campo ML, Soler G, Modolell M. Determination of arginase activity in macrophages: a micromethod. J Immunol Methods. 1994;174:231–235. doi: 10.1016/0022-1759(94)90027-2. [DOI] [PubMed] [Google Scholar]
- 46.Roncal T, Oviedo A, Lopez de Armentia I, Fernandez L, Villaran MC. High yield production of monomer-free chitosan oligosaccharides by pepsin catalyzed hydrolysis of a high deacetylation degree chitosan. Carbohydr Res. 2007;342:2750–2756. doi: 10.1016/j.carres.2007.08.023. [DOI] [PubMed] [Google Scholar]
- 47.Kahnert A, Seiler P, Stein M, Bandermann S, Hahnke K, Mollenkopf H, Kaufmann SHE. Alternative activation deprives macrophages of a coordinated defense program to Mycobacterium tuberculosis. European Journal of Immunology. 2006;36:631–647. doi: 10.1002/eji.200535496. [DOI] [PubMed] [Google Scholar]
- 48.Bauernfeind F, Bartok E, Rieger A, Franchi L, Núñez G, Hornung V. Cutting Edge: Reactive Oxygen Species Inhibitors Block Priming, but Not Activation, of the NLRP3 Inflammasome. The Journal of Immunology. 2011;187:613–617. doi: 10.4049/jimmunol.1100613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 50.Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14:1583–1589. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 51.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, Carter AB, Rothman PB, Flavell RA, Sutterwala FS. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A. 2008;105:9035–9040. doi: 10.1073/pnas.0803933105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Bruggen R, Koker MY, Jansen M, van Houdt M, Roos D, Kuijpers TW, van den Berg TK. Human NLRP3 inflammasome activation is Nox1–4 independent. Blood. 2010;115:5398–5400. doi: 10.1182/blood-2009-10-250803. [DOI] [PubMed] [Google Scholar]
- 54.van de Veerdonk FL, Smeekens SP, Joosten LA, Kullberg BJ, Dinarello CA, van der Meer JW, Netea MG. Reactive oxygen species-independent activation of the IL-1beta inflammasome in cells from patients with chronic granulomatous disease. Proc Natl Acad Sci U S A. 2010;107:3030–3033. doi: 10.1073/pnas.0914795107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meissner F, Seger RA, Moshous D, Fischer A, Reichenbach J, Zychlinsky A. Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood. 2010;116:1570–1573. doi: 10.1182/blood-2010-01-264218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kumar MN, Muzzarelli RA, Muzzarelli C, Sashiwa H, Domb AJ. Chitosan chemistry and pharmaceutical perspectives. Chem Rev. 2004;104:6017–6084. doi: 10.1021/cr030441b. [DOI] [PubMed] [Google Scholar]
- 58.Nel AE, Madler L, Velegol D, Xia T, Hoek EMV, Somasundaran P, Klaessig F, Castranova V, Thompson M. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8:543–557. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 59.Dostert C, Guarda G, Romero JF, Menu P, Gross O, Tardivel A, Suva ML, Stehle JC, Kopf M, Stamenkovic I, Corradin G, Tschopp J. Malarial Hemozoin Is a Nalp3 Inflammasome Activating Danger Signal. PLoS ONE. 2009;4:e6510. doi: 10.1371/journal.pone.0006510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Da Silva CA, Hartl D, Liu W, Lee CG, Elias JA. TLR-2 and IL-17A in chitin-induced macrophage activation and acute inflammation. J Immunol. 2008;181:4279–4286. doi: 10.4049/jimmunol.181.6.4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shibata Y, Foster LA, Bradfield JF, Myrvik QN. Oral Administration of Chitin Down-Regulates Serum IgE Levels and Lung Eosinophilia in the Allergic Mouse. The Journal of Immunology. 2000;164:1314–1321. doi: 10.4049/jimmunol.164.3.1314. [DOI] [PubMed] [Google Scholar]