

Abstract
Background
Economically, Leuconostoc lactis is one of the most important species in the genus Leuconostoc. It plays an important role in the food industry including the production of dextrans and bacteriocins. Currently, traditional molecular typing approaches for characterisation of this species at the isolate level are either unavailable or are not sufficiently reliable for practical use. Multilocus sequence typing (MLST) is a robust and reliable method for characterising bacterial and fungal species at the molecular level. In this study, a novel MLST protocol was developed for 50 L. lactis isolates from Mongolia and China.
Results
Sequences from eight targeted genes (groEL, carB, recA, pheS, murC, pyrG, rpoB and uvrC) were obtained. Sequence analysis indicated 20 different sequence types (STs), with 13 of them being represented by a single isolate. Phylogenetic analysis based on the sequences of eight MLST loci indicated that the isolates belonged to two major groups, A (34 isolates) and B (16 isolates). Linkage disequilibrium analyses indicated that recombination occurred at a low frequency in L. lactis, indicating a clonal population structure. Split-decomposition analysis indicated that intraspecies recombination played a role in generating genotypic diversity amongst isolates.
Conclusions
Our results indicated that MLST is a valuable tool for typing L. lactis isolates that can be used for further monitoring of evolutionary changes and population genetics.
Background
Leuconostoc species are usually found in association with plant, dairy, meat or other food products. Like other lactic acid bacteria (LAB), Leuconostoc species are important industrial starter microbes that are used in several industrial and food fermentation processes, such as the production of cheese, butter, buttermilk, kefir, sourdough and kimchi [1,2]. These species are closely related to heterofermentative species in the genus Lactobacillus[3]. Phenotypically, the genus Leuconostoc and Lactobacillus are often isolated from the same habitats and share many characteristics [4].
The genus Leuconostoc was first described by Van Tieghem [5]. In recent years, several species have been reclassified within the genus; some new species have been added and new genera have been erected from species previously considered to belong to Leuconostoc. For example, the species L. mesenteroides was reclassified into three subspecies: L. mesenteroides subsp. mesenteroides, L. mesenteroides subsp. dextranicum and L. mesenteroides subsp. cremoris[6]. A new species, L. fallax was identified from sauerkraut [7] and subsequently a number of L. fallax isolates have been found in the heterofermentative stage of sauerkraut fermentation [7,8]. The L. paramesenteroides group of species have been reclassified into a new genus, Weisella[8]; L. oenos has been reclassified into the genus Oenococcus as O. oeni[9] and L. durionis, L. ficulneum, L. pseudoficulneum and L. fructosum have been assigned to a new genus, Fructobacillus[10]. Furthermore, L. argentinum has been reclassified as a synonym of L. lactis following numerical analysis of repetitive extragenic palindromic-PCR patterns, whole-cell protein profiles (SDS-PAGE) and fluorescent amplified fragment length polymorphism (FAFLP) band patterns [11]. New species, including L. holzapfelii, L. palmae and L. miyukkimchii, have also been identified from wine and kimchi [12-14].
Typing methods for intraspecies identification of pathogens are essential epidemiological tools in infection prevention and control [15] and have also been applied to LAB. Typing methods are divided into two major categories i.e., phenotypic and genotypic methods. Traditional phenotyping methods, such as the use of serotypes, biotypes, phage-types and antibiograms, have been used for many years to isolate and characterise LAB and, sometimes, to distinguish between species and subspecies. Compared with phenotypic typing methods, genotypic typing methods have some advantages as they have more general applicability and greater discriminatory power. Currently, several molecular typing approaches, such as random amplified polymorphic DNA (RAPD)-PCR, pulsed-field gel electrophoresis (PFGE), restriction fragment length polymorphism (RFLP), protein fingerprinting, and repetitive element palindromic PCR (Rep-PCR), have been used to characterise Leuconostoc species [16-23].
Multilocus sequence typing (MLST) is a technique for distinguishing accurately between different isolates within a species. MLST is based on the principles of phenotypic multi-locus enzyme electrophoresis (MLEE). MLEE is a typing method that relies on differences in electrophoretic mobility of different enzymes present within a bacterium [15]. Maiden et al.,[24] first used the MLST method to identify virulent lineages of 107 isolates of Neisseria meningitides, a naturally transformable Gram-negative pathogenic bacterium [24]. Shortly thereafter, the method was used to analyse nonpathogenic food production bacteria including LAB. For example, Tanigawa and Watanabe [25] used MLST to compare seven housekeeping genes in 41 isolates of Lactobacillus delbrueckii and demonstrated that MLST was efficient for identification of isolates to subspecies level [25]. De Las Rivas et al.[26] compared the genetic diversity and genetic relationships amongst 18 O. oeni isolates using the gyrB, pgm, ddl, recP and mleA genes and MLST [26]. Bilhère et al. [27] found that MLST and pulsed-field gel electrophoresis (PFGE) were both useful for identifying 43 isolates of O. oeni, although the MLST method was more efficient [27]. Although the population biology of some LAB species has been characterised by MLST methods, to date, there is no MLST protocol available for Leuconostoc species.
The aim of the present study was to develop an effective MLST protocol for characterisation of L. lactis isolates and use this to explore the population structure and evolutionary relationships amongst isolates of this species.
Results
Assignment of sequence types
Fifty L. lactis isolates were typed using the MLST protocol. Isolates could be divided into 20 sequence types (STs) using combined data from eight loci. ST14 was the most frequent (21 isolates), followed by ST11 (four isolates), ST3 (three isolates), ST4 (three isolates), ST1 (two isolates), ST8 (two isolates) and ST12 (two isolates); there was only one isolate in each of the remaining 13 STs.
MLST protocol and allelic variation
Eight genes were successfully sequenced and analysed by MLST for all isolates in this study. Polymorphic sites, guanine-cytosine content, rate of non-synonymous (d N ) and synonymous (d S ) substitutions and the d N /d S for each locus (groEL, carB, recA, pheS, murC, pyrG, rpoB and uvrC ) were determined (Table 1). Fragment sizes of the eight selected loci ranged from 550 bp (recA) to 892 bp (groEL) (Table 2). The number of polymorphic sites per locus ranged from 3 (recA) to 9 (murC) and a total of 47 SNPs were identified (Table 1). The mean guanine-cytosine content of the partial sequence of the eight gene fragments ranged from 43.12% (pyrG) to 48.31% (recA), while it was 37.7% in the whole L. mesenteroides subsp. mesenteroides ATCC 8293 genome previously described [28]. The value of the non-synonymous (d N ) and synonymous (d S ) substitutions ranged from 0.0000 (groEL) to 0.0077 (murC) and 0.0556 (groEL) to 0.2852 (carB) respectively. The lowest d N /d S ratio (<1) calculated for all eight loci is suggestive of weak purifying selection pressure.
Table 1.
Allelic variation in 8 housekeeping genes
| Locus | Polymorphic sites | GC% content (mol%) | d N | d S | d N /d S * |
|---|---|---|---|---|---|
|
carB
|
4 |
44.09% |
0.0100 |
0.2852 |
0.0349 |
|
groEL
|
5 |
46.24% |
0.0000 |
0.0556 |
0.0000 |
|
murC
|
9 |
44.90% |
0.0077 |
0.2467 |
0.0313 |
|
pheS
|
5 |
45.26% |
0.0012 |
0.0900 |
0.0130 |
|
pyrG
|
8 |
43.12% |
0.0016 |
0.1356 |
0.0114 |
|
recA
|
3 |
48.31% |
0.0025 |
0.2399 |
0.0104 |
|
rpoB
|
7 |
43.97% |
0.0018 |
0.0715 |
0.0245 |
| uvrC | 6 | 43.68% | 0.0028 | 0.2684 | 0.0103 |
*The ratio of non-synonymous (d N ) and synonymous (d S ) substitutions is indicative of selective pressure on loci.
Table 2.
Genes and sequencing primers used
| Gene | Protein | PCR primers | Amplicon size (bp) | Location* |
|---|---|---|---|---|
|
pyrG |
CTP synthase |
5′-AGCAAACACCCAAGAACG-3′ |
598 |
481322 to 482935 |
| |
|
5′-TGGTGAAGCGAAGACAAA-3′ |
|
|
|
rpoB |
DNA-directed RNA polymerase subunit beta |
5′-CACTGTGCGGTCGTCTTCC-3′ |
608 |
1798123 to 1801731 |
| |
|
5′-GCGTTCTCCTGGTATCTATT-3′ |
|
|
|
groEL |
Chaperonin GroEL |
5′-CGGTGATAAGGCTGCTGT-3′ |
892 |
1734716 to 1736335 |
| |
|
5′-TTTGTTGGGTCCACGATA-3′ |
|
|
|
recA |
Recombinase A |
5′-GGAGTCGTTTCTGGGTTAC-3′ |
550 |
555064 to 556221 |
| |
|
5′-GTTGCTTTAGGCGTTGGTG-3′ |
|
|
|
uvrC |
Excinuclease ABC subunit C |
5′-AGAAATACAAGCCGTACTACAA-3′ |
560 |
483053 to 484852 |
| |
|
5′-TCTTCATCAGCGGAACCAA-3′ |
|
|
|
carB |
Carbamoyl phosphate synthase large subunit |
5′-ATGGGTTGTGGGAGTTGTA-3′ |
833 |
1202174 to 1205353 |
| |
|
5′-ACTTGTTGCGTCGTGGTGT-3′ |
|
|
|
murC |
UDP-N-acetylmuramate-L-alanine ligase |
5′-TTTCATAGGCGAACTCAT-3′ |
619 |
679802 to 681136 |
| |
|
5′-GTGCCATTGTTTGGTCAG-3′ |
|
|
|
pheS |
Phenylalanyl-tRNA synthetase subunit alpha |
5′-TTTCTTAGGTTTAGGCTTTG-3′ |
665 |
406737 to 407813 |
| 5′-CCTTTCGGTTAAATTGTGA-3′ |
*Positions correspond to the complete genome sequence of Leu. mesenteroides subsp. mesenteroides ATCC 8293.
Recombination in L. lactis
The level of linkage disequilibrium between all alleles of the isolates evaluated was high as the calculated I A S was 0.4264 (p = 0.000) and significantly different from the I A S value of 0 expected for a population with linkage equilibrium, indicating the genes investigated in this study were close to linkage disequilibrium.
Split decomposition analysis to examine evolutionary relationships amongst the isolates revealed different structures in the split graphs for all eight loci (Figure 1A). In the split graphs for murC, pheS, pyrG and uvrC, the parallelogram-shaped structures detected indicated that intergenic recombination had occurred during the evolution of these four genes. The split graphs obtained for carB, groEL, recA and rpoB loci revealed tree-like structures, suggesting that the descent of these genes was clonal and not significantly affected by intergenic recombination. The split graphs of the recA and carB genes were a polygonal line and columnar respectively because only three (recA) or four (carB) alleles were analysed.The combined split graph of alleles for all eight MLST loci displayed a network-like structure (Figure 1B). The 20 STs representing all isolates were divided into two main subpopulations and each subpopulation was completely disconnected. ST1 to ST10 were clustered together as one subpopulation, where some parallelogram-shaped groupings were detected. The split graphs for the remaining STs, clustered into a second subpopulation. This suggests that recombination had not occurred between isolates from the two subpopulations, but that intergenic recombination may occur between isolates from the same subpopulation during their evolution. ST19, which contained only isolate MAU80137 from non-traditional dairy production, was clearly disconnected from the others isolates, indicating no recombination had occurred between this isolate and other isolates from either of the two subpopulations.
Figure 1.
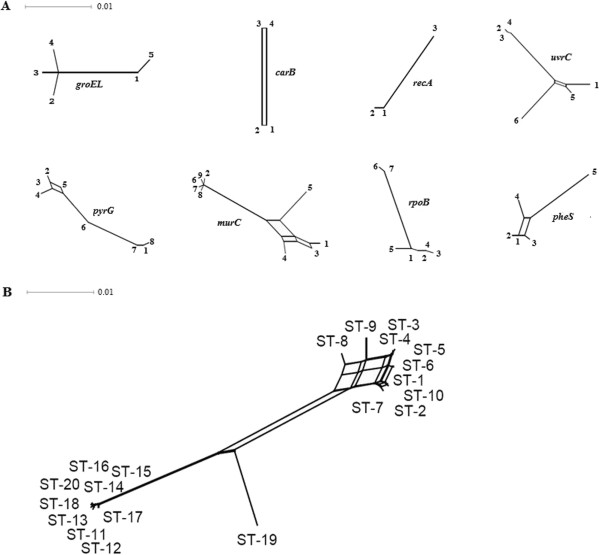
Split-decomposition analysis based on concatenated sequences of eight housekeeping genes from 50 L. lactis isolates. Multi-parallelogram formations indicate recombination events. (A) Split-decomposition analysis of individual MLST loci. (B) Combined split-decomposition analysis of all eight MLST loci.
Cluster analysis of the MLST data
Clustering by region amongst the isolates was evident in the minimum-spanning tree (Figure 2). The 50 L. lactis isolates evaluated were assigned to 20 STs that resolved into eight clonal complexes (CCs). Among these CCs, 14 STs were clustered together to form two CCs and there were six singleton STs that could not be assigned to any group.
Figure 2.
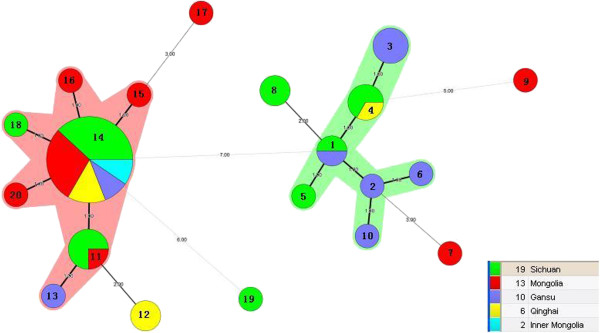
Minimum-spanning tree analysis of 50 L. lactis isolates based on MLST date according to region. Each circle indicates a sequence type, the size of the circle is proportional to the number of isolates and the type of line between isolates indicates the strength of the genetic relationship between these isolates (black line = strong relationship, grey line = intermediate relationship and dotted line = weak relationship).
The largest CC was comprised of ST11, ST13, ST14, ST15, ST16, ST18 and ST20, which included 30 isolates, mainly from Sichuan province and Mongolia. Within this CC (colour-coded pink) ST14 was the predicted primary founder surrounded by single-locus (ST11, ST15, ST16, ST18, and ST20), or two-locus variants (ST13). These STs have been connected by solid black lines indicating they are closely related. The second CC included ST1 to ST6 and ST10, which included 16 isolates mainly from Sichuan and Gansu provinces. ST1 from Sichuan and Gansu province located in the centre of the second clonal complex. Single-locus variants were ST2, ST4 and ST5, which contained isolates from Gansu, Qinghai and Sichuan provinces. Two-locus variants were ST3, ST6 and ST10 and included isolates from Gansu province. ST7, ST8, ST9, ST12, ST17 and ST19 were singletons unlinked to the other CCs. However, they are connected to two primary founders, either ST1 or ST14, by grey or dotted lines, indicating they had a distant relationship with the two predicted ancestors. ST7 and ST8 were two and four-locus variants of ST1 and connected with grey lines. ST12 and ST17 were three- and four-locus variants of ST14 and also connected by grey lines. Isolate IMAU20185 belonging to ST9 was a six-locus variant of ST1 to which it was connected by a dotted line. Isolate IMAU80137 belonging to ST19 was a six-locus variant of ST14 to which it was also connected by a dotted line.
UPGMA tree based on MLST data
Genetic relatedness amongst the L. lactis isolates investigated in this study showed they were well clustered within two major groups, A and B. Group A was comprised of 34 isolates and group B of only 16 isolates. Group A was the better supported group and included two subgroups. Group B was a weakly supported group that included four subgroups (Figure 3). With the exception of ST19, isolates in group A were closely related only differing in two out of the eight loci from the primary founder, ST14. The isolate that belonged to ST19 was a six-locus variant of the primary founder. Isolates in Group B were distantly related and differed in between two and six of the eight loci from the primary founder ST1.
Figure 3.
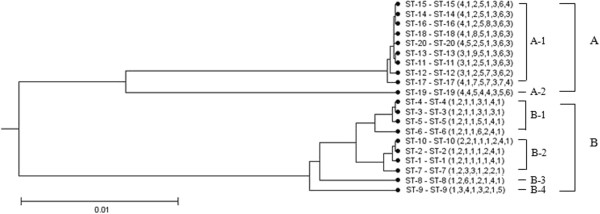
UPGMA dendrogram showing the genetic relationships between the 20 STs that belong to L. lactis through MLST typing in this study. The Phylogenetic tree was produced using START 2.0 software and the UPGMA method. The numbering in the figure refers to the ST. Two major phylogroups were designated as A and B.
Discussion
MLST is considered to be the best method for studying molecular epidemiology and population structure of bacteria [29-31]. Although this approach has been developed for several LAB, such as Lb. plantarum, Lb. delbrueckii, Lb. casei, and O. oeni[25,26,32], until this study there had been no MLST protocol used for L. lactis. In this study, we used MLST with eight housekeeping genes on 50 L. lactis isolates from a relatively large geographic area including Mongolia, a number of Chinese Provinces and an Autonomous region. These representative isolates are unique in their diversity of sources and provide the relevant information required for a better understanding of genetic diversity, persistence and movement.
The first step in development of a MLST typing method required analysis of the sequence diversity of eight housekeeping genes from the 50 L. lactis isolates under evaluation, to ensure that the MLST protocol had the discriminatory power to type isolates within a single species. The two loci that had low polymorphism, contained three and four polymorphic sites in the recA and carB loci respectively (Table 1). The low level of biodiversity in recA and carB suggested they had similar sequences at the species level and would, therefore, have a lower discriminatory ability than the other housekeeping loci used in this study. The remaining six loci, groEL, pheS, uvrC, rpoB, pyrG, murC had more polymorphic sites (between five and nine), suggesting that they would have a good discriminatory ability when used in MLST. A total of 47 polymorphic sites were detected in the eight loci giving a polymorphism rate of 0.88% of the 5,325 nucleotides present. The relatively low number of polymorphic sites suggests that the partial sequences of the eight housekeeping genes were somewhat conserved amongst the 50 L. lactis isolates. Although housekeeping genes evolve slowly, however, we still consider that the eight housekeeping genes selected provide sufficient discriminatory power for typing. In our genetic analysis, the eight housekeeping loci had a very low dN/dS ratio (<1), implying strong purifying selection. This was particularly the case in groEL where five substitutions were synonymous and the amino acid composition did not change. The d N /d S ratio was close to zero and similar to that seen by Madslien et al.[33], which supports our estimation that the housekeeping loci are all under stabilizing selection [33].
Leuconostoc lactis isolates are important industrially in the production of fermented foods. However, their population structure has not been investigated fully before. We used linkage disequilibrium expressed as the index of association (IA) by the equation, IA = VO/VE - 1 (VO, observed variance; VE, expected variance) to investigate population structure. This index of association is a generalised measure of linkage disequilibrium; does not rely on the number of loci analysed; has an expected value of zero if there is no association between loci, i.e. alleles are in linkage equilibrium (free recombination) [34,35]; and was pioneered to describe population structure in Hordeum spontaneum[36]. In our study, the value of IA and I A S for eight loci were calculated as 1.8 and 0.4264 (p = 0.000), respectively. These high values are indicative of a strong clonal population and similar to reports for other bacteria. For example, in Lb. plantarum, where seven selected housekeeping genes were analysed and assigned to 17 different STs, the I A S value was 0.444 [37]. In Bacillus licheniformis, where six housekeeping loci were analysed from 53 diverse isolates, the value of I A S was 0.4365 [33]. These results are similar to our study on L. lactis and, therefore, support our hypothesis that these are clonal populations and that allelic selection is close to linkage disequilibrium.
In general, Leuconostoc species are used as starter cultures for dairy fermentations. All isolates initiate lactose fermentation and lactic acid production and here we have shown that some essential housekeeping genes are highly conserved. However, the value of I A S and the number of unique STs reflect the genetic diversity amongst isolates that have each adapted to specialised environments during their evolution. Similar results have been reported for other LAB isolated from dairy products; for example 197 isolates of Lactococcus lactis isolated from homemade yogurt were assigned to 72 different STs and their I A S value was 0.3038 [38]. Uniformly, a clonal structure was also found in Streptococcus thermophilus, where eight housekeeping loci were analysed from 26 isolates from different dairy products [39].
Split-decomposition analysis based on the allelic profiles of isolates evaluated provided evidence of intraspecies recombination that could play a role in generating genotypic diversity amongst isolates. Parallelogram-shaped structures were commonly found in the split graphs of the partial housekeeping genes (murC, pheS, pyrG, and uvrC) and the combined alleles, illustrating recombination had occurred in some MLST loci. Previous studies have provided evidence that recombination could occur frequently in Leuconostoc species because mobile elements, such as bacteriophages, genomic islands and transposable elements, were found in the genome sequence [40,41]. In addition, some plasmids from Leuconostoc species have been identified [42,43]. In O. oeni isolates, a similar recombination phenomenon has been found including the presence of plasmids, bacteriophages and insertion sequences [44-46]. Furthermore, the presence of parallelogram-shaped structures were also found in the ddl, pgm and recP split graphs of O. oeni isolates [26].
Although this study on the population structure of L. lactis has made important steps forward, e.g. the split-decomposition analysis based on concatenated sequences of housekeeping genes (Figure 1), the UPGMA tree based on the MLST data (Figure 3) and the I A S values, we could still not confirm any association between ST and the original source of each isolate. Similar results have been reported in Lactococcus lactis and Lactobacillus sanfranciscensis, where no significant associations between STs and the various sources of the isolates could be found [47,48]. The absence of such an association in L. lactis may be because of the genetic diversity of individual L. lactis isolates. At the gene level, MLST analysis indicated two CCs and six singletons. The majority of L. lactis isoaltes from dairy products were found in these two CCs; the remaining isolates from various sources including yogurt, kurut, yak’s milk and pickle, were scattered into unique STs. This characterisation was also reflected in the UPGMA dendrogram, with isolates clustering as two groups that could be further divided into several subgroups (Figure 3). These unique STs (ST7, ST8,ST9, ST12, ST17 and ST19) illustrate the genetic diversity within the subspecies.
Conclusions
A MLST protocol for L. lactis isolates, based on eight housekeeping genes and 50 L. lactis isolates was developed. In this study, we demonstrated biodiversity, clonal population structure and genetic recombination in the isolates evaluated. All of these isolates could be separated into two distinct groups that had evolved independently from each other, except isolate MAU80137 from ST19. This isolate was the only one from a nontraditional dairy and was only distantly related to all the other isolates analysed. Future work will target other sources of L. lactis by examining environmental samples to obtain a better understanding of the evolution and population genetics of L. lactis.
Methods
Leuconostoc lactis isolates
In this study, a total of 50 L. lactis isolates, preserved in the Lactic Acid Bacteria Collection Center of the Inner Mongolia Agricultural University (LABCC), were examined and characterised (Additional file 1: Table S1). These isolates originated from various sources including yogurt, kurut, qula and other traditional foods from Mongolia, the P.R. of China Provinces Sichuan, Qinghai, Gansu and the P.R. China Inner Mongolia Autonomous Region. Leuconostoc lactis isolate MAU80137 was the only isolate from pickle (Sichuan province). All isolates were identified as L. lactis based on standard physiological and biochemical tests, and sequence analysis of the 16S rRNA gene [32,49]. Stock cultures were stored in 10% glycerol at -80°C. Working cultures were retrieved from storage and activated by two subcultures through de Man Rogosa Sharpe (MRS) broth (Becton, Dickinson Co., Sparks, Md., USA). Isolates were incubated at 30°C for 24 h under anaerobic conditions prior to evaluation.
DNA extraction
Genomic DNA was extracted from all isolates as described previously [50]. Briefly, after overnight incubation in MRS broth at 37°C, the bacterial cells were collected by centrifugation (8,000 × g, 3 min, 4°C) and subjected to freeze-thaw cycles for cell lysis. Next, 10% sodium dodecyl sulphate (SDS) and proteinase-K solution (20 mg/ml) were added, mixed well, and incubated in a shaking incubator at 200 rpm and 37°C overnight. This was following by addition of 0.7 M NaCl and 10% cetyltrimethyl ammonium bromide (CTAB) and further incubation at 65°C for 20 minutes. Protein contaminants were removed by the addition of phenol/chloroform/isoamyl alcohol (25/24/1). The DNA was precipitated as a pellet by the addition of an equal volume of ice-cold isopropanol, and then washed in 70% (v/v) ice-cold ethanol and dissolved in sterile ultrapure water. The purity of the extracted DNA was quantified by recording its optical density at 260 and 280 nm, respectively, using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA).
Selection of housekeeping genes for the MLST protocol
Eight loci representing housekeeping genes were selected for MLST on L. lactis isolates from those already described from the variable regions of the L. mesenteroides subsp. mesenteroides ATCC 8293 genome sequence [28]: pyrG encoding CTP synthetase (accession no. YP_818007), rpoB, encoding DNA-directed RNA polymerase subunit beta (YP_819285.1), groEL encoding chaperonin GroEL(YP_819222.1), recA encoding recombinase A (YP_818071.1), uvrC encoding excinuclease ABC subunit C (YP_818008.1), carB encoding carbamoyl phosphate synthase large subunit (YP_818678.1), murC encoding UDP-N-acetylmuramate-L-alanine ligase (YP_818192.1), pheS encoding phenylalanyl-tRNA synthetase subunit alpha (YP_817936.1).
Amplification and nucleotide sequencing
Primers for the polymerase chain reaction (PCR) were designed based on the internal fragments of the eight loci using Primers 5 software (Table 2). In this study, the MLST protocol was modified in two ways; firstly, the primers targeting internal fragments of each gene were extended from 450–500 to 500–700 bp and secondly, although MLST protocols generally only use five to seven genes, in this study, eight housekeeping genes were used to analyse the population structure of L. lactis. The eight housekeeping gene fragments (carB, groEL, murC, pheS, pyrG, recA, rpoB, uvrC) were amplified from chromosomal DNA from each isolate using amplification and sequencing primers (Table 2). The PCR procedure for the pyrG, carB, murC and pheS genes was done under the following conditions: 94°C for 5 sec, 30 cycles of amplification which included 95°C for 60 sec, 50°C for 45 sec, 72°C for 60 sec and then annealing at 72°C for 10 min. PCR for the remaining genes followed the same experimental conditions except for the annealing temperature which was 54°C. PCR reactions were made in a 10 μl reaction mixture containing 0.08 μl Taq polymerase (5 U/μl, Takara, Tokyo), 1 μl 10 × PCR Buffer (Mg2+ free), 0.8 μl dNTPs (2.5 mM each), 0.8 μl MgCl2 (25 mM), 0.4 μl forward primer (10 μM), 0.4 μl reverse primer (10 μM), 1 μl genomic DNA (10–50 ng/μL), and 5.52 μl dH2O. The PCR products were separated by electrophoresis on a 1.2% agarose gel and then visualised using ethidium bromide staining. Sequencing of the PCR products was done by the Shanghai Sangni Biosciences Corporation (Shanghai, China) and the sequences deposited in the GenBank/EMBL databases under accession numbers KJ149820 to KJ150219.
Data analysis
The sequences obtained for the eight housekeeping genes in the MLST protocol from all isolates were imported into BioNumerics software (version 6.0, Applied-Maths, Sint Maartens-Latem, Belgium) and the number of unique alleles per locus obtained. In date analysis, all unique sequences were assigned an allele number and each unique combination of eight allele numbers per isolate was assigned a ST [27]. The guanine-cytosine content, d N /d S ratio (d S is the number of synonymous substitutions per synonymous site and d N is the number of non-synonymous substitutions per non-synonymous site) and the number of polymorphic sites and single nucleotide polymorphisms (SNPs) of the eight housekeeping genes for each isolate were calculated using LIAN-Linkage analysis [51]. The level of linkage disequilibrium between all alleles of the isolates was investigated by determining the standardised index of association (I A S) [34]. Phylogenetic trees were constructed by the neighbour-joining (N-J) method in MEGA version 5.0 software (version 5.0, http://www.megasoftware.net). The relationships between MLST STs and analysis of CCs were revealed using eBURST (Based Upon Related Sequence Types) V 3.0 software ( http://eburst.mlst.net). CCs are typically composed of a single predominant genotype with a number of much less common close relatives of that genotype [52]; the isolates of L. lactis that shared a minimum of five identical alleles with the central genotype in a given subpopulation were assigned to a different CC. An UPGMA dendrogram was constructed by START 2.0 software using the unweighted pair-group method and the arithmetic average method (UPGMA). The split decomposition was done with SplitsTree and START 2.0 software on the MLST website ( http://eburst.mlst.net/). Minimum-spanning tree analysis of the STs from all isolates was done using Prims’s algorithm in the BioNumerics software according to region and source separation (version 6.0, Applied-Maths, Sint Maartens-Latem, Belgium).
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conceived and designed the experiments: TD WJL ZHS HPZ. Performed the experiments: QL HYX YQS. Analyzed the data: ZHS YQS. Contributed reagents/materials/analysis tools: ZHS QL HYX YQS. Wrote the paper: TD HPZ. All authors read and approved the final manuscript.
Supplementary Material
Allelic profiles of 50 Leuconostoc lactis isolates.
Contributor Information
Tong Dan, Email: dantong813218@aliyun.com.
Wenjun Liu, Email: wjliu168@163.com.
Zhihong Sun, Email: sunzhihong78@163.com.
Qiang Lv, Email: Lqiang0603@126.com.
Haiyan Xu, Email: weiliangxhy@163.com.
Yuqin Song, Email: songyqnd@163.com.
Heping Zhang, Email: hepingdd@vip.sina.com.
Acknowledgements
This research was supported by National Natural Science Foundation of China (Grant No. 31025019), Hi-Tech Research and Development Program of China (863 Planning, Grant No.2011AA100902), Synergetic Innovation Center of Food Safety and Nutrition, the China Agriculture Research System( Grant No.CARS-37), the Special Fund for Agro-scientific Research in the Public Interest (Grant No. 201303085), the Open Projects of Inner Mongolia Natural Science Foundation (No. 20102010), the Natural Science Foundation of Inner Mongolia (No. 2013MS1205; 2012MS1207), the Scientific Research Projects of Institution of Higher Education in Inner Mongolia (Grant No. NJZY12096).
References
- De Bruyne K, Schillinger U, Caroline L, Boehringer B, Cleenwerck I, Vancanneyt M, De Vuyst L, Franz CM, Vandamme P. Leuconostoc holzapfelii sp. nov., isolated from Ethiopian coffee fermentation and assessment of sequence analysis of housekeeping genes for delineation of Leuconostoc species. Int J Syst Evol Microbiol. 2007;57(Pt 12):2952–2959. doi: 10.1099/ijs.0.65292-0. [DOI] [PubMed] [Google Scholar]
- Hemme D, Foucaud-Scheunemann C. Leuconostoc, characteristics, use in dairy technology and prospects in functional foods. Int Dairy J. 2004;14:467–494. doi: 10.1016/j.idairyj.2003.10.005. [DOI] [Google Scholar]
- Ogier JC, Casalta E, Farrokh C, Saïhi A. Safety assessment of dairy microorganisms: the Leuconostoc genus. Int J Food Microbiol. 2008;126(3):286–290. doi: 10.1016/j.ijfoodmicro.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Sharpe ME, Garvie EI, Tilbury RH. Some slime-forming heterofermentative species of the genus Lactobacillus. Appl Microbiol. 1972;23(2):389–397. doi: 10.1128/am.23.2.389-397.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Tieghem P. Sur la gomme du sucerie (Leuconostoc mesenteroides) Ann Sci Nat Bot. 1878;7:180–203. [Google Scholar]
- Garvie EI. In: Methods in Microbiology, 16. Bergan, editor. London: Academic Press; 1984. Separation of species of the genus Leuconostoc and differentiation of the Leuconostocs from other lactic acid bacteria; pp. 147–178. [Google Scholar]
- Martinez-Murcia AJ, Collins MD. A phylogenetic analysis of an atypical Leuconostoc: description of Leuconostoc fallax sp. nov. FEMS Microbiol Lett. 1991;82:55–60. doi: 10.1111/j.1574-6968.1991.tb04839.x. [DOI] [PubMed] [Google Scholar]
- Collins MD, Samelis J, Metaxopoulos J, Wallbanks S. Taxonomic studies on some Leuconostoc-like organisms from fermented sausages: description of a new genus Weisella for the Leuconostoc parame:renteroides group of species. J Appl Bacteriol. 1993;75(6):595–603. doi: 10.1111/j.1365-2672.1993.tb01600.x. [DOI] [PubMed] [Google Scholar]
- Dicks LM, Dellaglio F, Collins MD. Proposal to reclassify Leuconostoc oenos as Oenococcus oeni [corrig.] gen. nov., comb. nov. Int J Syst Bacteriol. 1995;45(2):395–397. doi: 10.1099/00207713-45-2-395. [DOI] [PubMed] [Google Scholar]
- Endo A, Okada S. Reclassification of the genus Leuconostoc and proposals of Fructobacillus fructosus gen. nov., comb. nov., Fructobacillus durionis comb. nov., Fructobacillus ficulneus comb. nov. and Fructobacillus pseudoficulneus comb. nov. Int J Syst Evol Microbiol. 2008;58(Pt 9):2195–2205. doi: 10.1099/ijs.0.65609-0. [DOI] [PubMed] [Google Scholar]
- Vancanneyt M, Zamfir M, de Wachter M, Cleenwerck I, Hoste B, Rossi F, Dellaglio F, de Vuyst L, Swings J. Reclassification of Leuconostoc argentinum as a later synonym of Leuconostoc lactis. Int J Syst Evol Microbiol. 2006;56:213–216. doi: 10.1099/ijs.0.63898-0. [DOI] [PubMed] [Google Scholar]
- Jeong SH, Lee SH, Jung JY, Choi EJ, Jeon CO. Microbial succession and metabolite changes during long-term storage of Kimchi. J Food Sci. 2013;78(5):M763–769. doi: 10.1111/1750-3841.12095. [DOI] [PubMed] [Google Scholar]
- Ehrmann MA, Freiding S, Vogel RF. Leuconostoc palmae sp. nov., a novel lactic acid bacterium isolated from palm wine. Int J Syst Evol Microbiol. 2009;59(Pt 5):943–947. doi: 10.1099/ijs.0.005983-0. [DOI] [PubMed] [Google Scholar]
- Lee SH, Park MS, Jung JY, Jeon CO. Leuconostoc miyukkimchii sp. nov., isolated from brown algae (Undaria pinnatifida) kimchi. Int J Syst Evol Microbiol. 2012;62(Pt 5):1098–1103. doi: 10.1099/ijs.0.032367-0. [DOI] [PubMed] [Google Scholar]
- Sabat AJ, Budimir A, Nashev D, Sá-Leão R, van Dijl J, Laurent F, Grundmann H, Friedrich AW. Overview of molecular typing methods for outbreak detection and epidemiological surveillance. Euro Surveill. 2013;18(4):20380. doi: 10.2807/ese.18.04.20380-en. [DOI] [PubMed] [Google Scholar]
- Pérez G, Cardell E, Zárate V. Random amplified polymorphic DNA analysis for differentiation of Leuconostoc mesenteroides subspecies isolated from Tenerife cheese. Lett Appl Microbiol. 2002;34(2):82–85. doi: 10.1046/j.1472-765x.2002.01050.x. [DOI] [PubMed] [Google Scholar]
- Cibik R, Lepage E, Talliez P. Molecular diversity of Leuconostoc mesenteroides and Leuconostoc citreum isolated from traditional french cheeses as revealed by RAPD fingerprinting, 16S rDNA sequencing and 16S rDNA fragment amplification. Syst Appl Microbiol. 2000;23(2):267–278. doi: 10.1016/S0723-2020(00)80014-4. [DOI] [PubMed] [Google Scholar]
- Villani F, Moschetti G, Blaiotta G, Coppola S. Characterization of strains of Leuconostoc mesenteroides by analysis of soluble whole-cell protein pattern, DNA fingerprinting and restriction of ribosomal DNA. J Appl Microbiol. 1997;82(5):578–588. doi: 10.1111/j.1365-2672.1997.tb03588.x. [DOI] [PubMed] [Google Scholar]
- Alegría Á, Delgado S, Flórez AB, Mayo B. Identification, typing, and functional characterization of Leuconostoc spp. strains from traditional, starter-free cheeses. Dairy Sci Technol. 2013;93:657–673. doi: 10.1007/s13594-013-0128-3. [DOI] [Google Scholar]
- Nieto-Arribas P, Seseña S, Poveda JM, Palop L, Cabezas L. Genotypic and technological characterization of Leuconostoc isolates to be used as adjunct starters in Manchego cheese manufacture. Food Microbiol. 2010;27:85–93. doi: 10.1016/j.fm.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Sánchez JI, Martínez B, Rodríguez A. Rational selection of Leuconostoc strains for mixed starters based on the physiological biodiversity found in raw milk fermentations. Int J Food Microbiol. 2005;105:377–387. doi: 10.1016/j.ijfoodmicro.2005.04.025. [DOI] [PubMed] [Google Scholar]
- Vihavainen EJ, Björkroth KJ. Diversity of Leuconostoc gasicomitatum associated with meat spoilage. Int J Food Microbiol. 2009;136(1):32–36. doi: 10.1016/j.ijfoodmicro.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Björkroth KJ, Geisen R, Schillinger U, Weiss N, De Vos P, Holzapfel WH, Korkeala HJ, Vandamme P. Characterization of Leuconostoc gasicomitatum sp. nov., associated with spoiled raw tomato-marinated broiler meat strips packaged under modified-atmosphere conditions. Appl Environ Microbiol. 2000;66(9):3764–3772. doi: 10.1128/AEM.66.9.3764-3772.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant DA, Feavers IM, Achtman M, Spratt BG. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A. 1998;95(6):3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanigawa K, Watanabe K. Multilocus sequence typing reveals a novel subspeciation of Lactobacillus delbrueckii. Microbiol. 2011;157:727–738. doi: 10.1099/mic.0.043240-0. [DOI] [PubMed] [Google Scholar]
- De Las RB, Marcobal A, Muñoz R. Allelic diversity and population structure in Oenococcus oeni as determined from sequence analysis of housekeeping genes. Appl Environ Microbiol. 2004;70(12):7210–7219. doi: 10.1128/AEM.70.12.7210-7219.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilhère E, Lucas PM, Claisse O, Lonvaud-Funel A. Multilocus sequence typing of Oenococcus oeni: detection of two subpopulations shaped by intergenic recombination. Appl Environ Microbiol. 2009;75(5):1291–1300. doi: 10.1128/AEM.02563-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova K, Slesarev A, Wolf Y, Sorokin A, Mirkin B, Koonin E, Pavlov A, Pavlova N, Karamychev V, Polouchine N, Shakhova V, Grigoriev I, Lou Y, Rohksar D, Lucas S, Huang K, Goodstein DM, Hawkins T, Plengvidhya V, Welker D, Hughes J, Goh Y, Benson A, Baldwin K, Lee JH, Díaz-Muñiz I, Dosti B, Smeianov V, Wechter W, Barabote R. Comparative genomics of the lactic acid bacteria. Proc Natl Acad Sci U S A. 2006;103(42):15611–15616. doi: 10.1073/pnas.0607117103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Ducatelle R, Pasmans F, Smet A, Haesebrouck F, Flahou B. Multilocus sequence typing of the porcine and human gastric pathogen Helicobacter suis. J Clin Microbiol. 2013;51(3):920–926. doi: 10.1128/JCM.02399-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldo L, Dunning Hotopp JC, Jolley KA, Bordenstein SR, Biber SA, Choudhury RR, Hayashi C, Maiden MC, Tettelin H, Werren JH. Multilocus sequence typing system for the endosymbiont Wolbachia pipientis. Appl Environ Microbiol. 2006;72(11):7098–7110. doi: 10.1128/AEM.00731-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisharat N, Cohen DI, Harding RM, Falush D, Crook DW, Peto T, Maiden MC. Hybrid Vibrio vulnificus. Emerg Infect Dis. 2005;11(1):30–35. doi: 10.3201/eid1101.040440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diancourt L, Passet V, Chervaux C, Garault P, Smokvina T, Brisse S. Multilocus sequence typing of Lactobacillus casei reveals a clonal population structure with low levels of homologous recombination. Appl Environ Microbiol. 2007;73(20):6601–6611. doi: 10.1128/AEM.01095-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madslien EH, Olsen JS, Granum PE, Blatny JM. Genotyping of B. licheniformis based on a novel multi-locus sequence typing (MLST) scheme. BMC Microbiol. 2012;12:230. doi: 10.1186/1471-2180-12-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suerbaum S, Lohrengel M, Sonnevend A, Ruberg F, Kist M. Allelic diversity and recombination in Campylobacter jejuni. J Bacteriol. 2001;183(8):2553–2559. doi: 10.1128/JB.183.8.2553-2559.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH. SplitsTree: analyzing and visualizing evolutionary data. Bioinform. 1998;14:68–73. doi: 10.1093/bioinformatics/14.1.68. [DOI] [PubMed] [Google Scholar]
- Brown AH, Feldman MW, Nevo E. Multilocus structure of natural populations of HORDEUM SPONTANEUM. Genetics. 1980;96(2):523–536. doi: 10.1093/genetics/96.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Las RB, Marcobal A, Muñoz R. Development of a multilocus sequence typing method for analysis of Lactobacillus plantarum strains. Microbiol. 2006;152(Pt 1):85–93. doi: 10.1099/mic.0.28482-0. [DOI] [PubMed] [Google Scholar]
- Xu H, Sun Z, Liu W, Yu J, Song Y, Lv Q, Zhang J, Shao Y, Menghe B, Zhang H. Multilocus sequence typing of Lactococcus lactis from naturally fermented milk foods in ethnic minority areas of China. J Dairy Sci. 2014. doi:10.3168/jds.2013-7738. [DOI] [PubMed]
- Delorme C, Bartholini C, Bolotine A, Ehrlich SD, Renault P. Emergence of a cell wall protease in the Streptococcus thermophilus population. Appl Environ Microbiol. 2010;76(2):451–460. doi: 10.1128/AEM.01018-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meslier V, Loux V, Renault P. Genome sequence of Leuconostoc pseudomesenteroides strain 4882, isolated from a dairy starter culture. J Bacteriol. 2012;194(23):696–712. doi: 10.1128/JB.01696-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam SH, Choi SH, Kang A, Kim DW, Kim RN, Kim A, Park HS. Genome sequence of Leuconostoc argentinum KCTC 3773. J Bacteriol. 2010;192(24):6490–6491. doi: 10.1128/JB.01148-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JY, Chang HC. Identification of a replicon from pCC3, a cryptic plasmid from Leuconostoc citreum C4 derived from kimchi, and development of a new host-vector system. Biotechnol Lett. 2009;31(5):685–696. doi: 10.1007/s10529-009-9912-9. [DOI] [PubMed] [Google Scholar]
- Jeong SJ, Park JY, Lee HJ, Kim JH. Characterization of pFMBL1, a small cryptic plasmid isolated from Leuconostoc mesenteroides SY2. Plasmid. 2007;57(3):314–323. doi: 10.1016/j.plasmid.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Brito L, Pavela H. Presence and analysis of large plasmids in Oenococcus oeni. Plasmid. 1999;41:260–267. doi: 10.1006/plas.1999.1397. [DOI] [PubMed] [Google Scholar]
- Ze’-Ze’ L, Tenreiro R, Pavela H. The Oenococcus oeni genome: physical and genetic mapping of a strain GM and comparison with the genome of a “divergent” strain, PSU-1. Microbiol. 2000;146:3195–3204. doi: 10.1099/00221287-146-12-3195. [DOI] [PubMed] [Google Scholar]
- Zavaleta AI, Martı’nez-Murcia AJ, Rodrı’guez-Varela F. 16S-23S rDNA intergenic sequences indicate that Leuconostoc oenos is phylogenetically homogeneous. Microbiol. 1996;142:2105–2114. doi: 10.1099/13500872-142-8-2105. [DOI] [PubMed] [Google Scholar]
- Picozzi C, Bonacina G, Vigentini I, Foschino R. Genetic diversity in Italian Lactobacillus sanfranciscensis strains assessed by multilocus sequence typing and pulsed-field gel electrophoresis analyses. Microbiol. 2010;156:2035–2045. doi: 10.1099/mic.0.037341-0. [DOI] [PubMed] [Google Scholar]
- Passerini D, Beltramo C, Coddeville M, Quentin Y, Ritzenthaler P, Daveran-Mingot M-L, Le Bourgeois P. Genes but not genomes reveal bacterial domestication of Lactococcus lactis. PLoS ONE. 2010;5(12):e15306. doi: 10.1371/journal.pone.0015306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Q, Liu W, Yu J, Wang W, Qing M, Chen X, Wang F, Zhang J, Zhang W, Qiao J, Sun T, Zhang H. Isolation and identification of cultivable lactic acid bacteria in traditional yak milk products of Gansu Province in China. J Gen Appl Microbiol. 2012;58(2):95–105. doi: 10.2323/jgam.58.95. [DOI] [PubMed] [Google Scholar]
- Dan T, Cheng X, Bao QH, Liu WJ, Zhang HP. Effect of L-Threonine concentrations on acetaldehyde production and glyA gene expression in fermented milk by Streptococcus thermophilus. Food Biotechnol. 2012;26(3):280–292. doi: 10.1080/08905436.2012.699204. [DOI] [Google Scholar]
- Smith JM, Smith NH, O’Rourke M, Spratt BG. How clonal are bacteria? Proc Natl Acad Sci U S A. 1993;90(10):4384–4388. doi: 10.1073/pnas.90.10.4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil EJ, Cooper JE, Grundmann H, Robinson DA, Enright MC, Berendt T, Peacock SJ, Smith JM, Murphy M, Spratt BG, Moore CE, Day NP. How clonal is Staphylococcus aureus? J Bacteriol. 2003;185:3307–3316. doi: 10.1128/JB.185.11.3307-3316.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Allelic profiles of 50 Leuconostoc lactis isolates.