

Abstract
Prenatal arsenic exposure is associated with increased risk of disease in adulthood. This has led to considerable interest in arsenic’s ability to disrupt fetal programming. Many studies report that arsenic exposure alters DNA methylation in whole blood but these studies did not adjust for cell mixture. In this study, we examined the relationship between arsenic in maternal drinking water collected ≤ 16 weeks gestational age and DNA methylation in cord blood (n = 44) adjusting for leukocyte-tagged differentially methylated regions. DNA methylation was quantified using the Infinium HumanMethylation 450 BeadChip array. Recursively partitioned mixture modeling examined the relationship between arsenic and methylation at 473,844 CpG sites. Median arsenic concentration in water was 12 µg/L (range < 1- 510 µg/L). Log10 arsenic was associated with altered DNA methylation across the epigenome (P = 0.002); however, adjusting for leukocyte distributions attenuated this association (P = 0.013). We also observed that arsenic had a strong effect on the distribution of leukocytes in cord blood. In adjusted models, every log10 increase in maternal drinking water arsenic exposure was estimated to increase CD8+ T cells by 7.4% (P = 0.0004) and decrease in CD4+ T cells by 9.2% (P = 0.0002). These results show that prenatal exposure to arsenic had an exposure-dependent effect on specific T cell subpopulations in cord blood and altered DNA methylation in cord blood. Future research is needed to determine if these small changes in DNA methylation alter gene expression or are associated with adverse health effects.
Keywords: arsenic, DNA methylation, cord blood, immune function, leukocytes, developmental programming
Introduction
Arsenic is a common environmental contaminant. People are primarily exposed to arsenic from drinking water and industrial emissions and, to a lesser extent, from dietary sources.1 Data from exposure assessment studies demonstrate that arsenic readily crosses the placenta and that fetal exposure is highly correlated with maternal exposure.2-5 Evidence from epidemiological studies shows that early life exposure to arsenic is associated with increased risk of adverse health outcomes later in life. For instance, a large ecological study in Chile reported that people in Antofagasta who were exposed to high concentrations of arsenic either prenatally or in early childhood had significantly higher mortality from lung cancer and bronchiectasis compared with people from other parts of the country.6 A second study in Antofagasta reported diminished lung function in adults who were exposed to high levels of arsenic (> 800 µg/L) before age 10 compared with adults who had much lower arsenic exposures in childhood (0–250 µg/L).7 In Japan, a cohort study in Okayama Prefecture of infants that were potentially exposed to high levels of arsenic from contaminated milk powder had significantly higher cancer mortality rates compared with infants born in the same area 1 to 5 y after the contaminated infant formula was recalled.8 Finally, in Bangladesh, a prospective cohort study reported that infants who were exposed to higher levels of arsenic in utero had a greater risk of infectious diseases in the first year of life compared with infants with lower arsenic exposure.9
While it is not fully understood how early life exposure to arsenic would lead to increased susceptibility to disease later in life, experimental studies have shown that in utero exposure to arsenic alters DNA methylation in offspring.10-14 There is also evidence from epidemiological studies that arsenic exposure from drinking water alters DNA methylation in whole blood collected in adults.15-17 Recently, our group showed that maternal exposure to arsenic from drinking water was significantly associated with increased LINE-1 methylation in cord blood.17
Whole blood, however, is a mixture of cells with specific methylomic signatures based on morphology. Methylation profiles in DNA extracted from whole blood are strongly influenced by white blood cell composition, which is highly variable between subjects.18-22 Previous studies that examined the effect of arsenic on DNA methylation were unable to control for the heterogeneity of the cell population in the DNA extracted from whole blood making it difficult to interpret whether the observed effect were due to changes in specific blood cell populations. Fortunately, recently developed bioinformatics approaches can be employed that use leukocyte-tagging differentially methylated regions (DMRs) to identify cell types and adjust for white blood cell populations.19,23,24 This approach has been used successfully in epidemiological studies of cancer and rheumatoid arthritis to compare the direct effects of DNA methylation to those mediated by immune profile.19,20,25 Alternatively, this method provides a novel approach for using DNA to examine the association between environmental exposures and immune response.
Therefore, we examined the relationship between prenatal arsenic exposure and DNA methylation in cord blood of newborns that were born into a prospective birth cohort recruited in Bangladesh. We hypothesized that in utero exposure to arsenic would be associated with altered epigenome-wide methylation after controlling for cell mixture. Based on experimental evidence that arsenic alters the development, activation and proliferation of T-cells,26-37 we further hypothesized that prenatal arsenic exposure would independently influence the immune response, which would be associated with altered leukocyte subpopulations in cord blood.
Results
Forty-four newborns were included in this analysis. The median concentration of arsenic in their mother’s drinking water at ≤ 16 wk gestational age was 12 µg/L. The gestational age at recruitment ranged from 6 to 16 wk, with an average of 12.2 wk (Standard Deviation, [SD], 2.5 wk) and the gestational age at delivery ranged from 33 to 41 wk with an average of 37.6 wk (SD, 2.1). The average weight at birth was 2923 g (SD, 372 g).
Since DNA extracted from cord blood represents a mixture of cell types, we first reconstructed the distribution of white blood cells in the cord blood using leukocyte-tagged DMRs for B cells, granulocytes, monocytes, natural killer (NK) cells, CD4+ T cells, and CD8+ T cells. These linear regression models evaluated the association between arsenic exposure and the relative percentage of leukocytes in cord blood, adjusting for infant sex. We observed a significant association between arsenic and leukocyte distribution (P = 0.0118, 6 d.f. chi-sq test). By examining the linear relationship with individual leukocyte subpopulations, we observed that arsenic exposure was only associated with changes in the distribution of CD4+ and CD8+ T cells (Table 1). Specifically, for each increase in log10 arsenic the percentage of CD8+ increased by 7.4% (P = 0.0004) and the percentage of CD4+ cells decreased by 9.2% (P = 0.0002). A more modest association was also observed with B cells where each increase in log10 arsenic was estimated to decrease B cells by 1.4% (P = 0.056). These results suggested that in utero exposure to arsenic altered the distribution of white blood cell populations in cord blood.
Table 1. Results of linear regression models examining the relationship between maternal log10 water arsenic concentration and the percent composition of whole blood using methods described in Houseman et al.,20 adjusted models included infant sex.
| Effect Estimate (Raw)1 [% composition] |
Effect Estimate (Bias-Adj)2 [% composition] |
SE3 | P value4 | |
|---|---|---|---|---|
| B cell | -1.4 | -1.4 | 0.71 | 0.056 |
| Granulocyte | 1.4 | 1.6 | 1.84 | 0.430 |
| Monocyte | 0.5 | 0.5 | 0.50 | 0.310 |
| Natural Killer | -0.7 | -0.9 | 0.73 | 0.317 |
| T Cell (CD4+) | -7.4 | -9.2 | 1.97 | 0.0002 |
| T Cell (CD8+) | 5.5 | 7.4 | 1.54 | 0.0004 |
1 Raw estimates without bias-correction. 2Estimate corrected for measurement-error bias. 3Standard error based on double-bootstrap. 4P values based on raw effect estimates.
After adjusting for white cell mixture, we identified the top ten CpG sites whose methylation levels had the strongest association with arsenic exposure (Table 2). The magnitude and the direction of the effect of arsenic on measured DNA methylation in cord blood depended on each CpG site. After adjusting for the false discovery rate, log10 arsenic was associated with a significant increase in methylation at only one CpG site (q-value < 0.05), cg00498691. The other nine CpG sites were suggestive of an exposure-response relationship with log10 arsenic but did not reach statistical significance (q-value < 0.07). Specifically, log10 arsenic was modestly associated with increased methylation at cg04597393, cg16321474 (Ref Gene, VDR), cg01783894, cg03783410 (Ref Gene, SOX2OT), cg06448705 (Ref Gene, TRPC7), cg15346830 (Ref Gene, COL11A2), cg15641060, and a decrease in methylation at cg00122779 (Ref Gene, TNXB) and cg02095504 (Ref Gene, CIB4).
Table 2. Top 10 list of the most significant CpG sites based on ordinary-least squares regression analysis modeling the association between M-value and log10 arsenic, adjusted for infant sex and 6 imputed leukocytes cell mixture coefficients.
| CpG Site |
Effect estimate (M-val / log10 As) |
Unadjusted P value | q-value | Location |
Infinium Type |
Nearby SNPs* |
RefGene Name |
RefGene Group |
Chromosome |
RefGene Accession |
| cg00498691 | 0.26 | 5.89 x 10−8 | 0.021 | Island | II | 19 | ||||
| cg04597393 | 0.34 | 4.54 x 10−7 | 0.056 | I | Yes | 8 | ||||
| cg16321474 | 0.23 | 6.99 x 10−7 | 0.058 | Island | I | Yes | VDR | TSS1500 | 12 | NM_001017535 |
| cg00122779 | -0.66 | 4.25 x 10−7 | 0.056 | II | TNXB | Body | 6 | NM_019105 | ||
| cg01783894 | 0.28 | 7.87 x 10−7 | 0.058 | S_Shore | I | 1 | ||||
| cg03783410 | 0.21 | 9.49 x 10−7 | 0.058 | N_Shore | II | SOX2OT | Body | 3 | NR_004053 | |
| cg02095504 | -0.24 | 1.16 x 10−6 | 0.061 | II | CIB4 | 3′UTR | 2 | NM_001029881 | ||
| cg06448705 | 0.25 | 1.34 x 10−6 | 0.062 | II | TRPC7 | 5′UTR | 5 | NM_001167576 | ||
| cg15346830 | 0.22 | 1.79 x 10−6 | 0.065 | II | COL11A2 | Body | 6 | NM_080679 | ||
| cg15641060 | 0.21 | 1.96 x 10−6 | 0.065 | N_Shore | I | 11 |
CpGs with polymorphism in Infinium probe or target CpG.
Table 3 displays the omnibus P values for the overall epigenome-wide association with prenatal arsenic exposure. Note that the P values based on the average nominal F-statistic were attenuated after adjustment for leukocyte distribution, but P values based on the maximum F-statistic remained significant even after adjusting for leukocyte composition. This was consistent for the epigenome-wide analysis using all autosomal CpGs including SNPs and the epigenome-wide analysis excluding SNPs. This observation suggested the existence of a small number of CpGs with substantial stable alterations associated with arsenic exposure. Additionally, we analyzed the data with and without adjusting for cell mixture (Fig. 1). This depiction illustrates the epigenome-wide association results before adjusting for cell mixture effects (Fig. 1A) and after methylation values were adjusted for leukocyte composition (Fig. 1B). When adjusting for cell mixture, there is a reduction in the number and magnitude of observed associations, which is consistent with the results from Table 3.
Table 3. Overall association between maternal log10 water arsenic concentration and omnibus EWAS p-values with and without adjusting for 6 imputed leukocyte cell mixture coefficients.
| Unadjusted for Leukocyte1 | Adjusted for Leukocyte2 | ||||
|---|---|---|---|---|---|
| # CpGs | Mean F | Max F | Mean F | Max F | |
| All autosomal CpGs (II.a, II.b) |
473929 | 0.002 | 0.002 | 0.065 | 0.015 |
| Autosomal CpGs without polymorphisms (II.c, II.d) | 384474 | 0.002 | < 0.001 | 0.059 | 0.013 |
1 Estimated following statistical method described in IIa and IIc. 2Estimated following statistical method described in IIb and IId.
Figure 1.

Volcano plots illustrating the relationship between log10 arsenic and methylation at all 384 474 CpGs sites analyzed in Approach III, without adjusting for cell mixture (A) and after adjusting for cell mixture (B). Dashed lines represent q-value thresholds of q < 0.05 for genome-wide associations (P = 0.01 for A, and P = 4.54 x 10−7 for (B) and a dotted line for the Bonferroni correction of P = 1.30 x 10−7. Yellow = CpG Island, Black = CpG shore, Blue = everything else. Circle = Infinium Type II, Square = Infinium Type I.
A list of genes that included CpG sites that demonstrated an exposure-response relationship with arsenic exposure are provided in Table S1. At a gene level, arsenic exposure was associated with increased methylation at one or more CpG sites within 71 genes. Given that increased methylation is related to gene silencing we ranked genes based on the number of CpG sites within each gene that were significantly associated with exposure. We chose to describe our findings this way to account for the unequal coverage of measured CpG sites within genes in the Infinium HumanMethylation 450 BeadChip array. For instance, the array measured 6 CpG sites within patched domain-containing protein 4 (PTCHD4). In this gene, 2 of the 6 measured CpG sites had increased methylation that was associated with arsenic exposure (e.g., the first and third CpG sites increased methylation which is noted by “+”; the other 4 CpG sites had no association as noted by “.”). Of note, the gene that had the most CpG sites that were differentially methylated in proportion to arsenic exposure (e.g., three of the five measured CpG sites) was zinc finger protein 710 (ZNF710). Other genes with increased methylation relative to arsenic exposure at 2 of the 3 measured CpG sites were tumor necrosis factor receptor superfamily member 10b (TNFRSF10B), sulfotransferase Family 4A, Member 1 (SULT4A1), CD151 molecule (CD151), Acetoacetyl-CoA synthetase pseudogene 1 (AACSP1) and LIM Homeobox (LHX8).
Finally, we conducted a gene-set analysis to identify KEGG biological pathways that contained CpG sites whose methylation was influenced by arsenic exposure. Twenty-four biological pathways were differentially methylated in relationship to arsenic after adjusting for leukocyte distributions (Table 4). The pathways with the strongest association with arsenic exposure were maturity onset diabetes of the young (KEGG hsa04950), hematopoietic cell lineage (KEGG hsa04640), and renin-angiotensin system (KEGG hsa04614). Results using all autosomal CpGs for analysis including SNPs were similar (data not shown).
Table 4. Gene set analysis of the association of arsenic exposure and DNA methylation changes on biological pathways from the Kyoto Encyclopedia of Genes and Genomes (KEGG).
| Blood (unadjusted) | Blood (WBC-Adjusted) | ||||
|---|---|---|---|---|---|
| mean F | max F | mean F | max F | ||
| EWAS | Entire 450K Array | 0.004 | <0.001 | 0.06 | < 0.001 |
| Any Kegg | Any CpG mapped to any gene in a KEGG pathway | 0.001 | <0.001 | 0.066 | 0.075 |
| Any Gene | Any CpG mapped to any gene | 0.001 | <0.001 | 0.065 | 0.082 |
| hsa04950 | Maturity onset diabetes of the young | 0.001 | <0.001 | 0.014 | 0.054 |
| hsa04640 | Hematopoietic cell lineage | <0.001 | <0.001 | 0.023 | 0.023 |
| hsa04614 | Renin-angiotensin system | <0.001 | <0.001 | 0.024 | 0.002 |
| hsa04514 | Cell adhesion molecules (CAMs) | <0.001 | <0.001 | 0.028 | 0.036 |
| hsa05412 | Arrhythmogenic right ventricular cardiomyopathy (ARVC) | <0.001 | <0.001 | 0.027 | 0.029 |
| hsa04612 | Antigen processing and presentation | <0.001 | 0.01 | 0.053 | 0.036 |
| hsa04350 | TGF-β signaling pathway | <0.001 | <0.001 | 0.039 | 0.005 |
| hsa05410 | Hypertrophic cardiomyopathy (HCM) | <0.001 | <0.001 | 0.04 | 0.023 |
| hsa04020 | Calcium signaling pathway | <0.001 | <0.001 | 0.042 | 0.012 |
| hsa05414 | Dilated cardiomyopathy | <0.001 | <0.001 | 0.05 | 0.024 |
| hsa05218 | Melanoma | <0.001 | 0.002 | 0.053 | 0.283 |
| hsa04320 | Dorso-ventral axis formation | <0.001 | <0.001 | 0.036 | 0.02 |
| hsa04920 | Adipocytokine signaling pathway | <0.001 | <0.001 | 0.05 | 0.026 |
| hsa05200 | Pathways in cancer | <0.001 | <0.001 | 0.054 | 0.097 |
| hsa04512 | ECM-receptor interaction | <0.001 | <0.001 | 0.077 | 0.007 |
| hsa04670 | Leukocyte transendothelial migration | <0.001 | <0.001 | 0.054 | 0.013 |
| hsa04810 | Regulation of actin cytoskeleton | <0.001 | <0.001 | 0.063 | 0.038 |
| hsa04914 | Progesterone-mediated oocyte maturation | 0.001 | 0.001 | 0.054 | 0.039 |
| hsa04650 | Natural killer cell mediated cytotoxicity | <0.001 | <0.001 | 0.060 | 0.005 |
| hsa03320 | PPAR signaling pathway | <0.001 | <0.001 | 0.057 | 0.026 |
| hsa04150 | mTOR signaling pathway | 0.001 | 0.003 | 0.062 | 0.004 |
| hsa04510 | Focal adhesion | <0.001 | <0.001 | 0.083 | 0.016 |
| hsa04520 | Adherens junction | 0.001 | <0.001 | 0.051 | 0.135 |
| hsa04330 | Notch signaling pathway | <0.001 | 0.002 | 0.07 | 0.048 |
Discussion
In this study, we observed that prenatal exposure to arsenic was associated with altered DNA methylation patterns in cord blood after adjusting for white blood cell mixtures. Additionally, these results suggested that prenatal arsenic exposure had an exposure-dependent effect on specific T cell subpopulations and, to a lesser extent, B cell subpopulations in cord blood. While these observations have not been confirmed in an experimental model and the biological effects of these epigenetic changes are unknown, these findings complement in vitro and in vivo studies that show that arsenic reduces lymphocyte proliferation, CD4+ cell counts, CD4+:CD8+ cell ratios, and reduced T-regulatory cells in exposed adults and children38,34 and that arsenic alters DNA methylation.10-14 Conceptually, white blood cell populations could be acting as a confounder or a mediator for arsenic-related toxicity (Fig. 2). Since each leukocyte subtype contributes to the DNA extracted from whole blood, the measured DNA methylation could be the product of changes in upstream cellular activities that produce subtle shifts in cell type, i.e., leukocyte subtypes. The fluctuation in leukocyte distributions would be captured as changes in DNA methylation because cell differentiation is governed by DNA methylation. Thus, the upstream epigenetic alteration could only be revealed upon more detailed examination of the blood.39-42
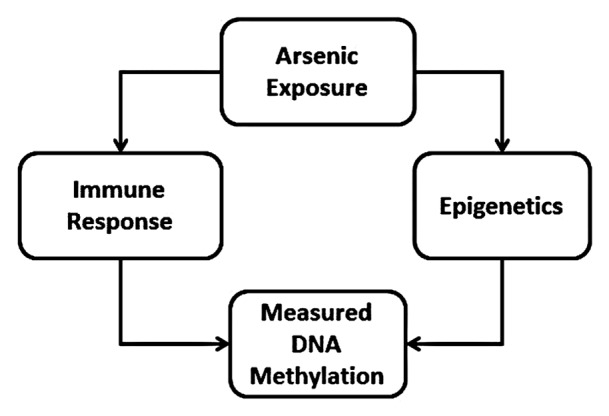
Figure 2. Conceptual effect of arsenic exposure on immune response and epigenetic processes that would contribute to measured DNA methylation changes.
Our agnostic approach identified several genes and biological pathways containing CpG sites that were differentially methylated in relationship to increasing prenatal arsenic exposure. While our study did not examine gene expression, it is noteworthy that many toxicological studies report arsenic interacting with many of these same genes and biological pathways. For instance, methylation at 3 out of 5 measured CpG sites in the promoter region of ZNF710 and the one measured CpG site in the 1st exon of ZNF679 were significantly associated with increasing arsenic exposure. Both of these genes encode for zinc finger proteins that have been identified as potential targets for toxic metals including arsenic.43,44
We also noticed that methylation of CpG sites within the maturity onset diabetes of the young biological pathway were significantly associated with arsenic exposure. This pathway contains five genes (HNF4alpha, HNF1alpha, PDX1, HNF1beta, and NEUROD1). There is some evidence from experimental models that arsenic alters expression of genes in this pathway. For instance, an in vitro study in a hepatoblastoma cell line demonstrated that high levels of arsenite trioxide reduced the expression of HNF4.45 Additionally, data from a transplacental in vivo study in apolipoprotein E-knockout mice showed increased gene expression from a cluster of 51 genes (including HNF4alpha) in the liver of offspring exposed to high levels of arsenic compared with controls.46 In regards to the renin-angiotensin system (RAS) pathway, it contains genes that are associated with regulating blood pressure. Overexpression of the RAS pathway in physiological conditions like hypertension has been shown to increase the production of reactive oxygen species (ROS).47 The overproduction of ROS is thought to be the first step of endothelial cell proliferation and apoptosis—two mechanisms that have been proposed as initiators for arsenic-related atherosclerosis.48 There is also data from an in vivo study in rats showing that chronic exposure (200 days) to inorganic arsenic significantly decreased the angiotensin-converting-enzyme (ACE) activity in hepatic tissue, regulated by the ACE gene in the renin-angiotensin pathway compared with controls.49 The hematopoietic cell lineage is a complex pathway in humans that gives rise to many different mature blood cell types. Arsenic trioxide has been used as an anti-leukemia agent and for other hematological malignancies.50 An in vivo study demonstrated that induced hepatic collagenesis by chronic exposure to arsenic was strongly correlated with expression of interleukin 6 (IL-6) and the tumor necrosis factor α (TNF-α), both important genes in the hematopoietic cell lineage pathway.51
One of the major strengths of this study is that we adjusted for cell mixture when examining the association between arsenic exposure and DNA methylation in blood. This was also a prospective study where drinking water arsenic exposure was measured early in pregnancy. This analysis does rely on one personal drinking water measurement to assign exposure and subsequently we cannot rule out misclassification of exposure. Although other studies in rural Bangladesh populations have shown that current drinking water exposures are relatively constant and highly correlated with other biomarkers of internal dose such as urine and toenails.52,53 There are also several limitations to this study. We did not measure gene expression in the cord blood and cannot assess whether the observed changes in DNA methylation were associated with any biological responses. This was also a relatively small study. We were not able to validate the observed shift in white blood cells proportions with a complementary technology due to limited sample availability and lack of flow cytometry facilities in Bangladesh. Also, we cannot rule out potential confounding because many host and environmental factors are known to influence DNA methylation including exposure to air pollutants resulting from cooking with biomass which is common in this population.
In conclusion, this study showed that prenatal arsenic exposure was associated with altered DNA methylation in cord blood of newborns after adjusting for leukocyte distributions. It also showed that arsenic was significantly associated with leukocyte subpopulations, specifically CD+4 and CD+8 populations. Additional studies are needed to determine whether these changes persist over time or are associated with any negative health effects.
Methods
Participant selection and sample collection
Pregnant women ≤ 16 wk gestational age were enrolled in a prospective birth cohort recruited in Bangladesh by Dhaka Community Hospital (DCH) to evaluate the effects of low level arsenic exposure on reproductive health outcomes. Health care workers at Sirajdikhan and Birahimpur Community Health Clinics identified pregnant women who were 18 y of age or older, used a tubewell that supplied groundwater as their primary drinking water source, planned to live at their current residence for the duration of the pregnancy and continue prenatal health care with DCH, and agreed to deliver at DCH or at home with a DCH-trained midwife. Informed consent was obtained from all participants prior to enrollment or partaking in any study activity. An ultrasound at the time of enrollment confirmed gestational age and a singleton pregnancy. As an incentive, all participants were provided with free prenatal care from DCH and prenatal vitamins that were replenished during monthly checkups in the participant’s home.
This analysis uses information from 44 infants who were selected for inclusion based on the concentration of arsenic in their mother’s drinking water (Range, < 1–510 µg/L) and availability of cord blood DNA. This selection approach was used to insure that there was representation across a wide range of arsenic exposures.
This study was approved by the Human Research Committees at the Harvard School of Public Health, Dhaka Community Hospital Trust and Oregon State University.
Arsenic exposure
At the time of enrollment, a water sample was collected from the well that each participant identified as her primary source of drinking water. Briefly, water samples were collected in 50 mL polypropylene tubes (BD Falcon, BD Bioscience) and preserved with Reagent Grade HNO3 (Merck, Germany) to a pH < 2 . Samples were kept at room temperature prior to analysis by inductively coupled plasma-mass spectrometry following US EPA method 200.8 (Environmental Laboratory Services). The average percent recovery of As from PlasmaCAL multi-element QC standard #1 solution (SCP Science) was 102% ± 7%.
DNA Methylation Assay and quality control
Cord blood was collected at the time of delivery into an EDTA-coated vacutainer tube (B.D. Scientific). DNA was extracted from whole cord blood using Purgene DNA isolation solutions (Qiagen/Gentra Systems) following manufacturer’s instructions. DNA was shipped to the University of Minnesota’s Biomedical Genomic Center where DNA methylation was quantified using the Infinium HumanMethylation 450 BeadChip array (Illumina) following manufacturer’s instructions.
Samples were randomly distributed across 16 chips. Image files for DNA methylation data were processed using the methylumi package in R. The DNA methylation data were normalized and a background and dye bias correction was conducted using the methylumi package in R. For each array, the methylumi package provides an “average beta” quantity for each of the 485 577 CpG sites interrogated by the Infinium 450K platform and for 834 additional controls. The average β was calculated as M/(M + U + ε), where M is the signal from the probe corresponding to the methylated target CpG, U is the signal from the probe corresponding to the unmethylated target, and ε = 100, a small number used to protect against division-by-zero. Thus, average β is an interval-scaled quantity between zero and one that is interpreted as the fraction of DNA molecules whose target CpG is methylated. We excluded 11 648 CpG sites that were located on sex chromosomes to circumvent confounding by subject sex. Multivariate characteristics of control probes were investigated graphically through the use of clustering heatmaps to examine the overall characteristics of the array data (total signal, distribution of detection P values, and fraction of missing values). We also identified 89 455 of the autosomal CpGs that had SNPs at the target CpG site or within the probe based on attributes provided in the Illumina annotation files as well as additional SNP information (data available upon request). For quality control purposes, we analyzed DNA from one participant in duplicate. The overall correlation between all CpG sites for this one duplicate sample was 99.5%. Models were evaluated with either 473 929 CpG sites (with SNPs) or 384 474 CpG sites (without SNPs) as described below.
Statistical analysis
The final data set consisted of 45 cord blood specimens (from 44 unique subjects). Technical replicates were accounted for as described below. Average β quantities were logit-transformed to “M-values” prior to analysis.54 Beta values were used only for the cell mixture analysis and the direct adjustment of leukocyte composition on DNA methylation described below. M-values were used for all other analyses presented. In a manner similar to the ComBat,55 M-values were adjusted for chip effects using a linear mixed effects (LME) model via the R function lme in the package nlme, with a fixed regression coefficient modeling the effect of exposure on methylation for each CpG, and random intercept term to account for chip effect. All chip effects were subtracted from M-values on a CpG-by-CpG basis. The LME procedure was used in place of ComBat in order to keep the size of the fixed-effects model parsimoniously small.
To evaluate the hypothesis that exposure (log10 arsenic concentration in maternal drinking water ≤ 16 wk gestational age) was significantly associated with changes in DNA methylation across the epigenome and that exposure influenced cell mixture in blood, we employed several complementary analytical approaches.
Cell mixture analysis - approach I
To estimate leukocyte composition, β values were used in placed of M-values to adequately account for linearity of cell mixture coefficients. We employed the indirect, two-stage method of Houseman et al. to estimate the effects of exposure on distribution of leukocytes.20 This method uses DNA methylation measurements on the average β at 100 select DMRs as a surrogate for leukocyte distribution, or cell mixture. The coefficients of exposure-methylation association are combined with reference mean methylation estimates measured from isolated leukocytes of specific cell type to obtain estimated associations between exposure and percent composition of individual cell types.
Epigenome-wide association analysis- approach IIa
We conducted a CpG-by-CpG analysis of adjusted M-values in relationship to exposure (log10 water arsenic). We used limma models to estimate the linear association between the M-values and exposure.56 We stratified CpGs by Infinium Type (I or II) so that the empirical Bayes estimates of standard deviation were not influenced by differences in the dynamic range between the two types of biochemistry. Omnibus tests for association, adjusting for multiple comparisons, were obtained by comparing a summary of association over all CpG sites with the corresponding permutation-distribution summary. The permutation-distribution was obtained by permuting the exposure with respect to sex and methylation data (1000 permutations). Note that exposure was permuted by subject (not specimen) in order to account for technical replication. A summary of genome-wide association was constructed using two different objective test statistics: (1) a maximum nominal F statistic over the array (akin to minimum P value) and (2) an average nominal F-statistic. The maximum nominal F statistic would be expected to be more powerful for detecting a small number of strong associations while the average nominal F-statistic would be expected to be more powerful for detecting a large number of weak or variable associations.
Epigenome-wide association analysis adjusted for cell mixture - approach IIb
The epigenome-wide association analysis (Approach IIb) was conducted on a CpG-by-CpG basis of adjusted M-values in relationship to arsenic exposure similar to approach IIa, except that M-values were first adjusted for leukocyte composition in the following manner. Leukocyte composition was determined from DNA methylation values measured at the 100 DMRs used in the previous cell mixture analysis (Approach I), employing the constrained linear projection described in Houseman et al.20 For each CpG, the effects of leukocyte composition on the chip-adjusted average-β were determined by shrinking cell-specific means toward a common value. An optimum constraint (tuning parameter) was determined using a LME.57 Adjusted average betas were obtained by antilogit-transforming adjusted M-values. Leukocyte effects were subtracted from the adjusted average betas with the resulting values truncated to fall between 0 and 1 and subsequently logit-transformed back to leukocyte-adjusted M-values.
Epigenome-wide association analysis excluding SNPs: unadjusted(IIc) and adjusted(IId) for cell mixture
Approach IIc and IId were similar to IIa and IIb respectively, except that CpGs with known polymorphisms at the target CpG or within the Infinium probe were excluded from consideration in limma analyses and omnibus permutation tests.
KEGG biological pathway analysis- approach III
We conducted omnibus permutation tests over subsets of CpGs defined by their mapped association with genes in specific biological pathways. XML files from the Kyoto Encyclopedia of Genes and Genomes (KEGG) were used to compile gene sets characterizing specific biological pathways using the Entrez IDs matched to those documented on the KEEG XML files with the Bioconductor library org.Hs.eg.db. The four sub-analyses within this approach (IIIa-IIId) correspond to approaches IIa-IId described above, except that permutation tests were conducted individually over subsets of CpGs defined by pathways. Note that the “pathway analysis” P values represent neither the Fisher test approach to over-representation analysis, nor the permutation tests used in the classical gene-set enrichment analysis (GSEA).58 We chose to analyze pathways in this relatively simple but tractable manner because of the difficulties in assumed sampling frame and null hypothesis documented in Goeman et al.59 and because of complications in biological interpretation arising from heterogeneity on the array in the number of CpGs per stratum defined by CpG Island status, Infinium type, and gene region.
All statistical analyses were performed using the R statistical package version 2.15.1 (http://www.R-project.org)
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by US NIH (NIEHS) grants K01 ES017800, R01 ES015533, R01 ES016454, P30 ES000210, P30 ES000002, and P42 ES016454. The authors declare no competing financial interests.
Glossary
Abbreviations:
- As
arsenic
- CpG
cytosine-guanine nucleotide sequence
- DCH
Dhaka Community Hospital
- DMR
differentially methylated region
- EWAS
epigenome-wide association study
- ICP-MS
inductively coupled plasma mass spectrometry
- NK
natural killer cells
- SD
standard deviation
References
- 1.Mandal BK, Suzuki KT. Arsenic round the world: a review. Talanta. 2002;58:201–35. doi: 10.1016/S0039-9140(02)00268-0. [DOI] [PubMed] [Google Scholar]
- 2.Concha G, Vogler G, Lezcano D, Nermell B, Vahter M. Exposure to inorganic arsenic metabolites during early human development. Toxicol Sci. 1998;44:185–90. doi: 10.1093/toxsci/44.2.185. [DOI] [PubMed] [Google Scholar]
- 3.Concha G, Nermell B, Vahter MV. Metabolism of inorganic arsenic in children with chronic high arsenic exposure in northern Argentina. Environ Health Perspect. 1998;106:355–9. doi: 10.1289/ehp.98106355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guan H, Piao F, Zhang X, Li X, Li Q, Xu L, Kitamura F, Yokoyama K. Prenatal exposure to arsenic and its effects on fetal development in the general population of Dalian. Biol Trace Elem Res. 2012;149:10–5. doi: 10.1007/s12011-012-9396-7. [DOI] [PubMed] [Google Scholar]
- 5.Hall M, Gamble M, Slavkovich V, Liu X, Levy D, Cheng Z, van Geen A, Yunus M, Rahman M, Pilsner JR, et al. Determinants of arsenic metabolism: blood arsenic metabolites, plasma folate, cobalamin, and homocysteine concentrations in maternal-newborn pairs. Environ Health Perspect. 2007;115:1503–9. doi: 10.1289/ehp.9906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith AH, Marshall G, Yuan Y, Ferreccio C, Liaw J, von Ehrenstein O, Steinmaus C, Bates MN, Selvin S. Increased mortality from lung cancer and bronchiectasis in young adults after exposure to arsenic in utero and in early childhood. Environ Health Perspect. 2006;114:1293–6. doi: 10.1289/ehp.8832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dauphiné DC, Ferreccio C, Guntur S, Yuan Y, Hammond SK, Balmes J, Smith AH, Steinmaus C. Lung function in adults following in utero and childhood exposure to arsenic in drinking water: preliminary findings. Int Arch Occup Environ Health. 2011;84:591–600. doi: 10.1007/s00420-010-0591-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yorifuji T, Tsuda T, Doi H, Grandjean P. Cancer excess after arsenic exposure from contaminated milk powder. Environ Health Prev Med. 2011;16:164–70. doi: 10.1007/s12199-010-0182-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rahman A, Vahter M, Ekström EC, Persson LA. Arsenic exposure in pregnancy increases the risk of lower respiratory tract infection and diarrhea during infancy in Bangladesh. Environ Health Perspect. 2011;119:719–24. doi: 10.1289/ehp.1002265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie Y, Liu J, Benbrahim-Tallaa L, Ward JM, Logsdon D, Diwan BA, Waalkes MP. Aberrant DNA methylation and gene expression in livers of newborn mice transplacentally exposed to a hepatocarcinogenic dose of inorganic arsenic. Toxicology. 2007;236:7–15. doi: 10.1016/j.tox.2007.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waalkes MP, Ward JM, Liu J, Diwan BA. Transplacental carcinogenicity of inorganic arsenic in the drinking water: induction of hepatic, ovarian, pulmonary, and adrenal tumors in mice. Toxicol Appl Pharmacol. 2003;186:7–17. doi: 10.1016/S0041-008X(02)00022-4. [DOI] [PubMed] [Google Scholar]
- 12.Waalkes MP, Liu J, Germolec DR, Trempus CS, Cannon RE, Tokar EJ, Tennant RW, Ward JM, Diwan BA. Arsenic exposure in utero exacerbates skin cancer response in adulthood with contemporaneous distortion of tumor stem cell dynamics. Cancer Res. 2008;68:8278–85. doi: 10.1158/0008-5472.CAN-08-2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waalkes MP, Liu J, Diwan BA. Transplacental arsenic carcinogenesis in mice. Toxicol Appl Pharmacol. 2007;222:271–80. doi: 10.1016/j.taap.2006.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tokar EJ, Qu W, Waalkes MP. Arsenic, stem cells, and the developmental basis of adult cancer. Toxicol Sci. 2011;120(Suppl 1):S192–203. doi: 10.1093/toxsci/kfq342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pilsner JR, Liu X, Ahsan H, Ilievski V, Slavkovich V, Levy D, Factor-Litvak P, Graziano JH, Gamble MV. Genomic methylation of peripheral blood leukocyte DNA: influences of arsenic and folate in Bangladeshi adults. Am J Clin Nutr. 2007;86:1179–86. doi: 10.1093/ajcn/86.4.1179. [DOI] [PubMed] [Google Scholar]
- 16.Chanda S, Dasgupta UB, Guhamazumder D, Gupta M, Chaudhuri U, Lahiri S, Das S, Ghosh N, Chatterjee D. DNA hypermethylation of promoter of gene p53 and p16 in arsenic-exposed people with and without malignancy. Toxicol Sci. 2006;89:431–7. doi: 10.1093/toxsci/kfj030. [DOI] [PubMed] [Google Scholar]
- 17.Kile ML, Baccarelli A, Hoffman E, Tarantini L, Quamruzzaman Q, Rahman M, Mahiuddin G, Mostofa G, Hsueh YM, Wright RO, et al. Prenatal arsenic exposure and DNA methylation in maternal and umbilical cord blood leukocytes. Environ Health Perspect. 2012;120:1061–6. doi: 10.1289/ehp.1104173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adalsteinsson BT, Gudnason H, Aspelund T, Harris TB, Launer LJ, Eiriksdottir G, Smith AV, Gudnason V. Heterogeneity in white blood cells has potential to confound DNA methylation measurements. PLoS One. 2012;7:e46705. doi: 10.1371/journal.pone.0046705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koestler DC, Marsit CJ, Christensen BC, Accomando W, Langevin SM, Houseman EA, Nelson HH, Karagas MR, Wiencke JK, Kelsey KT. Peripheral blood immune cell methylation profiles are associated with nonhematopoietic cancers. Cancer Epidemiol Biomarkers Prev. 2012;21:1293–302. doi: 10.1158/1055-9965.EPI-12-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lam LL, Emberly E, Fraser HB, Neumann SM, Chen E, Miller GE, Kobor MS. Factors underlying variable DNA methylation in a human community cohort. Proc Natl Acad Sci U S A. 2012;109(Suppl 2):17253–60. doi: 10.1073/pnas.1121249109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wieczorek G, Asemissen A, Model F, Turbachova I, Floess S, Liebenberg V, Baron U, Stauch D, Kotsch K, Pratschke J, et al. Quantitative DNA methylation analysis of FOXP3 as a new method for counting regulatory T cells in peripheral blood and solid tissue. Cancer Res. 2009;69:599–608. doi: 10.1158/0008-5472.CAN-08-2361. [DOI] [PubMed] [Google Scholar]
- 23.Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, Reinius L, Acevedo N, Taub M, Ronninger M, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol. 2013;31:142–7. doi: 10.1038/nbt.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koestler DC, Christensen B, Karagas MR, Marsit CJ, Langevin SM, Kelsey KT, Wiencke JK, Houseman EA. Blood-based profiles of DNA methylation predict the underlying distribution of cell types: a validation analysis. Epigenetics. 2013;8:816–26. doi: 10.4161/epi.25430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langevin SM, Koestler DC, Christensen BC, Butler RA, Wiencke JK, Nelson HH, Houseman EA, Marsit CJ, Kelsey KT. Peripheral blood DNA methylation profiles are indicative of head and neck squamous cell carcinoma: an epigenome-wide association study. Epigenetics. 2012;7:291–9. doi: 10.4161/epi.7.3.19134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burns LA, Munson AE. Gallium arsenide selectively inhibits T cell proliferation and alters expression of CD25 (IL-2R/p55) J Pharmacol Exp Ther. 1993;265:178–86. [PubMed] [Google Scholar]
- 27.Patterson R, Vega L, Trouba K, Bortner C, Germolec D. Arsenic-induced alterations in the contact hypersensitivity response in Balb/c mice. Toxicol Appl Pharmacol. 2004;198:434–43. doi: 10.1016/j.taap.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 28.Sikorski EE, McCay JA, White KL, Jr., Bradley SG, Munson AE. Immunotoxicity of the semiconductor gallium arsenide in female B6C3F1 mice. Fundam Appl Toxicol. 1989;13:843–58. doi: 10.1016/0272-0590(89)90338-2. [DOI] [PubMed] [Google Scholar]
- 29.Yoshida T, Shimamura T, Shigeta S. Enhancement of the immune response in vitro by arsenic. Int J Immunopharmacol. 1987;9:411–5. doi: 10.1016/0192-0561(87)90068-3. [DOI] [PubMed] [Google Scholar]
- 30.Lemarie A, Morzadec C, Mérino D, Micheau O, Fardel O, Vernhet L. Arsenic trioxide induces apoptosis of human monocytes during macrophagic differentiation through nuclear factor-kappaB-related survival pathway down-regulation. J Pharmacol Exp Ther. 2006;316:304–14. doi: 10.1124/jpet.105.092874. [DOI] [PubMed] [Google Scholar]
- 31.Lemarie A, Morzadec C, Bourdonnay E, Fardel O, Vernhet L. Human macrophages constitute targets for immunotoxic inorganic arsenic. J Immunol. 2006;177:3019–27. doi: 10.4049/jimmunol.177.5.3019. [DOI] [PubMed] [Google Scholar]
- 32.Bourdonnay E, Morzadec C, Sparfel L, Galibert MD, Jouneau S, Martin-Chouly C, Fardel O, Vernhet L. Global effects of inorganic arsenic on gene expression profile in human macrophages. Mol Immunol. 2009;46:649–56. doi: 10.1016/j.molimm.2008.08.268. [DOI] [PubMed] [Google Scholar]
- 33.Conde P, Acosta-Saavedra LC, Goytia-Acevedo RC, Calderon-Aranda ES. Sodium arsenite-induced inhibition of cell proliferation is related to inhibition of IL-2 mRNA expression in mouse activated T cells. Arch Toxicol. 2007;81:251–9. doi: 10.1007/s00204-006-0152-7. [DOI] [PubMed] [Google Scholar]
- 34.Hernández-Castro B, Doníz-Padilla LM, Salgado-Bustamante M, Rocha D, Ortiz-Pérez MD, Jiménez-Capdeville ME, Portales-Pérez DP, Quintanar-Stephano A, González-Amaro R. Effect of arsenic on regulatory T cells. J Clin Immunol. 2009;29:461–9. doi: 10.1007/s10875-009-9280-1. [DOI] [PubMed] [Google Scholar]
- 35.de la Fuente H, Portales-Pérez D, Baranda L, Díaz-Barriga F, Saavedra-Alanís V, Layseca E, González-Amaro R. Effect of arsenic, cadmium and lead on the induction of apoptosis of normal human mononuclear cells. Clin Exp Immunol. 2002;129:69–77. doi: 10.1046/j.1365-2249.2002.01885.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galicia G, Leyva R, Tenorio EP, Ostrosky-Wegman P, Saavedra R. Sodium arsenite retards proliferation of PHA-activated T cells by delaying the production and secretion of IL-2. Int Immunopharmacol. 2003;3:671–82. doi: 10.1016/S1567-5769(03)00049-3. [DOI] [PubMed] [Google Scholar]
- 37.Martin-Chouly C, Morzadec C, Bonvalet M, Galibert MD, Fardel O, Vernhet L. Inorganic arsenic alters expression of immune and stress response genes in activated primary human T lymphocytes. Mol Immunol. 2011;48:956–65. doi: 10.1016/j.molimm.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 38.Biswas D, Banerjee M, Sen G, Das JK, Banerjee A, Sau TJ, Pandit S, Giri AK, Biswas T. Mechanism of erythrocyte death in human population exposed to arsenic through drinking water. Toxicol Appl Pharmacol. 2008;230:57–66. doi: 10.1016/j.taap.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 39.Adalsteinsson BT, Gudnason H, Aspelund T, Harris TB, Launer LJ, Eiriksdottir G, Smith AV, Gudnason V. Heterogeneity in white blood cells has potential to confound DNA methylation measurements. PLoS One. 2012;7:e46705. doi: 10.1371/journal.pone.0046705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee ST, Xiao Y, Muench MO, Xiao J, Fomin ME, Wiencke JK, Zheng S, Dou X, de Smith A, Chokkalingam A, et al. A global DNA methylation and gene expression analysis of early human B-cell development reveals a demethylation signature and transcription factor network. Nucleic Acids Res. 2012;40:11339–51. doi: 10.1093/nar/gks957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bock C, Beerman I, Lien WH, Smith ZD, Gu H, Boyle P, Gnirke A, Fuchs E, Rossi DJ, Meissner A. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol Cell. 2012;47:633–47. doi: 10.1016/j.molcel.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ivascu C, Wasserkort R, Lesche R, Dong J, Stein H, Thiel A, Eckhardt F. DNA methylation profiling of transcription factor genes in normal lymphocyte development and lymphomas. Int J Biochem Cell Biol. 2007;39:1523–38. doi: 10.1016/j.biocel.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 43.Hartwig A, Blessing H, Schwerdtle T, Walter I. Modulation of DNA repair processes by arsenic and selenium compounds. Toxicology. 2003;193:161–9. doi: 10.1016/j.tox.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 44.Zhou X, Sun X, Cooper KL, Wang F, Liu KJ, Hudson LG. Arsenite interacts selectively with zinc finger proteins containing C3H1 or C4 motifs. J Biol Chem. 2011;286:22855–63. doi: 10.1074/jbc.M111.232926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu D, Wang ZH, Cheng SB, Li HK, Chan HB, Chew EC. The effect of arsenic trioxide on the expression of Hsc and HNF4 in nuclear matrix proteins in HepG2 cells. Anticancer Res. 2001;21(4A):2553–9. [PubMed] [Google Scholar]
- 46.States JC, Singh AV, Knudsen TB, Rouchka EC, Ngalame NO, Arteel GE, Piao Y, Ko MS. Prenatal arsenic exposure alters gene expression in the adult liver to a proinflammatory state contributing to accelerated atherosclerosis. PLoS One. 2012;7:e38713. doi: 10.1371/journal.pone.0038713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Griendling KK, Ushio-Fukai M. Reactive oxygen species as mediators of angiotensin II signaling. Regul Pept. 2000;91:21–7. doi: 10.1016/S0167-0115(00)00136-1. [DOI] [PubMed] [Google Scholar]
- 48.Navas-Acien A, Sharrett AR, Silbergeld EK, Schwartz BS, Nachman KE, Burke TA, Guallar E. Arsenic exposure and cardiovascular disease: a systematic review of the epidemiologic evidence. Am J Epidemiol. 2005;162:1037–49. doi: 10.1093/aje/kwi330. [DOI] [PubMed] [Google Scholar]
- 49.Yang HT, Chou HJ, Han BC, Huang SY. Lifelong inorganic arsenic compounds consumption affected blood pressure in rats. Food Chem Toxicol. 2007;45:2479–87. doi: 10.1016/j.fct.2007.05.024. [DOI] [PubMed] [Google Scholar]
- 50.Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood. 1997;89:3354–60. [PubMed] [Google Scholar]
- 51.Das S, Santra A, Lahiri S, Guha Mazumder DN. Implications of oxidative stress and hepatic cytokine (TNF-alpha and IL-6) response in the pathogenesis of hepatic collagenesis in chronic arsenic toxicity. Toxicol Appl Pharmacol. 2005;204:18–26. doi: 10.1016/j.taap.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 52.Kile ML, Hoffman E, Hsueh YM, Afroz S, Quamruzzaman Q, Rahman M, Mahiuddin G, Ryan L, Christiani DC. Variability in biomarkers of arsenic exposure and metabolism in adults over time. Environ Health Perspect. 2009;117:455–60. doi: 10.1289/ehp.11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kile ML, Houseman EA, Rodrigues E, Smith TJ, Quamruzzaman Q, Rahman M, Mahiuddin G, Su L, Christiani DC. Toenail arsenic concentrations, GSTT1 gene polymorphisms, and arsenic exposure from drinking water. Cancer Epidemiol Biomarkers Prev. 2005;14:2419–26. doi: 10.1158/1055-9965.EPI-05-0306. [DOI] [PubMed] [Google Scholar]
- 54.Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–27. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 56.Smyth GK. Limma: linear models for microarray data. New York: Springer, 2005. [Google Scholar]
- 57.Ruppert D, Wand MP, Carroll RJ. Semiparametric Regression. Cambridge: Cambridge University Press, 2003. [Google Scholar]
- 58.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goeman JJ, Bühlmann P. Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics. 2007;23:980–7. doi: 10.1093/bioinformatics/btm051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.