

Abstract
Cancer is as much an epigenetic disease as a genetic one; however, the interplay between these two processes is unclear. Recently, it has been shown that a large proportion of DNA methylation variability can be explained by allele-specific methylation (ASM), either at classical imprinted loci or those regulated by underlying genetic variants. During a recent screen for imprinted differentially methylated regions, we identified the genomic interval overlapping the non-coding nc886 RNA (previously known as vtRNA2-1) as an atypical ASM that shows variable levels of methylation, predominantly on the maternal allele in many tissues. Here we show that the nc886 interval is the first example of a polymorphic imprinted DMR in humans. Further analysis of the region suggests that the interval subjected to ASM is approximately 2 kb in size and somatically acquired. An in depth analysis of this region in primary cancer samples with matching normal adjacent tissue from the Cancer Genome Atlas revealed that aberrant methylation in bladder, breast, colon and lung tumors occurred in approximately 27% of cases. Hypermethylation occurred more frequently than hypomethylation. Using additional normal-tumor paired samples we show that on rare occasions the aberrant methylation profile is due to loss-of-heterozygosity. This work therefore suggests that the nc886 locus is subject to variable allelic methylation that undergoes cancer-associated epigenetic changes in solid tumors.
Keywords: imprinting, DNA methylation, miRNAs, vault RNAs, vtRNA2-1, nc886
Introduction
DNA methylation is assumed to be complementary on both alleles, except in the cases of parent-of-origin-dependent DNA methylation, which regulates imprinted gene clusters and the random silencing of one of the two X-chromosomes in females to ensure dosage compensation with males. A third type of allelic specific methylation (ASM) has recently been reported, which is associated with DNA sequences in cis (ie, single nucleotide polymorphisms, SNPs), and is termed methylation quantitative trait loci (mQTL).1 This form of allele-specific methylation has been reported to show tissue and population specificity, since it is dictated by underlying DNA sequence and acquired somatically.2 In contrast, ASM associated with imprinted differentially methylated regions (DMRs) are cis-acting regulatory elements that influence the allelic expression of neighboring genes in a parent-of-origin manner. Most imprinted DMRs acquire their differential methylation status during gametogenesis, when the two parental genomes are separated, resulting from the cooperation of the de novo methyltransferase DNMT3A and its co-factor DNMT3L.3,4 These primary, or germline, imprinted DMRs are stably maintained throughout somatic development by the UHRF1-DNMT1 complex,5 surviving the epigenetic reprogramming at the oocyte-to-embryo transition.6,7
Both types of ASM can shed light on disease susceptibility, including multifactorial phenotypes and cancer. For example, a constitutional epimutation of the MutL homolog 1 (MLH1) tumor suppressor gene in hereditary nonpolyposis colon cancer is associated with a specific polymorphic variant that modifies gene expression,8 whereas most tumor types present with frequent loss-of-imprinting (For a review see ref. 9).
During a genome-wide screen for regions of ~50% methylation in multiple tissues that could manifest as novel imprinted DMRs, we identified a region of partial methylation overlapping the non-coding RNA (ncRNA) nc886 region located on chromosome 5q31. The 102 bp nc886 transcript is a novel class of RNA. It was originally proposed to be a precursor microRNA (pri-miRNA) or a RNA component of the vault complex RNA (vtRNAs); however, based on the size of the RNA and its dicer-independent processing, it has been shown to be neither.10
The nc886 transcript was recently shown to be a tumor-suppressing ncRNA and key for tumor surveillance through its ability to bind and repress the double-stranded RNA (dsRNA)-dependent kinase (PKR).10,11 Tumor-associated repression of nc886 activates PKR, the phosphorylated form being the kinase that phosphorylates EIF2α, which in turn inhibits global protein synthesis and ultimately induces cell death. At least in acute myeloid leukemia (AML), hypermethylation of the nc886 promoter results in abolished expression, causing the inhibitory effect to disappear.12 Stress/chemotherapy-induced PKR-phosphorylation then switches to favor the activation of NFK-β and cancer cell survival at the expense of EIF2α-induced apoptosis.13
Here, we have characterized the methylation profile of the nc886 region in numerous normal tissues and observed variable methylation profile in which 76% of the normal samples analyzed were predominantly methylated on one allele and, when informative, this was the maternal chromosome. While this manuscript was in preparation, the region overlapping nc886 was shown to be polymorphic maternally methylated region in 83% of tissues, corroborating our observations.14 To ascertain whether this region is associated with aberrant methylation in solid tumors and could potentially be a marker for disease, we profiled tumor and normal paired samples from the Cancer Genome Atlas. Abnormal methylation within this interval occurred in 25% of bladder, 38% of breast, 21.5% of colorectal and 32% of lung primary tumor samples. Using additional paired samples sets, we showed that, occasionally, the aberrant epigenetic profile was explained by cancer-associated loss-of-heterozygosity (LOH), suggesting that deletion of 5q31 also occurs in solid tumors.
Results
Methylation profiling of the nc886 region by WGBS and Infinium Methylation Arrays
We have previously used whole genome bisulphite sequencing (WGBS) and the Illumina Infinium HumanMethylation450 BeadChip array to identify novel human imprinted loci.15 During this screen, we identified a region overlapping the nc886 transcript that was consistently partially methylated in DNA samples derived from leukocytes, liver, brain and placenta tissues. Twenty probes present on the Infinium array platform confirm this partially methylated profile (Fig. 1) in tissues and immune-selected blood lineages (Fig. S1). Despite appearing as a promising candidate for a novel imprinted DMR, this region did not show the correct parent-of-origin methylation profile in reciprocal genome-wide uniparental disomy (UPD) samples or hydatidiform moles (containing only paternal chromosomes), which would be expected for a bona fide imprinted DMR (Fig. 1).

Figure 1. Mapping the extent of allelic methylation at the nc886 interval in 4 human tissues. (A) Map of the nc886 locus, showing the location of the transcript, CpG islands, genetic variants and DNA repeat-elements. (B) Detailed methylation map of the nc886 interval for placenta as determined by WGBS and Infinium HumanMethylation BeadChip array. Vertical dark gray lines in the WGBS tracks represent the methylation value for individual CpG dinucleotides and each dot representing the methylation of a single array probe: normal placenta (black dots) and hydatidiform mole (blue dots). The pattern of methylation was confirmed using bisulphite PCR in heterozygous samples so that allelic origin could be ascertained. Each circle represents a single CpG dinucleotide on a DNA strand, a methylated cytosine (⚫) or an unmethylated cytosine (○). The methylation profiles for brain (C), liver (D) and leukocytes (E) also reveal partial methylation over the nc886 transcript. The lower panel also shows the DNA methylation profile for normal leukocytes (black), genome-wide pUPD (blue) and mUPD (red) samples as determined by the Infinium methylation array.
Allelic methylation of the nc886 promoter
Despite the contradictory evidence for parent-of-origin methylation within the nc886 locus, we decided to pursue the mechanism leading to the partially methylated profile. We utilized bisulphite PCR and subsequent sequencing of sub-cloned DNA strands incorporating the SNP rs9327740, which is located ~200 bp upstream of CpG island 24 in the promoter of nc886. This semi-quantitative technique was used rather than quantitative pyrosequencing, as the latter does not allow for allelic discrimination or assessment of PCR clonality. We observed that in 87% of heterozygous samples (n = 7), irrespective of tissue type analyzed, the A allele was more methylated (Fig. 1; Table S1). In informative samples, the methylation was always on the maternal chromosome (Fig. 2). Using this genetic variant to assay for allelic methylation, we could not distinguish between polymorphic imprinted and SNP-associated methylation. To exclude that the methylation pattern we observe was directly due to this SNP variant, we analyzed samples that were homozygous for rs9327740. Most homozygous tissue samples (9/16) had both methylated and unmethylated strands consistent with imprinting (Fig. 1).

Figure 2. Confirmation that nc886 is a maternally methylated DMR. Examples of bisulphite PCRs confirming the presence of methylation on the maternal allele in cord blood and placental-derived DNA samples heterozygous for the SNPs (A) rs2346018, (B) rs2346019 and (C) rs9327740.
Variable maternal methylation across the interval nc886
To determine the extent of the allelic methylation we performed a similar bisulphite PCR strategy for 5 additional regions that contained highly informative SNPs. The genomic interval overlapping the rs2346019 polymorphism also presented with allelic methylation in 83% of samples, with the G allele being methylated in 9/10 heterozygous samples (Table S1). When informative, the maternal chromosome was more methylated (Figs. 1 and 2). For only one placenta and cord blood pair (sample set 060) the A allele was inherited maternally and was methylated, confirming that the methylation in this domain is inherited in a parent-of-origin manner and not dictated by SNP genotype (Fig. 2). The next PCR amplicon targeted SNP rs2346018 near CpG island 18 and presented with a gradient methylation profile. The maternal allele was heavily methylated, whereas the paternal allele gained methylation in the centromeric direction in 63% of samples, confirming the WGBS data that indicated this was the boundary of the allelic methylation. To determine the extent of the ASM we performed allelic bisulphite PCR on the regions surrounding rs2215012 and rs4976364. These SNPs are located within MER5B-hAT sequences and a LINE L2a-SINE MIRb element, respectively. Both of these repeats were hypermethylated on both chromosomes in all analyzed somatic tissues (Fig. 1; Table S1). These observations are concordant with the discrete boundaries of the DMR identified by Paliwal et al. using high throughput bar-code bisulfite PCR Fluidigm AccessArrays.14
ASM associated with nc886 is somatically acquired
To address whether the DNA methylation within the nc886 domain is germline- or somatically-acquired, we performed bisulphite analysis on DNA extracted from sperm, stem cells derived from parthenogenetically-activated metaphase-2 oocyte blastocysts (phES)16,17 and human embryonic stem (hES) cell lines generated from both seven-cell blastomeres and the inner cell mass (ICM) of blastocysts. We have previously shown that low passage phES cells are good surrogates for primary oocytes since they maintain the correct methylation profile at imprinted loci.15
The bisulphite analysis of a sperm sample heterozygous for rs2346018 revealed that spermatozoa with either chromosome 5 homolog were completely unmethylated. This interval was also devoid of methylation in the phES and hES cells generated from seven-cell blastomere, but allelic in hES cells derived from the ICM. This strongly suggests that the methylation at nc886 is a post-fertilization event, occurring early in development at a time point between post-fertilization days 4–6 (Fig. 3).
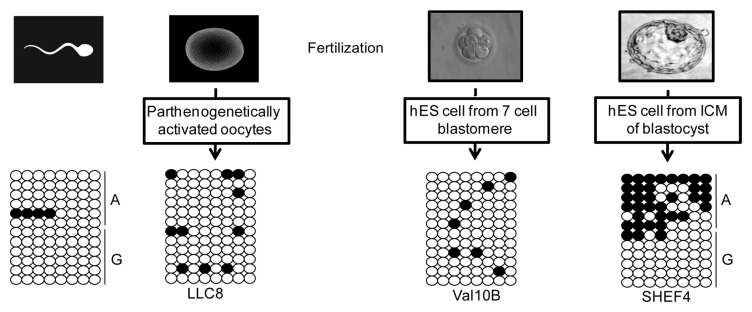
Figure 3. Timing of the acquisition of DNA methylation at nc886. The methylation profiles for the bisulphite PCR incorporating the SNP rs2346019 in DNA extracted from mature sperm, phES and hES cells derived from different aged pre-implantation embryos.
Tumor samples show aberrant methylation changes at nc886 with occasional loss-of-heterozygosity
Recent studies have shown that nc886 has tumor suppressing function, with hypermethylation and concomitant decreased expression predicting outcome in AML patients.12 To determine if the methylation of this interval has prognostic value in solid tumors, we determined the methylation index for a range of tumors and adjacent normal tissues using the Infinium HumanMethylation450 BeadChip array. This analysis revealed that 65, 71, 66.5 and 56.5% of normal bladder, breast, colon and lung tissues, respectively, were ~50% methylated and that the corresponding tumors samples maintained this profile. In some instances, we observed samples with ~50% in the normal tissues but either hyper- or hypo-methylation in the matched tumor sample (Fig. 4; Table S2). We confirmed these observations using bisulphite PCR and sub-cloning in samples for which we had DNA. Three colorectal sample sets were heterozygous for the rs2346019 variant. Interestingly, one clearly presented with hypermethylation on the normally unmethylated allele (colon 1184), whereas the remaining two sample pairs (colon 326 and 973) were associated with loss of the unmethylated allele, consistent with a cancer-associated heterozygous deletion (Fig. 4). In addition, we observed tumor-associated hypermethylation when the normal sample was constitutionally unmethylated. For the colon sample set 1404, three different tumor sections presented with various degrees of hypermethylation.

Figure 4. Profiling the methylation status of the nc886 locus in human tumor samples. Circular heat map of Infinium array probes mapping to the nc886 locus in (A) breast, (B) colorectal, (C) lung and (D) bladder tumors. In each case the inner circle represents the methylation values of the normal tissue and the outer circle the corresponding tumor. (E) Confirmation of the methylation profile overlapping the rs2346019 interval in various CRC samples sets. Each circle represents a single CpG dinucleotide on a DNA strand, a methylated cytosine (⚫) or an unmethylated cytosine (○).
Discussion
Through the combined use of genome-wide methylation screening, we have identified a region of partial methylation overlapping the nc886 region. Allelic methylation analysis using various SNPs throughout the interval revealed that the methylation is predominantly on one allele in ~73% of all samples, suggesting it is an atypical imprinted DMR. To date, all known human imprinted DMRs are stably maintained in somatic tissues, irrespective of gene expression levels, by the DNA methyltransferase activity of DNMT1 during semi-conservative DNA replication.5 Polymorphic imprinting has been observed in human placental samples at the IGF2R locus.18,19 In-depth studies trying to delineate the mechanism for polymorphic imprinting at this locus failed to identify an epigenetic signature that differentiates imprinted and biallelic expressing samples.18 Both categories maintained a maternally methylated DMR within intron 2 of IGF2R, overlapping the promoter of the ncRNA AIRN. Therefore, our observations, substantiated by those noted by Paliwai et al.,14 are the first evidence for a polymorphic imprinted DMR. Unfortunately, the ~102 bp nc886 transcript does not contain any genetic variants that would allow us to directly determine whether the presence of maternal methylation within this interval directly influences allele-specific expression.
The nc886 gene is located at chromosome 5q31. This region is frequently associated with LOH in many tumor types. Deletions of chromosome 5q are associated with poor outcomes in AML.12 Heterozygous deletions encompassing 5q, identified by microsatellite polymorphisms and CGH array analyses, have also been identified in colorectal and breast tumors. While no significant association between LOH and recurrence or survival was observed in colorectal cancer,20,21 LOH of this interval often occurred with BRCA2 mutations in sporadic breast tumors.22 Furthermore, 5q31 LOH was described as a hallmark for basal-like breast tumors and was associated with estrogen-receptor status.23 Taken together, these observations suggest the presence of one or more tumor-suppressor genes at this locus. The nc886 RNA is a confirmed tumor suppressor ncRNA that acts by downregulating the cancer survival factor pPKR.12,24 Our results suggest that abolished expression of nc886 can be achieved by either promoter hypermethylation or LOH, as seen in 25, 36, 16.5 and 29% of bladder, breast, colon and lung tumors, respectively. However, in addition to hypermethylation, ~5% tumors, respective of tissue of origin, presented with decreased methylation and, presumably, overexpression of the nc886 transcript, suggesting that survival of some tumors is not governed by the nc886-pPKR pathway.
Conclusions
Our results strongly suggest that the ~2 kb region overlapping the nc886 RNA locus somatically acquires DNA methylation of the maternal allele in the majority of individuals, with approximately 27% of the population not associated with ASM. In solid tumors, this region is subject to cancer-associated methylation changes as well as LOH. Further work is now required to determine if, as in AML, aberrant nc886 expression disrupts the nc886-PKR tumor surveillance pathway in these tumor types, and whether abnormal epigenetic regulation is responsible.
Materials and Methods
Tissue-sample and patient material
A cohort of 100 normal placental tissue, paired cord blood and maternal peripheral blood samples were collected from the Hospital St. Joan De Deu collection. Normal peripheral blood was collected from adult volunteers aged between 19–60 y old. A selection of normal brain samples was obtained from BrainNet Europe/Barcelona Brain Bank and normal kidney samples were obtained from XBTC tissue bank. DNA and RNA extraction and cDNA synthesis were performed as previously described.25 Ethical approval for adult blood and placenta tissue collection was granted by the Hospital St Joan De Deu Ethics Committee (Study number 35/07) and IDIBELL (PR251/12).
Paired tumor and normal samples sets
The methylation profiles determined by the Illumina Infinium HumanMethylation450 BeadChip arrays for 20 paired normal and bladder tumor samples (urothelial carcinomas), 92 paired breast sample sets (invasive carcinomas), 42 colorectal tumors with adjacent normal tissues (adenocarcinomas) and 69 lung cancers pairs (40 squamous cell carcinomas and 29 adenocarcinomas) were obtained from the Cancer Genome Atlas. Surplus bisulphite DNA from 4 in-house hybridized colorectal cancer sample sets were used for allelic bisulphite PCRs.
Allelic bisulphite sequencing
Prior to methylation analysis all samples were genotyped by PCR and direct sequencing. Sequence traces were interrogated using Sequencher v4.6 (Gene Codes Corporation) to distinguish heterozygous and homozygous samples. Approximately 1 μg DNA was subjected to sodium bisulphite treatment and purified using the EZ GOLD methylation kit (ZYMO). Bisulphite specific primers for each region were used with Hotstar Taq polymerase (Qiagen) for 40 PCR cycles. All PCR products were cloned into pGEM-T Easy vector (Promega) with a minimum of 12 clones were selected for sequencing. All primers for genotyping and bisulphite PCR can found in Table S3.
WGBS and Illumina Infinium HumanMethylation450 BeadChip array analysis
We analyzed 6 publically available methylomes, including those derived from CD4+ lymphocytes (GSE31263), brain (GSM913595) liver and placenta (GSE46698). Matching samples were also hybridized to the Illumina Infinium HumanMethylation450 BeadChip arrays (GSE46698). In addition we used 3 leukocyte data sets from GSE30870 and the isolated cell populations published by Reinius and colleagues (GSE35069).26
Data filtering and analysis
WGBS: The sequence reads were aligned to either strand of the HG19 reference genome using a custom computational pipeline. The methylation level of each cytosine within CpG dinucleotides was estimated as the number of reads reporting a C, divided by the total number of reads reporting C or T. The location of the nc886 interval was determined by the sequence from the UCSC sequence browser.
Illumina Infinium HumanMethylation 450 BeadChip array: Before analyzing the data, we excluded possible sources of technical biases that could influence results. We applied signal background subtraction and inter-plate variation was normalized using default control probes in BeadStudio (version 2011.1_Infinium HD). We discarded probes with a detection P value > 0.01. We also excluded probes that lack signal values in one or more of the DNA samples analyzed. In addition we discarded 16,631 probes as they contained SNPs present in more than 1% of the population (dbSNP 137). We utilized in-house R-package scripts evaluate the methylation of the probes mapping within the chr5: 135414408–135417466 region. The circular heatmaps used to display the DNA methylation profiles of the paired normal and tumor samples were generated using Circos software.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Dr Joan Blanco and Dr Cristina Camprubi for supplying sperm DNA from normal individuals with proven fertility, Professor Hans Keirstead for the phES cell lines and Harry Moore for the SHEF hES cell DNAs. We would like to thank Professor Isidro Ferrer of the Barcelona Brain Bank for supplying human brain specimens. Additional normal tissues were obtained from the Xarxa tumor tissue bank which is sponsored by the Pla Director d’Oncologia de Catalunya (XBTC). This work was supported by the Spanish Ministerio de Educación y Ciencia (BFU2011–27658 to D.M); Fundació La Marató de TV3 (101130 to D.M); Research in the Neonatal Unit in HSJD is partially funded by an unrestricted grant from BBDue Spain. MV is a recipient of a FPU research fellowship and DM is a Ramón y Cajal research fellow (RYC-04548).
Glossary
Abbreviations:
- Nc
non-coding
- vtRNA
vault complex RNA
- miRNA
microRNA
- pri-miRNA
precursor microRNA
- dsRNA
double stranded RNA
- CRC
colorectal carcinomas
- LOH
loss-of-heterozygosity
- UPD
uniparental disomy
- DMR
differentially methylated region
- ICM
inner cell mass
- hES
human embryonic stem cells
- pa-oocytes
parthenogenetically-activated metaphase II oocytes
- AML
acute myeloid leukemia
References
- 1.Kerkel K, Spadola A, Yuan E, Kosek J, Jiang L, Hod E, Li K, Murty VV, Schupf N, Vilain E, et al. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat Genet. 2008;40:904–8. doi: 10.1038/ng.174. [DOI] [PubMed] [Google Scholar]
- 2.Heyn H, Moran S, Hernando-Herraez I, Sayols S, Gomez A, Sandoval J, Monk D, Hata K, Marques-Bonet T, Wang L, et al. DNA methylation contributes to natural human variation. Genome Res. 2013;23:1363–72. doi: 10.1101/gr.154187.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bourc’his D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536–9. doi: 10.1126/science.1065848. [DOI] [PubMed] [Google Scholar]
- 4.Hata K, Okano M, Lei H, Li E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development. 2002;129:1983–93. doi: 10.1242/dev.129.8.1983. [DOI] [PubMed] [Google Scholar]
- 5.Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA, Shinga J, Mizutani-Koseki Y, Toyoda T, Okamura K, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;450:908–12. doi: 10.1038/nature06397. [DOI] [PubMed] [Google Scholar]
- 6.Smallwood SA, Tomizawa S, Krueger F, Ruf N, Carli N, Segonds-Pichon A, Sato S, Hata K, Andrews SR, Kelsey G. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat Genet. 2011;43:811–4. doi: 10.1038/ng.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith ZD, Chan MM, Mikkelsen TS, Gu H, Gnirke A, Regev A, Meissner A. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature. 2012;484:339–44. doi: 10.1038/nature10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hitchins MP, Rapkins RW, Kwok CT, Srivastava S, Wong JJ, Khachigian LM, Polly P, Goldblatt J, Ward RL. Dominantly inherited constitutional epigenetic silencing of MLH1 in a cancer-affected family is linked to a single nucleotide variant within the 5’UTR. Cancer Cell. 2011;20:200–13. doi: 10.1016/j.ccr.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Monk D. Deciphering the cancer imprintome. Brief Funct Genomics. 2010;9:329–39. doi: 10.1093/bfgp/elq013. [DOI] [PubMed] [Google Scholar]
- 10.Jeon SH, Lee K, Lee KS, Kunkeaw N, Johnson BH, Holthauzen LM, Gong B, Leelayuwat C, Lee YS. Characterization of the direct physical interaction of nc886, a cellular non-coding RNA, and PKR. FEBS Lett. 2012;586:3477–84. doi: 10.1016/j.febslet.2012.07.076. [DOI] [PubMed] [Google Scholar]
- 11.Jeon SH, Johnson BH, Lee YS. A tumor surveillance model: a non-coding RNA senses neoplastic cells and its protein partner signals cell death. Int J Mol Sci. 2012;13:13134–9. doi: 10.3390/ijms131013134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Treppendahl MB, Qiu X, Søgaard A, Yang X, Nandrup-Bus C, Hother C, Andersen MK, Kjeldsen L, Möllgård L, Hellström-Lindberg E, et al. Allelic methylation levels of the noncoding VTRNA2-1 located on chromosome 5q31.1 predict outcome in AML. Blood. 2012;119:206–16. doi: 10.1182/blood-2011-06-362541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blalock WL, Bavelloni A, Piazzi M, Faenza I, Cocco L. A role for PKR in hematologic malignancies. J Cell Physiol. 2010;223:572–91. doi: 10.1002/jcp.22092. [DOI] [PubMed] [Google Scholar]
- 14.Paliwal A, Temkin AM, Kerkel K, Yale A, Yotova I, Drost N, Lax S, Nhan-Chang CL, Powell C, Borczuk A, et al. Comparative anatomy of chromosomal domains with imprinted and non-imprinted allele-specific DNA methylation. PLoS Genet. 2013;9:e1003622. doi: 10.1371/journal.pgen.1003622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Court F, Tayama C, Romanelli V, Martin-Trujillo A, Iglesias-Platas I, Okamura K, Sugahara N, Simón C, Moore H, Harness JV, et al. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of the human imprinting and suggests a germline methylation independent mechanism of establishment. Genome Res. 2014 doi: 10.1101/gr.164913.113. Forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mai Q, Yu Y, Li T, Wang L, Chen MJ, Huang SZ, Zhou C, Zhou Q. Derivation of human embryonic stem cell lines from parthenogenetic blastocysts. Cell Res. 2007;17:1008–19. doi: 10.1038/cr.2007.102. [DOI] [PubMed] [Google Scholar]
- 17.Harness JV, Turovets NA, Seiler MJ, Nistor G, Altun G, Agapova LS, Ferguson D, Laurent LC, Loring JF, Keirstead HS. Equivalence of conventionally-derived and parthenote-derived human embryonic stem cells. PLoS One. 2011;6:e14499. doi: 10.1371/journal.pone.0014499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Monk D, Arnaud P, Apostolidou S, Hills FA, Kelsey G, Stanier P, Feil R, Moore GE. Limited evolutionary conservation of imprinting in the human placenta. Proc Natl Acad Sci U S A. 2006;103:6623–8. doi: 10.1073/pnas.0511031103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim AL, Ng S, Leow SC, Choo R, Ito M, Chan YH, Goh SK, Tng E, Kwek K, Chong YS, et al. Epigenetic state and expression of imprinted genes in umbilical cord correlates with growth parameters in human pregnancy. J Med Genet. 2012;49:689–97. doi: 10.1136/jmedgenet-2012-100858. [DOI] [PubMed] [Google Scholar]
- 20.Gerdes H, Chen Q, Elahi AH, Sircar A, Goldberg E, Winawer D, Urmacher C, Winawer SJ, Jhanwar SC. Recurrent deletions involving chromosomes 1, 5, 17, and 18 in colorectal carcinoma: possible role in biological and clinical behavior of tumors. Anticancer Res. 1995;15:13–24. [PubMed] [Google Scholar]
- 21.Kambara T, Sharp GB, Nagasaka T, Takeda M, Sasamoto H, Nakagawa H, Isozaki H, MacPhee DG, Jass JR, Tanaka N, et al. Allelic loss of a common microsatellite marker MYCL1: a useful prognostic factor of poor outcomes in colorectal cancer. Clin Cancer Res. 2004;10:1758–63. doi: 10.1158/1078-0432.CCR-0779-3. [DOI] [PubMed] [Google Scholar]
- 22.Johannsdottir HK, Jonsson G, Johannesdottir G, Agnarsson BA, Eerola H, Arason A, Heikkila P, Egilsson V, Olsson H, Johannsson OT, et al. Chromosome 5 imbalance mapping in breast tumors from BRCA1 and BRCA2 mutation carriers and sporadic breast tumors. Int J Cancer. 2006;119:1052–60. doi: 10.1002/ijc.21934. [DOI] [PubMed] [Google Scholar]
- 23.Wang ZC, Lin M, Wei LJ, Li C, Miron A, Lodeiro G, Harris L, Ramaswamy S, Tanenbaum DM, Meyerson M, et al. Loss of heterozygosity and its correlation with expression profiles in subclasses of invasive breast cancers. Cancer Res. 2004;64:64–71. doi: 10.1158/0008-5472.CAN-03-2570. [DOI] [PubMed] [Google Scholar]
- 24.Kunkeaw N, Jeon SH, Lee K, Johnson BH, Tanasanvimon S, Javle M, Pairojkul C, Chamgramol Y, Wongfieng W, Gong B, et al. Cell death/proliferation roles for nc886, a non-coding RNA, in the protein kinase R pathway in cholangiocarcinoma. Oncogene. 2013;32:3722–31. doi: 10.1038/onc.2012.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Camprubí C, Iglesias-Platas I, Martin-Trujillo A, Salvador-Alarcon C, Rodriguez MA, Barredo DR, Court F, Monk D. Stability of genomic imprinting and gestational-age dynamic methylation in complicated pregnancies conceived following assisted reproductive technologies. Biol Reprod. 2013;89:50. doi: 10.1095/biolreprod.113.108456. [DOI] [PubMed] [Google Scholar]
- 26.Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlén SE, Greco D, Söderhäll C, Scheynius A, Kere J. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One. 2012;7:e41361. doi: 10.1371/journal.pone.0041361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.