

Abstract
Background
Treatment approaches for major depressive disorder (MDD) result in approximately one-third of patients achieving remission after a first treatment. Added treatment generally improves remission rates, but approximately one-third of all patients fail to respond after several treatments (sequential monotherapies or combined treatment). A pretreatment biomarker could help identify these patients. Over activity of the subcallosal cingulate has been associated with treatment non-response in MDD, and it is a potential candidate for such a biomarker.
Methods
Eighty-two currently untreated MDD patients were enrolled in a two-phase treatment study. A flurodeoxyglucose positron emission tomography scan was acquired. Following scanning, patients were randomized to 12 weeks of either escitalopram or cognitive behavior therapy (CBT). Patients not achieving remission after 12 weeks of initial treatment were treated with an additional 12 weeks of escitalopram plus CBT. Subcallosal cingulate metabolism was compared between non-responders and remitters to either Phase 1 or Phase 2 treatment. This analysis was followed by a whole brain analysis making the same comparison.
Results
After two phases of treatment (24 weeks), 36 patients achieved remission, 6 patients achieved response, and 9 patients were non-responders. Subcallosal cingulate metabolism was significantly higher in non-responders than remitters. In the follow-up whole brain analysis, increased superior temporal sulcus activity was also associated with two-treatment non-response.
Conclusions
Depressed patients who fail to remit to CBT or escitalopram, either alone or in combination, have a distinct brain metabolic pattern compared to patients who remit with CBT, escitalopram or their combination.
Registered at clinicaltrials.gov (NCT00367341)
Keywords: depression, biomarker, subcallosal cingulate, superior temporal sulcus, antidepressant medication, psychotherapy
INTRODUCTION
After more than 40 years of research on treatment outcomes in major depressive disorder, current standards for treatment selection remain imprecise and non-personalized. This has significant clinical repercussions as published remission rates are consistently less than 40% in depressed patients treated with first-line monotherapies such as antidepressant medications (e.g., SSRIs – serotonin reuptake inhibitors) or evidence-based psychotherapies (e.g., CBT- cognitive behavior therapy, IPT- interpersonal therapy) (1–6). After an initial treatment failure, subsequent steps generally involve switching between or combining first-line treatments. Common second-step treatment strategies include moving between psychotherapy and antidepressant medication, switching between antidepressant medications, or augmenting antidepressant medication treatment with psychotherapy or a second medication. However, such strategies result in additional remission rates of only 15–20% (5; 7–9). Critically, the lack of response to initial treatments increases the vulnerability of non-remitting patients to ongoing suicidal ideation, social dysfunction, and treatment dropout (10).
Initial choice of treatment for MDD is typically based on the judgment of the mental health professional delivering the intervention, patient preference, consideration of potential side effects, and drug interactions. Treatment guidelines have suggested that severity of the disorder should guide the choice of intervention, with antidepressant medications or the combination of medications and CBT as the first-line treatment for severely depressed patients (3; 11); however, data to support this recommendation are limited and inconsistent (12).
This lack of evidence-based guidance for optimizing treatment for depression has encouraged clinical researchers to evaluate various predictive markers that could be applied at the level of the individual patient. Toward such a “stratified medicine” approach, numerous strategies have been tested, including clinical (13), imaging (14–17), genetic (18; 19), electroencephalographic (20) and immune-related metrics (21). Many of these focus on a single treatment, however, and can only identify factors predicting good or poor outcome to one treatment modality. Such predictors of single treatment response have limited clinical application since the key clinical decision is to pick which of several treatment modalities is likely to be most successful for a given patient. Addressing this issue, our group recently reported that metabolic activity in the right anterior insula (among several identified candidates measured using flurodeoxyglucose positron emission tomography, FDG PET), best predicted differential remission and non-response to randomized initial treatment with either an antidepressant medication (escitalopram) or CBT (22). However, a more complex combination of regional patterns may be needed to fully characterize patients who require alternative treatments or who may be treatment resistant.
The next step from this dataset was therefore to examine potential predictors of patients who are unlikely to show meaningful improvement to either of these first-line treatments. Defining neural activity patterns predictive of failure to both a standard antidepressant medication and an evidence-based course of psychotherapy could help “fast-track” such patients to alternative treatments, partially circumventing the protracted trial-and-error process of current clinical care. Toward these goals, we examined regional cerebral glucose metabolism that characterized non-response to two MDD recommended treatments: evidence-based psychotherapy (P) and a selective serotonin reuptake inhibitor (SSRI) (1). These P+SSRI treatment non-responders are defined as those patients who fail to respond over 6 months of treatment; the first 3 months randomized to either CBT or escitalopram (sCIT), the second 3 months receiving combined sCIT + CBT.
Based on previous investigations of treatment failure in major depression (16; 23–25), we hypothesized that P+SSRI treatment non-responders would show increased pre-treatment subcallosal cingulate (SCC) metabolism as indexed by FDG-PET. Previous studies have shown hyperactivity in the SCC at baseline in patients who fail to respond to various treatments (16), especially among those patients who have already failed at least one treatment (23–25). Many of the prior studies included patients on active treatment or patients who previously demonstrated treatment resistance. We explored the pretreatment neural patterns associated with non-response in depressed patients following randomized, controlled, stepwise treatment with two antidepressant interventions with different presumed mechanisms of action.
METHODS AND MATERIALS
Participants
Study enrollment has been previously described (22; 26). Briefly, a primary diagnosis of MDD was assessed by the Structural Clinical Interview for DSM-IV Axis I Disorders (SCID-I) (27) and confirmed through psychiatric evaluation by a study psychiatrist. The Mood and Anxiety Disorders Program at Emory University recruited adult outpatients (ages 18 to 60) through clinician referrals and advertisement. Patient severity was defined by the Hamilton Depression Rating Scale (HDRS) (28); cutoffs for inclusions were a 17-item score ≥18 at screening and ≥15 at the baseline randomization visit. Exclusion criteria included: a non-MDD primary psychiatric condition; a medical or neurological condition potentially contributing to depression or interfering with response to treatment; psychotic features; current suicidal ideation requiring urgent clinical intervention; current substance abuse (past 3 months) or dependence (past 12 months); current obsessive compulsive disorder or eating disorder; current or intended pregnancy or breastfeeding; current treatment with antidepressant medication; or receipt of electroconvulsive therapy within six months of the screening visit. Additional exclusion criteria included lifetime history of failure to respond to adequate treatment with the treatments offered in the current study (minimum 4 sessions CBT; minimum 10 mg/day escitalopram for 6 weeks).
Written informed consent was obtained from all participants with the protocol conducted as approved by the Emory Institutional Review Board and as registered at clinicaltrials.gov (NCT00367341). For interpretation of identified differences in regional metabolism, a comparison group of 24 healthy volunteers were similarly screened with the additional exclusion criterion of no current or past MDD.
Treatment Protocol
Treatment consisted of two phases: monotherapy treatment (Phase 1) followed by combination treatment (Phase 2) (Figure 1) (22). In Phase 1, patients were randomly assigned (1:1) to receive 12 weeks of either escitalopram (sCIT) or manual-based, depression-focused cognitive behavior therapy (CBT). PET and MRI scans were acquired prior to treatment randomization. Patients were then randomized to sCIT or CBT if they continued to meet eligibility criteria. sCIT was started at 10 mg/day and increased to 20 mg/day at/after week 3 if the patient was not in remission and was tolerating the medication. If side effects were intolerable at the higher dose, dosage could be reduced to 10 mg/d. Sixteen CBT sessions were scheduled, twice weekly for the first 4 weeks and weekly for the subsequent 8 weeks. Raters—blinded to treatment group—assessed changes in symptom severity using the HDRS. Ratings were performed weekly for the first 6 weeks, then biweekly through week 12. Upon completion of their Phase 1 treatment, non-remitting patients (patients with an HDRS >7 at either week 10 or 12) were offered enrollment in Phase 2. Phase 2 treatment included an additional 12 weeks of treatment with combination sCIT+CBT. In Phase 2, patients initially randomized to sCIT continued on their current dosage with CBT sessions added twice weekly for the first 4 weeks, then weekly for the subsequent 8 weeks. Patients initially randomized to CBT received 3 booster sessions of CBT at monthly intervals, and sCIT was added, dosed as in Phase 1. Raters assessed changes in symptom severity using the HDRS: weekly for the first 6 weeks of phase 2, then biweekly until week 24.
Figure 1.

Study design and treatment outcomes. *11 sCIT remitters with FDG-PET scans (out of 12 sCIT remitters)
Clinical Metrics
Clinical outcomes were defined using the HDRS with remission as the target endpoint. An HDRS score ≤7 at both weeks 10 and 12 of treatment defined Phase 1 remission. Similarly, Phase 2 remission was defined as an HDRS score ≤7 at both weeks 22 and 24 of treatment. Patients remitting at the end of Phase 1 or Phase 2 treatments were included in the ‘remitter’ group. P+SSRI non-response was defined by an HDRS change of <50% from baseline to the end of Phase 2 (week 24). To avoid potential dilution of either the remission or P+SSRI non-response groups, dropouts and patients who achieved response but not remission (change in HDRS ≥50% but with an HDRS score >7) by the end of Phase 2 were not included in these main outcome groups, but were examined post-hoc. Other clinical measures included: Beck Depression Inventory (BDI) (29), Hamilton Anxiety Scale (HAMA) (30), age, gender, age of MDD onset, duration of the current episode, number of previous episodes, previous treatment, MDD type, family history of mood disorder, education, marital status, race, employment status, and Childhood Trauma Questionnaire (31). T-tests were performed to compare P+SSRI non-responders to remitters on these variables as well as comorbid psychiatric disorders (current anxiety disorder, lifetime PTSD, and lifetime substance abuse) independent of the primary imaging analyses described below.
Imaging Acquisition
Prior to treatment randomization, brain glucose metabolism was measured using standard positron emission tomography (PET) methods (Siemens HRRT, Nashville, TN) as previously described (22). A 10mCi dose of 2-[18F]-fluoro-2-deoxy-D-glucose (FDG) was administered intravenously for each scan. A 40-minute uptake period where patients remained supine, awake, and resting with eyes closed and ears uncovered was followed by a 20-minute 3-D image acquisition without arterial blood sampling. During FDG uptake, patients were given no explicit cognitive instructions but were asked to avoid ruminating on any one topic. Raw emission images were corrected for injected dose and attenuation (using Cs-137, 6 minute transmission scan), reconstructed, and smoothed to an in-plane resolution of 4.0 mm. For image preprocessing procedures and anatomical reference, a high-resolution T1-weighted structural magnetic resonance imaging (MRI) scan was separately acquired (Siemens Tim Trio 3T whole body scanner, 3D MP-RAGE optimized at TE/TR=5/35, matrix=256×208×196, 1mm isotropic resolution). Healthy control data were acquired using identical methods.
Image Preprocessing
FDG-PET images were attenuation corrected (32). Each individual’s FDG-PET scan was then co-registered with their T1-weighted MRI anatomical image using a six degree of freedom linear transform. Co-registered FDG-PET images were written into standard space using a non-linear transform calculated from the T1-weighted image and smoothed with an 8mm FWHM Gaussian kernel (DARTEL,(33) SPM8; Welcome Department of Imaging Neuroscience, London, England; http://www.fil.ion.ucl.ac.uk/spm/). Four patients were missing anatomical scans and were alternatively normalized using a study specific FDG-template, then smoothed to match the DARTEL normalized images. All spatially normalized images were corrected for differences in whole brain global mean activity (34). Relative glucose metabolic rates were used for all analyses.
Image Analysis
A priori region of interest analysis: subcallosal cingulate (SCC)
Based on the anatomical variability of the SCC in published reports of this region (variously incorporating Brodmann Areas 25,24,32) (16; 23–25), the entire subcallosal cingulate was surveyed using small volume correction methods (http://afni.nimh.nih.gov/pub/dist/doc/program_help/3dClustSim.html). The subcallosal cingulate volume was defined using FSL’s Harvard-Oxford atlas (35), SCC thresholded at 50% probability (Figure 2) centered on MNI coordinates - x=3, y=−17, z=10. Within this bilateral subcallosal cingulate volume, the P+SSRI non-response and remitter groups were contrasted using a voxel-wise t-test. Results were considered statistically significant at a family-wise error (FWE) corrected p< 0.05 (p < 0.005 uncorrected, small volume cluster size 0.14 mL).
Figure 2.
Atlas derived subcallosal cingulate (SCC) region of interest
Whole Brain Analysis
To probe other regions predictive of non-response to combined treatment, a whole-brain voxel-wise t-test was performed using the same P+SSRI non-response and remitter groups. Resulting clusters were considered statistically significant at a FWE corrected p< 0.05 (p < 0.001 uncorrected, cluster size 2.3 mL). Average metabolism was extracted in statistically significant regions of interest and post-hoc tests were performed.
Remitter by Phase, Remitter by Treatment Follow-up Analyses
The remitter group included all patients remitting to Phase 1 monotherapy treatment or Phase 2 combination treatment. To ensure that findings attributed to differences between P+SSRI non-responder and remitter groups were not due to systematic differences within the remitter group, two additional post-hoc t-tests were performed on all statistically significant regions. First, Phase 1 and Phase 2 remitters were compared to test for bias from different numbers of treatments (monotherapy vs. combination treatment). Second, to test for treatment specific effects, Phase 1 CBT monotherapy remitters were compared to Phase 1 sCIT monotherapy remitters.
Effect Size and Correlational Analyses
Effect sizes were calculated from the regions identified in the P+SSRI non-responder vs. remitter contrasts. To evaluate the relationship between regional metabolism and two-treatment outcome, percent change in HDRS from baseline to the Phase 2 endpoint was correlated with metabolism in each extracted region. Patients achieving remission during Phase 1, and those who did not enter or dropped out of Phase 2, were treated for a shorter period of time than Phase 2 completers and received only one treatment. In Phase 1 completers, metabolism was separately correlated with the percent change in HDRS from baseline to week 12 (Phase 1 endpoint). These correlations allow for inclusion of patients with unclear outcomes in addition to those in the P+SSRI non-responder and remitter groups.
Comparisons with healthy controls
To further characterize the nature of identified patient group differences, mean metabolism in regions identified in the P+SSRI non-responder vs. remitter contrasts was extracted in the healthy control group (N=24,12 male/12 female; age mean±SD= 34.13± 7.74). A 3 group one-way ANOVA was performed, with post-hoc comparisons contrasting each patient group with controls.
RESULTS
Clinical Outcome
Phase 1 clinical outcomes (26) and the corresponding Phase 1 treatment-specific biomarkers (22) were previously reported. To summarize, 82 patients were randomized to treatment, with 42 randomized to CBT, and 40 to sCIT. Sixty-five patients completed Phase 1, sixty-three with baseline FDG-PET scans appropriate for analysis. Phase 1 remission rates were similar for both treatments: CBT: 12/33 (36.3%), sCIT: 12/30 (40.0%) (Figure 1). Only 11 of 12 sCIT remitters had usable PET scans. Based on Phase 1 outcomes, 40 patients were offered enrollment in Phase 2. Thirty patients entered Phase 2 with 27 completing 24 weeks of treatment. Of these 27 patients, 12 remitted to combination treatment, 6 achieved clinical response but not remission, and 9 were P+SSRI non-responders. Therefore, the outcome groups analyzed included 36 remitters (35 with usable PET scans) and 9 P+SSRI non-responders.
Remitter vs. P+SSRI non-responder comparisons of clinical variables
There were no demographic or behavioral differences between the remitter and P+SSRI non-responder groups (Table 1).
Table 1.
Demographic and behavioral comparisons between P+SSRI non-responders and remitters
| Variable | P+SSRI non-responders (n = 9) | Remitters (n = 36*) | Test | p Value |
|---|---|---|---|---|
| Age, Years | 43.8(8.0) | 41.1(9.0) | t=−.830 | .411 |
| Duration of Current Episode, Weeks | 156.6(250.7) | 132.3(113.5) | t=.791 | .791 |
| Baseline HDRS | 19.0(3.5) | 18.5(3.1) | t=−.441 | .661 |
| Age of MDD Onset, Years | 28.2(8.5) | 27.8(12.1) | t=−.094 | .926 |
| Baseline HAMA | 14.7(6.3) | 14.0(2.9) | t=−.456 | .650 |
| Baseline BDI | 21.6(3.0) | 20.0(7.4) | t=−.611 | .544 |
| CTQ Total | 40.3(10.3) | 46.4(14.6) | t=.1.173 | .247 |
| Education, Years | 15.0(1.8) | 16.0(1.7) | t=1.357 | .183 |
| Gender, Female/Male | 4/5 | 17/19 | χ2=.200 | .655 |
| Treatment arm, CBT/sCIT | 4/5 | 19/17 | χ2=.200 | .655 |
| Current anxiety disorder No/Yes | 7/2 | 26/10 | χ2=.114 | .736 |
| Lifetime Substance Use Absent/Subthreshold/Threshold | 4/3/2 | 21/10/5 | χ2=.648 | .723 |
| Current MDD, None/Melancholic/Atypical | 3/4/2 | 12/16/7 | χ2=.022 | .989 |
| Previous Medication, No/Yes | 1/8 | 10/26 | χ2=1.083 | .298 |
| Previous Psychotheraphy, No/Yes | 5/4 | 17/19 | χ2=.200 | .655 |
| Lifetime PTSD, No/Yes | 9/0 | 33/3 | χ2=.804 | .370 |
| Married or Cohabitating, No/Yes | 5/3 | 14/21 | χ2=1.337 | .248 |
| Employed Full Time, No/Yes | 4/4 | 15/20 | χ2=.135 | .714 |
| First Degree Family History, No/Yes | 3/5 | 24/11 | χ2=2.691 | .101 |
| Race, White/Black/Hispanic | 8/0/0 | 26/5/4 | χ2=2.602 | .272 |
| Lifetime Episodes, 1/2/3+ | 3/3/3 | 10/11/14 | χ2=.145 | .930 |
Data are mean (SD) except as noted. Included only patients with available PET scans. HDRS, Hamilton Depression Rating Scale; HAMA, Hamilton Anxiety Scale; BDI, Beck Depression Inventory; CBT, cognitive behavior therapy; sCIT, escitalopram; PTSD, Post traumatic stress disorder
Demographic and behavior data were available for all 36 remitters, PET scans were only available for 35 remitters.
Subcallosal cingulate metabolism t-test results
Relative to the remitter group, significantly higher baseline left subcallosal cingulate (SCC) metabolism was identified in the P+SSRI non-response group (FWE corrected p ≤ 0.05) (Figure 3A, Table 2).
Figure 3.
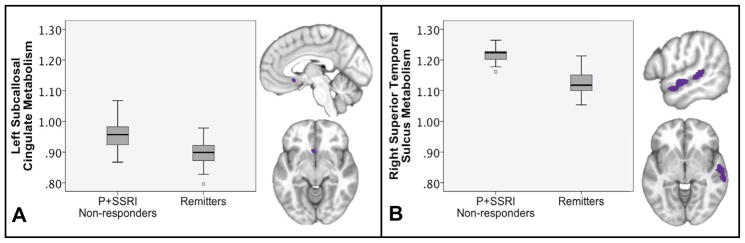
Subcallosal (SCC) region of interest and Whole brain t-test results of P+SSRI non-responders compared with remitters. Boxplots represent mean metabolism for each region of interest.
Table 2.
Subcallosal Cingulate and Whole Brain T-Test Results
| Anatomical Location | Cluster Size (voxels) | MNI coordinates | T | Effect Size (N=44) | ||
|---|---|---|---|---|---|---|
| x | y | z | ||||
| Subcallosal Cingulate T-Test | ||||||
| Subcallosal Cingulate (L) | 46 | −1.5 | −16.5 | −9.0 | 2.963 | 1.15 |
| Whole Brain T-Test | ||||||
| Superior Temporal Sulcus (R) | 1268 | 59.8 | 20.9 | −3.0 | 3.530 | 1.76 |
MNI coordinates are center of mass. Cluster size reported at p 0.005 for small volume correction, p 0.001 for whole brain, p values FWE corrected at p< 0.05. L=left, R= right
Whole brain T-test of FDG-PET results
Only one region, the right superior temporal sulcus (STS), met FWE corrected statistical significance (p < .05) in the whole brain t-test (Figure 3B, Table 2). Similar to the SCC, the right STS showed relative hypermetabolism in the P+SSRI non-response patients compared with the remitters.
Follow up T-tests between remitter groups
There were no differences in SCC or STS metabolism between Phase 1 and Phase 2 remitters. There were also no metabolic differences in these regions between CBT and sCIT monotherapy remitters; indicating no compound treatment or treatment specific effects on these regions.
Full sample effect sizes and correlations with outcome
Effect sizes are reported in Table 2. STS showed the largest effect size (1.7 SD); the SCC effect size also exceeded 1 SD. To verify that the regions defined by the P+SSRI non-responders were applicable to the complete sample and not just the extremes, correlations with percent change in HDRS were performed. We first tested the response in Phase 2 completers (n=27) to determine if the association of brain activity with response was consistent with the ANOVA results. The STS showed a strong correlation of metabolism with percent change in Phase 2 completers (r= 0.655, p<0.0005) while the SCC showed a less strong correlation (r= 0.364, p<0.06) (Figure 4).
Figure 4.

Subcallosal cingulate (SCC) and superior temporal sulcus (STS) metabolism correlated with outcome measured by percent change in Hamilton Depression Rating Scale (HDRS). Phase 1 Completers not entering/completing Phase 2 and Phase 2 Completers were correlated separately.
We also examined both regions for predictive potential in Phase 1 outcome; we limited these correlations to the 36 Phase 1 completers who did not go on to Phase 2. Although there was no significant correlation of percent change in HDRS with STS in this group (r= −0.261, p<0.124), the correlation with SCC was significant (r= 0.422, p<0.01).
Comparisons with healthy controls
The one-way ANOVA of P+SSRI non-responder, remitter, and healthy control groups was significant for the SCC (F 4.767, p< 0.012) and STS (F 16.392 p <0.001) regions. Post-hoc t-tests showed higher SCC metabolism in P+SSRI non-responders compared with healthy controls (p< 0.022) but no difference between remitters and healthy controls (p< 0.854). STS metabolism was higher in P+SSRI non-responders compared to controls (p< 0.001) with remitters showing the opposite pattern; low STS metabolism in remitters compared with healthy controls (p<0.036). Although there was a significant difference in age between the control and patient groups, age did not correlate with SCC or STS metabolism (STS R= −0.087 p<0.388: SCC R=0.079 p< 0.434; N=101 (sample includes 77 patients with usable baseline FDG-PET scans and 24 controls)).
DISCUSSION
This study identified two candidate pretreatment FDG-PET biomarkers that distinguish P+SSRI non-responders from patients who remit to treatment with CBT or sCIT, delivered as monotherapy or in combination. These results extend our previous CBT/escitalopram treatment-selection biomarker work to identify brain states of patients who are potentially inappropriate for either first-line intervention. As hypothesized, baseline subcallosal cingulate (SCC) metabolism was higher in patients who went on to fail both treatments compared to those that remitted to either. Further, SCC activity in P+SSRI non-responders was uniquely increased compared with healthy controls. In the whole brain analysis, relative hypermetabolism was seen in the right superior temporal sulcus of P+SSRI non-responders.
The a priori hypothesis targeting the SCC was based on previous research identifying a relationship between treatment non-response and greater subcallosal cingulate activity, particularly in samples that have already failed at least one treatment (23–25; 36). Baseline SCC hyperactivity has been reported in eventual non-responders to CBT or venlafaxine (16) in a randomized 16-week monotherapy treatment study. SCC hyperactivity is also present in patients with multiple depression treatment failures, including ECT, compared with controls (23). This suggests over activity in the SCC is a core characteristic of eventual treatment non-response in depression. Functional connectivity fMRI studies further support over activity of the SCC, showing increased connectivity of the subcallosal cingulate to the resting-state default mode network in recurrently depressed patients with past treatment failures (36). Lastly, the SCC is a surgical target for deep brain stimulation of treatment-resistant depression, with baseline SCC hyperactivity characterizing this extreme treatment resistant group (23). Chronic DBS in TRD is associated with a decrease in SCC activity with treatment, a similar change to that seen with successful response to SSRI, SNRI, rTMS, VNS, and ECT (23; 37–42). The resting-state SCC hyperactivity seen here is unique to P+SSRI non-responders, compared with both remitting patients and healthy controls. SCC hyperactivity in P+SSRI non-responders may represent a non-responsive or ‘stuck’ sad mood state that cannot be modulated by typical first-line interventions (23; 43).
In addition to the SCC finding, right superior temporal sulcus (STS) showed significant metabolic differences between P+SSRI non-responders and remitters. The STS showed both a larger effect size and stronger correlation with two-treatment outcome suggesting it is a better discriminator of P+SSRI non-response than the hypothesized SCC region. STS and SCC metabolism were not significantly correlated (r = .192 p> 0.095, N=77) suggesting the two measures, while possibly related, are not redundant (Figure S1 in Supplemental Information). Although a role for the STS in MDD is not strongly established, it has been identified in some fMRI connectivity and structural network studies of MDD, though without definitive interpretations (44; 45). More generally, the STS and other sites within the lateral temporal lobe are involved in evaluation of emotional valence (46), prospection (47), and default mode network activity (48). Alterations in the default mode network have been identified in MDD (49) as have changes in emotional self-evaluation (50) potentially linking these STS findings to MDD more broadly. We speculate that STS hyperactivity may be related to the over-engagement of the default mode at rest in treatment-resistant MDD, contributing specifically to alterations in how the resting brain contextualizes emotion.
The results presented here contribute to the growing literature on predictors of response to treatments in patients with MDD. Fully integrating the current results with those of previous studies is challenging due to differences in patient samples and research methodology. One notable absence from the whole brain analysis was the rostral anterior cingulate. Rostral anterior cingulate activity has been repeatedly demonstrated to predict depression treatment outcome (51) primarily in studies of a single medication. We failed to identify a rostral anterior cingulate discriminator in the initial whole brain analysis to define CBT-sCIT outcome differences. A post hoc analysis within each treatment did, however, identify a non-significant correlation between rostral anterior cingulate metabolism and treatment outcome in escitalopram treated patients, but not CBT treated patients (22). This escitalopram specific trend is consistent with published studies demonstrating this same association of activity in Brodmann area 24a (rostral anterior cingulate) with hypermetabolism associated with response and hypometabolism associated with non-response to a single pharmacological treatment (17; 39). None of these published studies used remission as an outcome variable or examined patterns that specifically discriminated across different classes of treatments. However, to further explore this potential relationship, a post-hoc comparison of P+SSRI non-responders and remitters was performed using a rostral anterior cingulate region that included the entire perigenual anterior cingulate cortex (Figure S2 in Supplemental Information). There were no differences in rostral anterior cingulate metabolism between patients remitting to either treatment compared with those failing both treatments.
Some limitations may affect the interpretation of these findings. The P+SSRI non-response group was not intentionally powered and is small. Patients achieving response but not remission at some point during the study (N=6) were excluded from t-tests identifying potential biomarker candidates further decreasing our non-responder sample size (N=9). To ensure detection of a difference that would represent the biology of unambiguous two-treatment nonresponders, we intentionally avoided including patients responding but not remitting. Additionally, we did not group responders with remitters because of the known relationship between residual symptoms and greater likelihood of clinical relapse (52). Although this decision provides for the most unambiguous biological signal detection, a next-step treatment choice would still need to be made for these patients. While SCC and STS metabolism show consistency across all levels of two-treatment response (Figure 2), our data do not address a specific strategy for choosing a next-step treatment.
A second limitation concerns the age difference between patients and healthy controls. While the age difference may be a factor in the metabolic differences between patient groups and controls, the lack of correlation between age and either SCC or STS metabolism, indicates that age is unlikely to be driving the identified differences. Age was consistent between P+SSRI non-responder and remitter groups, with SCC results showing a difference between P+SSRI non-responders and healthy controls and no difference between remitters and healthy controls. More importantly, analysis of the control subject data provides some context for understanding the nature of the regional hypermetabolism identified here, but the comparison does not directly influence the interpretation of the biomarker itself for discriminating the two patient groups.
Although the SCC and STS show promise as biomarkers of P+SSRI non-response, replication in additional studies will be necessary before these patterns could be considered reliable for clinical use. Notably, the Phase 2 P+SSRI non-responders were a subset of those patients in the Phase 1 analysis (22) with hyperactivity of the right anterior insula which predicted response to sCIT and failure to CBT (7 of 9, Figure S3 in Supplemental Information). Taken together, a patient presenting with hypermetabolism of the anterior insula in combination with increased activity in the SCC and STS may benefit most from starting treatment with a non-SSRI antidepressant medication or an alternative therapy. This speculation will require explicit testing of alternative antidepressant medications and non-pharmacological treatments now reserved for more treatment-resistant patients. Such studies are a critical next step towards the systematic development of clinical algorithms for antidepressant treatment selection in individual patients.
Supplementary Material
Acknowledgments
This study was supported by NIH grants R01 MH073719 (HSM), T32 GM08695 (CLM), K23 MH086690 (BWD), K23 MH077869 (PEH). We thank the treating psychotherapists: Sheethal Reddy, PhD, Patrick Sylvers, PhD, Lorie Ritschel, PhD, Meredith Jones, PhD, Mary Heekin, LCSW, Maryrose Gerardi, PhD, and Jill Rosenberg, LCSW; treating physicians: Ebrahim Haroon, MD, Jeffrey Rakofsky, MD, Dylan Wint, MD, Corey Beck, MD; clinical coordinators: Ronald Chismar, Melanie Galanti, Rachelle Gibson, Lauren Marx, Melissa McKenzie, and Tanja Mletzko; Blinded Raters: Margo Aaron, Yara Betancourt, Cristina Velasquez Delgado, Novall Khan, Ximena Marincic, and Christopher Vaughan; and the imaging team: Rebecca DeMayo, Eundria Hill, Kiseung Choi, Justin Rajendra for their contributions.
Footnotes
FINANCIAL DISCLOSURES:
Dr. Dunlop has received honoraria for consulting work performed for Bristol-Myers Squibb, MedAvante, Pfizer and Roche. He has received research support from AstraZeneca, Bristol-Myers Squibb, Evotec, Forest, GlaxoSmithKline, Novartis, Ono Pharmaceuticals, Pfizer, Shire, Takeda, and Transcept. Dr. Holtzheimer has received consulting fees from St. Jude Medical Neuromodulation and Cervel Neurotech, research funding from Cervel Neurotech and Otsuka, and an honorarium from Johnson and Johnson. Dr. Craighead is a Board Member of Hugareill enf, an Icelandic company dedicated to the prevention of depression, and he receives book royalties from John Wiley & Sons. He is a Consultant to the George West Mental Health Foundation that oversees Skyland Trail, a residential treatment facility in Atlanta, GA. Dr. Mayberg has received consulting and IP licensing fees from St. Jude Medical, Inc. Dr. Kelley and Ms. McGrath report no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders - Text Revision. 4. Washington, DC: American Psychiatric Press; 2000. [Google Scholar]
- 2.Kennedy SH, Lam RW, Parikh SV, Patten SB, Ravindran AV. Canadian Network for Mood and Anxiety Treatments (CANMAT) Clinical guidelines for the management of major depressive disorder in adults. Introduction. J Affect Disord. 2009;117(Suppl):S1–2. doi: 10.1016/j.jad.2009.06.043. [DOI] [PubMed] [Google Scholar]
- 3.American Psychiatric Association. Treating major depressive disorder: a quick reference guide. Washington, DC: American Psychiatric Press; 2010. [Google Scholar]
- 4.National Collaborating Center for Mental Health. Depression: The treatment and management of depression in adults. Leicester (UK): British Psychological Society; 2009. [Google Scholar]
- 5.Gaynes BN, Dusetzina SB, Ellis AR, Hansen RA, Farley JF, Miller WC, Stürmer T. Treating depression after initial treatment failure: directly comparing switch and augmenting strategies in STAR*D. J Clin Psychopharmacol. 2012;32:114–119. doi: 10.1097/JCP.0b013e31823f705d. [DOI] [PubMed] [Google Scholar]
- 6.Perlis RH, Patrick A, Smoller JW, Wang PS. When is pharmacogenetic testing for antidepressant response ready for the clinic? A cost-effectiveness analysis based on data from the STAR*D study. Neuropsychopharmacology. 2009;34:2227–2236. doi: 10.1038/npp.2009.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thase ME, Friedman ES, Biggs MM, Wisniewski SR, Trivedi MH, Luther JF, et al. Cognitive therapy versus medication in augmentation and switch strategies as second-step treatments: a STAR*D report. Am J Psychiatry. 2007;164:739–752. doi: 10.1176/ajp.2007.164.5.739. [DOI] [PubMed] [Google Scholar]
- 8.Kocsis JH, Gelenberg AJ, Rothbaum BO, Klein DN, Trivedi MH, Manber R, et al. Cognitive behavioral analysis system of psychotherapy and brief supportive psychotherapy for augmentation of antidepressant nonresponse in chronic depression: the REVAMP Trial. Arch Gen Psychiatry. 2009;66:1178–1188. doi: 10.1001/archgenpsychiatry.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Craighead WE, Dunlop BW. Combination psychotherapy and antidepressant medication treatment for depression: For whom, when and how. Annu Rev Psychol. doi: 10.1146/annurev.psych.121208.131653. (In press) [DOI] [PubMed] [Google Scholar]
- 10.National Committee for Quality Assurance. The State of Health Care Quality 2007. Washington, DC: National Committee for Quality Assurance; 2007. pp. 20–21. [Google Scholar]
- 11.DeRubeis RJ, Hollon SD, Amsterdam JD, Shelton RC, Young PR, Salomon RM, et al. Cognitive therapy vs medications in the treatment of moderate to severe depression. Arch Gen Psychiatry. 2005;62:409–416. doi: 10.1001/archpsyc.62.4.409. [DOI] [PubMed] [Google Scholar]
- 12.Hollon SD, Beck AT. Cognitive and cognitive-behavioral therapies. In: Lambert MJ, editor. Bergin and Garfield’s handbook of psychotherapy behavior change. 6. Hoboken, NJ: John Wiley & Sons Inc; 2013. pp. 393–442. [Google Scholar]
- 13.Perlis RH. A Clinical Risk Stratification Tool for Predicting Treatment Resistance in Major Depressive Disorder. Biol Psychiatry. 2013;74:7–14. doi: 10.1016/j.biopsych.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siegle GJ, Carter CS, Thase ME. Use of FMRI to predict recovery from unipolar depression with cognitive behavior therapy. Am J Psychiatry. 2006;163:735–738. doi: 10.1176/ajp.2006.163.4.735. [DOI] [PubMed] [Google Scholar]
- 15.Siegle GJ, Thompson WK, Collier A, Berman SR, Feldmiller J, Thase ME, Friedman ES. Toward clinically useful neuroimaging in depression treatment: prognostic utility of subgenual cingulate activity for determining depression outcome in cognitive therapy across studies, scanners, and patient characteristics. Arch Gen Psychiatry. 2012;69:913–924. doi: 10.1001/archgenpsychiatry.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Konarski JZ, Kennedy SH, Segal ZV, Lau MA, Bieling PJ, McIntyre RS, Mayberg HS. Predictors of nonresponse to cognitive behavioural therapy or venlafaxine using glucose metabolism in major depressive disorder. J Psychiatry Neurosci. 2009;34:175–180. [PMC free article] [PubMed] [Google Scholar]
- 17.Mayberg HS, Brannan SK, Mahurin RK, Jerabek PA, Brickman JS, Tekell JL, et al. Cingulate function in depression: a potential predictor of treatment response. Neuroreport. 1997;8:1057–1061. doi: 10.1097/00001756-199703030-00048. [DOI] [PubMed] [Google Scholar]
- 18.Uher R, Huezo-Diaz P, Perroud N, Smith R, Rietschel M, Mors O, et al. Genetic predictors of response to antidepressants in the GENDEP project. Pharmacogenomics J. 2009;9:225–233. doi: 10.1038/tpj.2009.12. [DOI] [PubMed] [Google Scholar]
- 19.Ising M, Lucae S, Binder EB, Bettecken T, Uhr M, Ripke S, et al. A genomewide association study points to multiple loci that predict antidepressant drug treatment outcome in depression. Arch Gen Psychiatry. 2009;66:966–975. doi: 10.1001/archgenpsychiatry.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leuchter AF, Cook IA, Marangell LB, Gilmer WS, Burgoyne KS, Howland RH, et al. Comparative effectiveness of biomarkers and clinical indicators for predicting outcomes of SSRI treatment in Major Depressive Disorder: results of the BRITE-MD study. Psychiatry Res. 2009;169:124–131. doi: 10.1016/j.psychres.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 21.Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry. 2013;70:31–41. doi: 10.1001/2013.jamapsychiatry.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGrath CL, Kelley ME, Holztheimer PE, III, Dunlop BW, Craighead WE, Franco AF, et al. Toward a neuroimaging treatment selection biomarker for major depressive disorder. JAMA Psychiatry. 2013;70:821–829. doi: 10.1001/jamapsychiatry.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayberg HS, Lozano AM, Voon V, McNeely HE, Seminowicz D, Hamani C, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45:651–660. doi: 10.1016/j.neuron.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 24.Wu J, Buchsbaum MS, Gillin JC, Tang C, Cadwell S, Wiegand M, et al. Prediction of antidepressant effects of sleep deprivation by metabolic rates in the ventral anterior cingulate and medial prefrontal cortex. Am J Psychiatry. 1999;156:1149–1158. doi: 10.1176/ajp.156.8.1149. [DOI] [PubMed] [Google Scholar]
- 25.Dougherty DD, Weiss AP, Cosgrove GR, Alpert NM, Cassem EH, Nierenberg AA, et al. Cerebral metabolic correlates as potential predictors of response to anterior cingulotomy for treatment of major depression. J Neurosurg. 2003;99:1010–1017. doi: 10.3171/jns.2003.99.6.1010. [DOI] [PubMed] [Google Scholar]
- 26.Dunlop BW, Kelley ME, Mletzko TC, Velasquez CM, Craighead WE, Mayberg HS. Depression beliefs, treatment preference, and outcomes in a randomized trial for major depressive disorder. J Psychiatr Res. 2012;46:375–381. doi: 10.1016/j.jpsychires.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.First MB, Spitzer RL, Gibbon M, Williams JB. Structured Clinical Interview for DSM-IV Axis I Disorders, Clinician Version (SCID-CV) Washington, DC: American Psychiatric Press, Inc; 1996. [Google Scholar]
- 28.Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry. 1961;4:561–571. doi: 10.1001/archpsyc.1961.01710120031004. [DOI] [PubMed] [Google Scholar]
- 30.Hamilton M. The assessment of anxiety states by rating. Br J Med Psychol. 1959;32:50–5. doi: 10.1111/j.2044-8341.1959.tb00467.x. [DOI] [PubMed] [Google Scholar]
- 31.Bernstein DP, Fink L, Handelsman L, Foote J, Lovejoy M, Wenzel K, et al. Initial reliability and validity of a new retrospective measure of child abuse and neglect. Am J Psychiatry. 1994;151:1132–1136. doi: 10.1176/ajp.151.8.1132. [DOI] [PubMed] [Google Scholar]
- 32.de Jong HW, van Velden FH, Kloet RW, Buijs FL, Boellaard R, Lammertsma AA. Performance evaluation of the ECAT HRRT: an LSO-LYSO double layer high resolution, high sensitivity scanner. Phys Med Biol. 2007;52:1505–1526. doi: 10.1088/0031-9155/52/5/019. [DOI] [PubMed] [Google Scholar]
- 33.Ashburner J. A fast diffeomorphic image registration algorithm. Neuroimage. 2007;38:95–113. doi: 10.1016/j.neuroimage.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 34.Kennedy SH, Konarski JZ, Segal ZV, Lau MA, Bieling PJ, McIntyre RS, Mayberg HS. Differences in brain glucose metabolism between responders to CBT and venlafaxine in a 16-week randomized controlled trial. Am J Psychiatry. 2007;164:778–788. doi: 10.1176/ajp.2007.164.5.778. [DOI] [PubMed] [Google Scholar]
- 35.Desikan RS, Ségonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968–980. doi: 10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 36.Greicius MD, Flores BH, Menon V, Glover GH, Solvanson HB, Kenna H, et al. Resting-state functional connectivity in major depression: abnormally increased contributions from subgenual cingulate cortex and thalamus. Biol Psychiatry. 2007;62:429–437. doi: 10.1016/j.biopsych.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pardo JV, Sheikh SA, Schwindt GC, Lee JT, Kuskowski MA, Surerus C, et al. Chronic vagus nerve stimulation for treatment-resistant depression decreases resting ventromedial prefrontal glucose metabolism. Neuroimage. 2008;42:879–889. doi: 10.1016/j.neuroimage.2008.04.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayberg HS, Brannan SK, Tekell JL, Silva JA, Mahurin RK, McGinnis S, Jerabek PA. Regional metabolic effects of fluoxetine in major depression: serial changes and relationship to clinical response. Biol Psychiatry. 2000;48:830–843. doi: 10.1016/s0006-3223(00)01036-2. [DOI] [PubMed] [Google Scholar]
- 39.Kennedy SH, Evans KR, Krüger S, Mayberg HS, Meyer JH, McCann S, et al. Changes in regional brain glucose metabolism measured with positron emission tomography after paroxetine treatment of major depression. Am J Psychiatry. 2001;158:899–905. doi: 10.1176/appi.ajp.158.6.899. [DOI] [PubMed] [Google Scholar]
- 40.Goldapple K, Segal Z, Garson C, Lau M, Bieling P, Kennedy S, Mayberg H. Modulation of cortical-limbic pathways in major depression: treatment-specific effects of cognitive behavior therapy. Arch Gen Psychiatry. 2004;61:34–41. doi: 10.1001/archpsyc.61.1.34. [DOI] [PubMed] [Google Scholar]
- 41.Kennedy SH, Giacobbe P, Rizvi SJ, Placenza FM, Nishikawa Y, Mayberg HS, Lozano AM. Deep brain stimulation for treatment-resistant depression: follow-up after 3 to 6 years. Am J Psychiatry. 2011;168:502–510. doi: 10.1176/appi.ajp.2010.10081187. [DOI] [PubMed] [Google Scholar]
- 42.Nobler MS, Oquendo MA, Kegeles LS, Malone KM, Campbell CC, Sackeim HA, Mann JJ. Decreased regional brain metabolism after ect. Am J Psychiatry. 2001;158:305–8. doi: 10.1176/appi.ajp.158.2.305. [DOI] [PubMed] [Google Scholar]
- 43.Holtzheimer PE, Mayberg HS. Stuck in a rut: rethinking depression and its treatment. Trends Neurosci. 2011;34:1–9. doi: 10.1016/j.tins.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sheline YI, Barch DM, Price JL, Rundle MM, Vaishnavi SN, Snyder AZ, et al. The default mode network and self-referential processes in depression. Proc Natl Acad Sci U S A. 2009;106:1942–1947. doi: 10.1073/pnas.0812686106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu L, Zeng LL, Li Y, Ma Q, Li B, Shen H, Hu D. Altered cerebellar functional connectivity with intrinsic connectivity networks in adults with major depressive disorder. PLoS One. 2012;7:e39516. doi: 10.1371/journal.pone.0039516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peelen MV, Atkinson AP, Vuilleumier P. Supramodal representations of perceived emotions in the human brain. J Neurosci. 2010;30:10127–10134. doi: 10.1523/JNEUROSCI.2161-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schacter DL, Addis DR, Buckner RL. Remembering the past to imagine the future: the prospective brain. Nat Rev Neurosci. 2007;8:657–661. doi: 10.1038/nrn2213. [DOI] [PubMed] [Google Scholar]
- 48.Buckner RL, Andrews-Hanna JR, Schacter DL. The brain’s default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci. 2008;1124:1–38. doi: 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- 49.Hamilton JP, Chen MC, Gotlib IH. Neural systems approaches to understanding major depressive disorder: an intrinsic functional organization perspective. Neurobiol Dis. 2013;52:4–11. doi: 10.1016/j.nbd.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fossati P, Hevenor SJ, Graham SJ, Grady C, Keightley ML, Craik F, Mayberg H. In search of the emotional self: an fMRI study using positive and negative emotional words. Am J Psychiatry. 2003;160:1938–1945. doi: 10.1176/appi.ajp.160.11.1938. [DOI] [PubMed] [Google Scholar]
- 51.Pizzagalli DA. Frontocingulate dysfunction in depression: toward biomarkers of treatment response. Neuropsychopharmacology. 2011;36:183–206. doi: 10.1038/npp.2010.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paykel ES, Ramana R, Cooper Z, Hayhurst H, Kerr J, Barocka A. Residual symptoms after partial remission: an important outcome in depression. Psychol Med. 1995;25:1171–1180. doi: 10.1017/s0033291700033146. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.