

Abstract
Gastrointestinal (GI) anthrax results from the ingestion of Bacillus anthracis. Herein, we investigated the pathogenesis of GI anthrax in animals orally infected with toxigenic non-encapsulated B. anthracis Sterne strain (pXO1+ pXO2−) spores that resulted in rapid animal death. B. anthracis Sterne induced significant breakdown of intestinal barrier function and led to gut dysbiosis, resulting in systemic dissemination of not only B. anthracis, but also of commensals. Disease progression significantly correlated with the deterioration of innate and T cell functions. Our studies provide critical immunologic and physiologic insights into the pathogenesis of GI anthrax infection, whereupon cleavage of mitogen-activated protein kinases (MAPKs) in immune cells may play a central role in promoting dysfunctional immune responses against this deadly pathogen.
Introduction
Gastrointestinal (GI) anthrax, named for its primary route of infection, is an acute infectious disease resulting from the ingestion of the spore-forming, Gram-positive bacterium, Bacillus anthracis [1]. Anthrax can also be contracted via inhalation or cutaneous exposure, with inhalation anthrax having the highest mortality rate of the three clinical subtypes [2]. Disease-causing B. anthracis spores primarily infect grazing animals, but humans may be exposed to anthrax through the handling of infected animals and animal products, the consumption of tainted meat, or through intentional exposure [1]. Independent of the route of entry, unchecked infection rapidly becomes systemic and death occurs due to septicemia and/or toxemia [3].
Within fully virulent B. anthracis strains, two large plasmids, pXO1 and pXO2, are composed of the genes needed for toxin production and capsule formation, respectively, and both plasmids are necessary for complete virulence [4], [5]. The pXO1 encodes protective antigen (PA), lethal factor (LF), and edema factor (EF); lethal toxin (LT) comprises PA+LF, while edema toxin (ET) comprises PA+EF. Via these two toxins, B. anthracis evades and inhibits critical signals of the innate and adaptive immune systems [6]. The poorly immunogenic anthrax capsule is encoded on pXO2 and consists of poly-γ-D-glutamic acid, which protects B. anthracis from phagocytosis and complement binding [7], [8]. Several therapeutic strategies have targeted specific B. anthracis virulence factors [9], [10]; however, development of next generation vaccines and therapeutics against B. anthracis requires a better understanding of disease pathogenesis in humans. In particular, insufficient data exist regarding the pathogenesis of GI anthrax [11]–[13]. GI B. anthracis infection is not only a persistent and major problem in developing countries, but also poses a threat in biological warfare, whereby intentional contamination of food sources may occur [1].
Here, we report that GI B. anthracis spore infection results in swift morbidity and mortality and is associated with pathogen dissemination throughout visceral organs by induction of leakage in the intestinal barrier and significant changes in the gut’s microbial composition, all of which may orchestrate dysfunctional immune responses. A greater understanding of the pathogenesis of GI anthrax and molecular studies of the “microorganism-mammalian immune defense interface” [14] is imperative and may result in improvement of a protective vaccine in man.
Materials and Methods
Mice and Ethics Statement
A/J mice were purchased from the Jackson Laboratory and bred in-house in the animal facility at the College of Veterinary Medicine, University of Florida. For microbiota composition experiments, mice were tested after a minimum of two generations of in-house breeding. Mice were used at 6–8 weeks of age in accordance with the Animal Welfare Act and the Public Health Policy on Humane Care. All procedures were approved by the Institutional Animal Case and Use Committee (IACUC) at the University of Florida under protocol number 201107129, and all efforts were made to minimize animal suffering. Infected mice were monitored every 24 hours and were humanely euthanized when signs of advanced infection (e.g., difficulty breathing) were noted; in some cases, mice died as a direct result of the infection before euthanasia could take place. Euthanasia was performed by prolonged inhalation of isoflurane and confirmed by cervical dislocation.
B. anthracis Spore Preparation and Mouse Infections
Spores were prepared with a toxigenic non-encapsulated strain of B. anthracis (Sterne), as described previously [15] with the approval of the Institutional Biosafety Committee (IBC) at the University of Florida. To calculate final concentrations, serial dilutions were grown in triplicate on lysogeny broth agar plates and colonies counted. For survival studies, mice were orally infected with 105 spores (n = 10), 107 spores (n = 10), or 109 spores (n = 20) in a final volume of 100 µL with a reusable, 30 mm, 20 gauge, barrel-tipped feeding needle after fasting for 4 hours; infected mice were monitored, and deaths recorded. For immunologic and microbiota composition studies, A/J mice (n = 10/group) were orally infected with Sterne spores (109 spores/100 µL PBS/mouse) for the specified time points. Groups of A/J mice (n = 10/group) were also either orally gavaged or injected intraperitoneally (i.p.) with 125 µg LT (PA+LF) and monitored for morbidity and death.
Histopathology
Sterne-infected A/J mice were sacrificed at various days post-infection and the colon, spleen, liver, kidney, and lungs surgically excised for analyses. Tissues were fixed, sectioned, and stained with hematoxylin and eosin (H&E) by Histology Tech Services (Gainesville, FL). Histopathology and bacterial dissemination in infected mice were analyzed by a boarded veterinary pathologist (JLO). In some cases, cytocentrifugation of single cell suspensions from spleen, mesenteric lymph nodes (MLNs), and bronchoalveolar lavage (BAL) fluid were evaluated. Neither vegetative bacilli nor spores could be detected in the lungs prior to three days post-infection, confirming that spores were not accidently introduced into the respiratory tract during oral gavage.
Ex vivo Trans Epithelial Electrical Resistance (TEER), Short-circuit Current (ISC) and Trans-epithelial Conductance (GT) Measurements of Intestinal Tissues
Differences in TEER and electrogenic ion transport in the colons of Sterne-infected versus uninfected mice were quantified by measuring the short circuit current responses of isolated colonic tissues mounted in modified Ussing chambers, as previously described [16]. Briefly, Sterne-infected (day 3) and uninfected A/J mice were euthanized and colons quickly isolated. Segments were cut open along the mesenteric border into a flat sheet and flushed with ice-cold HEPES-Ringer solution; intact, full-thickness segments containing all of the layers of colons were used. The intestinal sheets were mounted between two halves of a modified Ussing chamber (Physiologic Instruments, San Diego, CA) and short-circuited by a voltage clamp (VCC MC6, Physiologic Instruments) with correction for solution resistance. The exposure area was 0.3 cm2. The mucosal and serosal surfaces of the tissues were bathed in reservoirs with 3 mL HEPES-Ringer solution containing 100 mM NaCl, 25 mM HCO3, 1.5 mM CaCl2, 1.5 mM MgCl2, 5 mM KCl, 10 mM glucose, and 22 mM HEPES, pH 7.4, maintained at 37°C and continuously bubbled with 95% O2 and 5% CO2. Tissues and solutions were maintained at 37°C by surrounding water jackets. Tissues were allowed a 30 min stabilization period before measurements were recorded at basal and challenged conditions. The delta current (ΔISC) before and after challenge was used to estimate electrogenic current stimulation by external stimuli; the post-challenge secretory current was measured 10 minutes after challenge as basolateral bumetanide-sensitive ISC. Throughout the experiments, tissues were constantly short-circuited by clamping the transepithelial potential at 0 mV, except for 1 second intervals every 20 seconds when tissues were clamped at±3 mV and GT determined. Data were acquired via DATAQ instruments (Akron, OH) and processed using the Acquire & Analyze software (Warner Instruments, Hamden, CT).
Real-time PCR
RNA was isolated from distal colons of mice with Aurum Total RNA Kit (Bio-Rad). iScript Select cDNA Synthesis Kit (Bio-Rad) was used for reverse transcription and cDNA used for quantitative PCR by SYBR Green Dye gene expression assay on a Bio-Rad CFX96 Real time system. mRNA levels are shown as fold-increase over uninfected mice; n = 10/group. For fecal bacteria detection, PowerSoil DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA) was used to extract total DNA according to the manufacturer’s instructions. Real-time PCR analyses were performed on 2 ng of total DNA template (SsoAdvanced SYBR Green Supermix, Bio-Rad) to target bacteria group-specific 16S rDNA sequences, as previously described [17]. Groups tested include Enterobacteriaceae family (Proteobacteria phyla) and Bifidobacterium genus (Actinobacteria phyla). Specific groups were normalized to the housekeeper Eubacteria group; n = 10/group. For bacterial dissemination, total DNA from murine organs was extracted by using gDNA MiniPrep kit (Zymoresearch, Irvine, CA). Ten ng of total DNA was used to assess the dissemination of Bifidobacterium and Enterobacteriaceae; n = 6/group. A list of primers used and their sequences can be found in Table S1. For statistical analyses of gene array data, unpaired t-tests were performed using the relative expression levels of each gene at the specified time point compared to the relative expression level of the same gene in uninfected mice.
16S Ribosomal DNA Sequencing
For microbiome analyses, fecal DNA samples were amplified by Illumina Miseq compatible primers, targeting the 16s rDNA V4–V5 region. Amplicons were purified by QIAquick Gel extraction kit (Qiagene, Madison, WI) and quantified by Qubit 2.0 Fluorometer (Invitrogen, Grand Island, NY) and Kapa SYBR fast qPCR kit (Kapa Biosystems, Inc., Woburn, MA). Equal amounts of amplicons were pooled with 10% of Phix control. Miseq v2 reagent kit (Illumina, Inc., San Diego, CA) was used to run the pooled samples on the Illumina Miseq machine. The Q score of this run was 86.59% and cluster density was 975±61. Data were analyzed, as previously described [18]. Primers used are found in Table S2.
Colonoscopy of Uninfected and Sterne-infected A/J Mice
Colons of uninfected or day 3 Sterne-infected A/J mice were imaged with a Multi-Purpose Rigid Telescope attached to a TELE PACK X (Karl Storz–Endoscope, Germany). A/J mice were fasted for 6h prior to visualization of the colons of live animals under appropriate anesthetic conditions.
Propria (LP) Leukocyte Preparation
Colons surgically excised from Sterne-infected and uninfected control A/J mice were flushed with ice-cold PBS to remove fecal contents, cut open longitudinally, washed, and cut into 1 cm pieces. Tissue pieces were incubated with agitation at 37°C for 25 min in 30 mL of RPMI 1640 Glutamine (GIBCO, Life Technologies, Grand Island, New York) containing 5 mM EDTA (Ambion, Life Technologies), 1 mM DTT (Sigma-Aldrich, St. Louis, MO), 10 mM HEPES (GIBCO, Life Technologies), and supplemented with 5% heat inactivated FBS (HyClone, ThermoFisher Scientific, Waltham, MA) to remove intraepithelial lymphocytes and epithelial cells. After incubation, colonic tissues were washed with ice-cold PBS, minced, and incubated at 37°C with pre-warmed DMEM (GIBCO, Life Technologies) containing 0.25 mg/mL collagenase type VII (Sigma-Aldrich), 0.125 U/mL Liberase TM Research Grade (Roche Applied Science, Indianapolis, IN), 10 mM HEPES, 0.1 M CaCl2 (Sigma-Aldrich), and 5% FBS (3×10 min digestions). After each digestion, cell suspensions were passed through a strainer, spun down, and resuspended in DMEM supplemented with 5% FBS. Cells obtained from the three digestions were combined and immediately counted for staining and flow cytometry-based analyses.
Flow Cytometry and Antibodies
To exclude dead cells, single cell suspensions obtained from processed spleens and LPLs were stained with LIVE/DEAD Aqua Dead Cell Stain Kit (Molecular Probes, Life Technologies). Cells were subsequently washed and incubated with Mouse Fc Blocking Reagent (Miltenyi Biotec, Auburn, CA) prior to staining with combinations of the following antibodies or their corresponding isotype controls purchased from eBioscience (San Diego, CA), Biolegend (San Diego, CA), BD Pharmingen, R&D Systems (Minneapolis, MN), or Cell Signaling Technology, Inc. (Danvers, MA): CD45 (30-F11), CD11c (N418), CD11b (M1/70), CD11b (M1/70), F4/80 (BM8), GR1 (RB6-8C5), I-A/I-E MHCII (2G9), CD3 (145-2C11), CD4 (RM4–5), CD8 (53–607), PD-1/Rat IgG2a, κ, Pro-IL-1β (NJTEN3)/Rat IgG1, κ, TNFα (MP6-XT22)/Rat IgG1, κ, IL-6 (MP5-20F3)/Rat IgG1, κ, IFNγ (XMG1.2)/Rat IgG1, κ, FoxP3 (FJK-16A)/Rat IgG2a, κ, phospho-p38 MAPK (28B10)/Mouse IgG1, phospho-p44/42 MAPK (Erk1/2) (D13.14.4E)/Rabbit. Prior to intracellular staining, cells were fixed and permeabilized with BD Cytofix/Cytoperm (BD Biosciences). Colonic T cells were stimulated with phorbol 12-myristate 13-acetate [(PMA) 50 ng/mL] and ionomycin (1 µg/mL) for 2.5 hrs for the detection of intracellular cytokines. After staining, a BD LSRFortessa (BD Biosciences) cell analyzer was used to acquire fixed cells. Data were analyzed with FlowJo software (Tree Star, Ashland, OR).
Ex vivo Evaluation of MAPKs
Colonic cells were isolated as described above. Equilibrated LP cells were incubated with 1 multiplicity of infection (MOI) of spores or left untreated for 1, 3 and 6 h. Cells were stained and analyzed, as described above.
Sera Analyses
Cytokines in the sera of Sterne-infected and uninfected A/J mice were measured using the Bio-Plex Pro Mouse Cytokine 23-plex immunoassay kit (Bio-Rad, Hercules, CA). Magnetic beads were acquired with a MAGPIX system (Luminex, Austin, TX) and data analyzed with Bio-Plex Data Pro Software (Bio-Rad).
Statistical Analyses
Unless stated otherwise, representative data indicate mean ± SEM. Significance was determined by two-tailed unpaired t-tests for two group comparisons (GraphPad Prism 5 for Mac OS X, La Jolla, CA).
Results
Lethality and Systemic Dissemination of GI B. anthracis Infection
To explore GI anthrax pathogenesis while circumventing the difficulties of working with biosafety level 3 (BSL3) B. anthracis strains, we employed an extensively used mouse model, A/J mice, which is highly susceptible to B. anthracis Sterne [19]. Given the route of infection utilized in our studies, and the potential excretion of gavaged Sterne spores, doses that have been previously shown to be lethal in experimental models of inhalational anthrax [20] resulted in little to no death of A/J mice when administered orally. The lethal dose 50 (LD50) for this bacterium via the respiratory route of infection has been reported to be as low as 1×103 CFU of spores/mouse [21], and the LD50 of the vegetative bacteria was previously found to be 2.3×107 CFU [13]. However, to better mimic the likely form of B. anthracis when ingested, we fed A/J mice varying doses of Sterne spores. We found that 105 spores given orally did not kill any mice and inoculation with 107 spores resulted in 80% survival, likely because of the myriad of enzymes and the extreme pH levels of the gut milieu. The oral LD50 was approximately 5×108 spores/mouse (Fig. 1A), and 80% of infected mice died when orally gavaged with 109 Sterne spores (Fig. 1A). These mice were lethargic and showed signs of dyspnea as early as 2 days post-infection. After 3 days of infection, long chains of vegetative bacilli with the characteristic “bamboo-like appearance” were observed (marked with asterisk for ease of visualization) not only in the colon (Fig. 1B), but also systemically (Fig. 1C–I). Vegetative bacilli were seen in numerous organs, including the mesenteric lymph nodes (MLNs) (Fig. 1C), spleen (Fig. 1D, E), liver (Fig. 1F), kidneys (Fig. 1G), lungs (Fig. 1H), and in BAL fluid collected from the lungs (Fig. 1I). In some mice, vegetative bacilli were found in the liver after only 1 day of infection (Fig. S1A), suggesting a hematogenous component in the dissemination process, considering the portal vein carries blood from the GI tract to the liver for nutrition and detoxification. Anthrax-induced death in A/J mice is thought to be due to bacteremia and toxemia resulting from the high number of B. anthracis microbes in the periphery [19]. Indeed, we noted focal and diffuse areas of lympholysis in the spleens of infected mice (Fig. 1D, E), consistent with the known necrotizing effects of the toxins. Establishment of active infection is required for morbidity and mortality in our model, as A/J mice that were given LT (125 µg) orally did not show any signs of illness, likely due to proteolytic enzymes within the GI tract (Fig. S1B). In contrast, the same LT dose, when given i.p., resulted in the death of over 50% of the mice (Fig. S1B). Nonetheless, both routes of injection resulted in decreased expression of interleukin (IL)-1β in the colonic tissues (Fig. S1C).
Figure 1. Lethality and Systemic B. anthracis Sterne Dissemination in A/J Mice.
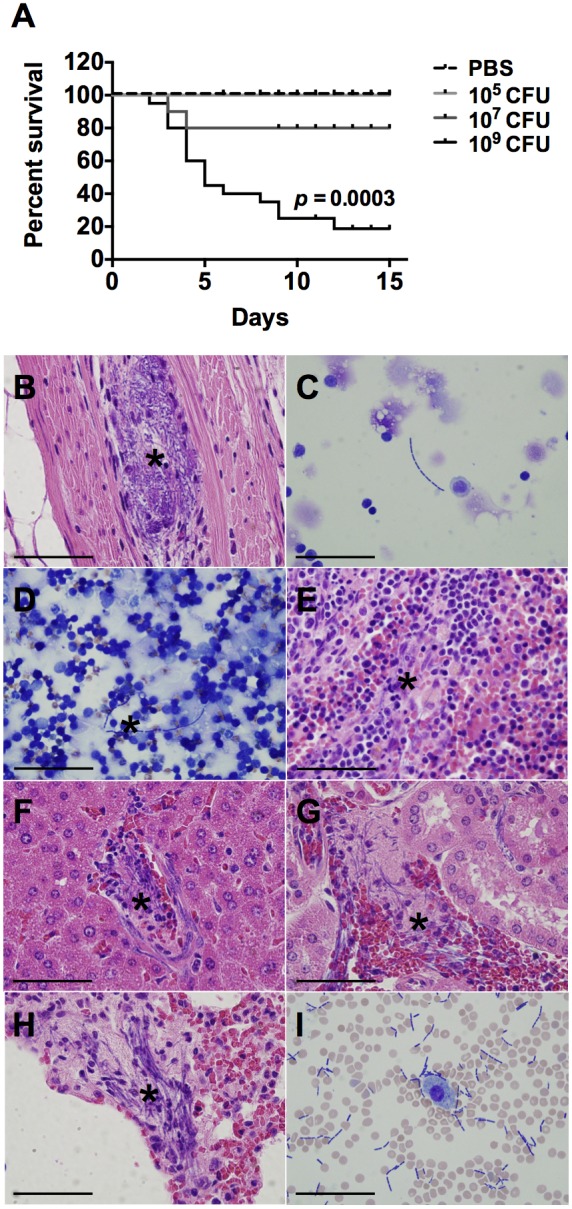
A. A/J mice were orally gavaged with 105, 107, or 109 spores of the Sterne strain of B. anthracis. Lethal infection was established within 3 days in A/J mice receiving 109 spores; n = 10 mice/group (105 and 107), n = 20 mice/group (109). Experiments were performed a minimum of three times. Statistical significance was calculated using the log-rank test. After 3 days of infection, A/J mice were sacrificed; both spores and vegetative bacilli (marked with *) were observed in the colon (B), MLNs (C), spleen (D, E), liver (F), kidneys (G), lungs (H), and in bronchoalveolar lavage (BAL) fluid (I). Bar = .
GI Epithelial Barrier Dysfunction and Dysbiosis in Sterne-infected A/J Mice
Observing the dissemination of B. anthracis Sterne in various mouse organs led us to investigate the intestinal barrier function of infected mice. We tested the epithelial barrier integrity of colons isolated from mice infected for 3 days. Infected colons showed a significant breakdown of intestinal barrier integrity, as evidenced by a significantly lower transepithelial electrical resistance (TEER) (Fig. 2A). Consistent with barrier disruption, infected mice also exhibited higher transepithelial conductance (GT), and short-circuit current (ISC) (Fig. 2B, C). Additionally, infection altered transcellular ion transport, which is manifested as abnormal electrogenic ion transport responses to cholinergic/muscarinic and histamine challenges (Fig. 2D, E, respectively) and abnormally high post-challenge secretory currents (Fig. 2F). Despite these signs of colonic mucosal damage, colonoscopies performed 3 days post-infection indicated no signs of hemorrhagic lesions within the colons of the majority of the mice studied (Fig. 2G). However, a very small number of mice (<10%) did show gross intestinal hemorrhage (Fig. 2H), vascular congestion, and trace microscopic evidence of hemorrhage in the colons (Fig. 2I) and in the small intestines of infected mice (Fig. 2J).
Figure 2. GI Epithelial Barrier Dysfunction Induced by Sterne Infection.

A/J mice were orally gavaged with 109 spores of the Sterne strain of B. anthracis and intestinal barrier integrity was analyzed ex vivo three days post-infection; n = 5 mice/group. TEER (A), trans-epithelial conductance (B), and short-circuit current (C) of Sterne-infected versus uninfected A/J mice. D and E. Delta current (ΔISC) before and after cholinergic (D) and histamine (E) challenges. F. Post-challenge secretory current of Sterne-infected versus uninfected A/J mice. Data are shown as mean +/− SEM. *P<0.05, **P<0.01, ***P<0.001 compared with PBS. G. Colonoscopies were performed in groups of uninfected and 109 Sterne spores-infected A/J mice three days post infection with a Multi-Purpose Rigid Telescope attached to a TELE PACK X. H. Gross hemorrhage in the small intestines. I and J. Hemorrhagic lesions in the colon (I) and small intestine (J) of Sterne-infected A/J mice. Bar = 200 µm.
A mutualistic relationship exists between the intestinal microbiota and the epithelial cells that comprise the single cell barrier between the host and the intestinal lumen, and both populations can modulate the other [22]. Thus, we examined the composition of the gut microbiota of infected mice. In fact, significant changes in the composition of the microbiota were noted in the Sterne-infected mice (Fig. 3). Global changes in microbial community distribution were analyzed by the UniFrac method [23]. In principle coordinate analyses (PCoA), the gut microbes before and 3 days post-infection clustered separately (Fig. 3A), indicating that GI B. anthracis Sterne significantly promoted microbial dysbiosis, in which a significant reduction in species richness and changes in the dominant phyla were observed (Fig. 3B, C). Several algorithms were used to determine the species richness and diversity within our samples. For instance, Chao Richness represents an estimate of the total number of species in the samples analyzed as it considers the sequences obtained a subsample of the entire community. Pielou evenness describes how close in numbers species in an environment are; a value nearing 1 indicates an even distribution within the community. Finally, Shannon Diversity takes into account both abundance and evenness within the community.
Figure 3. GI Dysbiosis Subsequent to Sterne Infection.

A/J mice were orally gavaged with 109 spores of the Sterne strain of B. anthracis and changes in microbiota composition during the course of Sterne infection were monitored. A. Unweighted UniFrac analyses were used to calculate distances between samples obtained from Sterne-infected A/J mice before infection and three days post-infection and three dimensional scatterplots were generated by using principal coordinate analysis (PCoA); n = 9 mice/group. B. Average abundance values of indicated phyla. C. Decreased microbial diversity, evenness, and species richness. Left: The Chao richness index was used as a measure of species richness. Middle: The Shannon diversity index was used to estimate microbial diversity for each group. Right: The species evenness index was calculated using the formula J’ = H’/H’max, where H’ is the Shannon diversity index and H’max is the maximal value of H’. Data are shown as mean +/− SEM. *P<0.05, ***P<0.001 compared with PBS-treated or day 0 mice. D. Bacteria genera most enriched or depleted in Sterne-infected mice at day 3 versus day 0, as measured by linear discriminant analysis (LDA). E. Reduced relative abundance of Enterobacteriaceae and Bifidobacterium in Sterne-infected A/J mice. F. Presence of Enterobacteriaceae in MLNs, spleens, and livers of Sterne-infected mice. G. Persistence of B. anthracis spores in the feces of Sterne-infected mice.
Subsequently, the algorithm linear discriminant analysis (LDA) effect size (LEfSe) [24] was used to determine which bacteria taxa were differentially represented with infection. Differentially depleted and enhanced genera were mostly composed of unculturable bacteria (Fig. 3D); however, infected mice were enriched for the novel genus Anaerotruncus, of which A. colihominis is the only described species and has been previously shown to cause bacteremia [25]. Additionally, to overcome PCR primer bias leading to underrepresentation of Actinobacteria, in particular bifidobacteria [26], real-time PCR was used to quantify Bifidobacterium in the colons. We noted a significant decrease in the relative abundance of Bifidobacterium with infection (Fig. 3E); the relative abundance of Enterobacteriaceae was also decreased (Fig. 3E). Because of the breach in intestinal epithelial integrity with infection, we then determined the presence of Enterobacteriaceae systemically. Indeed, members of this bacterial family were detected in the spleen, liver, and MLNs of infected mice (Fig. 3F). However, Bifidobacterium was not detected in these tissue samples (data not shown), indicating that its reduction in the colon may represent an actual depletion subsequent to infection. Not surprisingly, fecal shedding of Sterne was observed in all infected mice after one day of infection (Fig. 3G).
Innate Immune Responses in B. anthracis Sterne-infected Mice
It was previously demonstrated that innate cells, including dendritic cells (DCs) are the first targets for B. anthracis toxins such as LT [27], [28]. The internalized toxin complexes in the cytosol function as Zn+2-dependent metalloproteases that cleave members of the mitogen-activated protein kinase kinase (MAPKK) family, consequently blocking critical signals for cell activation [29]–[33], all of which could contribute to a failure in microbial clearance [28], [34], [35]. In line with these findings, it was recently observed that both toxins, LT and ET, significantly impair protective innate immune responses [28], [34]–[36].
To demonstrate whether GI anthrax infection induces the same effects in DCs derived from orally infected mice, colonic DCs were isolated and studied. Indeed, the activation of colonic CD45+MHCIIhiCD11c+F4/80−CD11b+ DCs (Fig. 4A), co-stimulatory molecules, CD80/CD86, and co-inhibitory, B7-H1, were unchanged or significantly depressed during infection (Fig. 4B), suggesting inhibition of immune functions. It is worth noting that B7-H1 is tightly regulated by ERK/p38 MAPK signaling [37], which is known to be disrupted by LF [38]. Accordingly, we addressed the question whether GI Sterne infection induces the cleavage of MAPKs in colonic DCs by evaluating the activation of downstream kinases, p38 and Erk1/2, in vivo and ex vivo. Data clearly show that infection with Sterne resulted in the impairment of p38 and Erk1/2 phosphorylation in colonic DCs of mice that were orally gavaged with 1×109 spores (Fig. 4C). To strengthen this observation, we then isolated and infected colonic cells with 1 MOI of B. anthracis Sterne spores for 1, 3, or 6 hours. A transient increase in p38 and Erk1/2 phosphorylation in total colonic cells (Fig. S2) and more specifically phospho-p38 in DCs was observed at earlier time points; however, after 6 hours, cleavage of p38 and Erk1/2 phosphorylation was measured (Fig. 4D, and Fig. S2). Furthermore, B. anthracis Sterne also downregulated IL-1β, tumor necrosis factor (TNF)-α, and IL-6 by DCs (Fig. 5A), a consequence that is attributed to cleavage of MAPKs [28], [34], [35].
Figure 4. Cleavage of MAPKs in DCs of Sterne-Infected A/J Mice.

A/J mice were orally gavaged with 109 spores of the Sterne strain of B. anthracis and DCs analyzed at various time points. A. Gating strategy for the analysis of CD45+MHCIIhiCD11c+F4/80−CD11b+ colonic DCs. B. Cell surface expression of CD86, CD80, and B7-H1 by colonic DCs was analyzed by flow cytometry. Gray tinted line = isotype control; black line = PBS group; green line = day 1 Sterne-infected A/J mice; magenta line = day 3 Sterne-infected A/J mice; blue line = day 5 Sterne-infected mice. C. Activity of MAPKs in colonic DCs of infected versus uninfected mice was analyzed by flow cytometry. Gray tinted line = isotype control; black line = PBS group; blue line = day 5 Sterne-infected mice. D. Colonic LP cells were isolated from uninfected A/J mice and incubated with 1 MOI of B. anthracis spores for 1, 3, or 6 hours. Activity of p38 and Erk1/2 was subsequently analyzed in colonic DCs. Gray tinted line = isotype control; black line = PBS group; green line = 1 hour treatment; magenta line = 3 hour treatment; blue line = 6 hour treatment. Data represent observations from three independent experiments and are shown as mean +/− SEM. *P<0.05, **P<0.01, ***P<0.001 compared with PBS.
Figure 5. Suppression of Innate Immune Responses in Sterne-Infected A/J Mice.

A/J mice were orally gavaged with 109 spores of the Sterne strain of B. anthracis and innate immune responses analyzed at various time points. A. DCs isolated from the colons of Sterne-infected A/J mice were analyzed by flow cytometry for the production of the pro-inflammatory cytokines IL-1β, TNF-α, and IL-6. Representative plots indicate cytokine production of uninfected and day 3 infected mice. Corresponding isotype controls were utilized for gating of intracellular cytokines. B. Gene expression profile of the distal colon of Sterne-infected A/J mice. Data represent observations from four independent experiments and are shown as mean +/− SEM. *P<0.05, **P<0.01, ***P<0.001 compared with PBS.
Having noted inhibition of colonic DC function, transcriptional changes in genes encoding pattern recognition receptors and inflammatory mediators were analyzed in the colonic tissues of infected mice as a measure of local induced inflammation and immune activation by B. anthracis Sterne. Inflammation-associated genes were found to be transiently upregulated early during Sterne infection (day 1, Fig. 5B); however, this gene upregulation was rapidly downregulated on days 3 and 5 post-infection, possibly due to the release of bacterial toxins (Fig. 5B). Therefore, the GI milieu mirrored the phenomenon of immune dysfunction observed in colonic DCs. Similar to the lack of colonic innate immune activation observed, systemic innate responses were also suppressed in infected mice, as both splenic DCs and macrophages produced less pro-inflammatory cytokines than those of uninfected mice (Fig. S3A, B). Not unexpectedly, given the aforementioned immune defects, with the exception of IL-1β at day 1 post-infection, no major increases in pro-inflammatory cytokine levels were detected in the sera of infected mice compared to control animals (Fig. S4); instead, we found that circulating levels of interferon (IFN)γ were reduced after 3 days of infection (Fig. S4).
Local and Systemic T Cell Immune Responses in Sterne-infected A/J Mice
To better understand the impact of deteriorated innate cell function resulting from GI anthrax on local and systemic T cell responses, we evaluated Th1, Th17, and regulatory (Treg) T cell activation in the colonic LP (Fig. 6A) and in the spleens of infected mice. Colonic and splenic T cell responses in infected mice were mostly limited to the CD4+ T cell subset. No significant changes were observed in day 1 and day 3-infected mice (Data Not Shown). However, a significant increase in the frequency of IL-17A+CD4+ T cells (day 5) was noted in the colons of Sterne infected mice (Fig. 6B, top). Systemically, splenic CD4+ T cells from infected mice showed increased intracellular IFNγ and a transient increase in IL-17A+CD4+ T cells (Fig. S5A, B). Recently, it has been shown that depending on the levels of ET, B. anthracis can suppress T cell proliferation, skew CD4+ T cells toward Th17 differentiation, and/or potentiate Th2 polarization [39]. Moreover, concurrent with increased colonic Th17 responses, Tregs were also enhanced in day 5-infected mice (Fig. 6B, bottom), suggesting that these induced levels of Tregs might potentially regulate protective Th17 immunity, which has been demonstrated to be critical for protection in inhalational anthrax infection [40].
Figure 6. T Cell Responses in Sterne-Infected A/J Mice.

A/J mice were orally gavaged with 109 spores of the Sterne strain of B. anthracis and adaptive immune responses in the colon analyzed at various time points by flow cytometry. A. Gating strategy for the analysis of colonic T cells. B. Th1, Th17, and regulatory T cells responses were tested by flow cytometry. Representative plots indicate cytokine production of uninfected and day 5-infected mice. C. Surface expression of PD1 in colonic T cells. D. Gene expression profile of the distal colon of Sterne-infected A/J mice. Data represent observations from four independent experiments and are shown as mean +/− SEM. *P<0.05, **P<0.01, ***P<0.001 compared with PBS.
Increased levels of Programmed Death-1 (PD-1) on T cells has recently been shown to be a critical “molecular signature” of T cell exhaustion in several models of chronic viral infection; this “molecular signature” also includes increased mRNA for cell-surface receptors suspected or known to have inhibitory function, such as 2B4, Ly49 family members, and GP49B [41]. Consistent with induction of immune dysfunction by GI anthrax infection, PD-1 receptor expression was significantly augmented on colonic CD4+ and CD8+ T cells of infected mice (Figure 6C), the transcription of which was also confirmed by gene expression analyses (Fig. 6D). We also observed increased gene expression of Gp49b, Ly49, and the 2B4 short isoform (Fig. 6D) in colonic tissues derived from B. anthracis infected mice. Moreover, colonic expression of Cxcr3, a chemokine receptor that is preferentially expressed on Th1 cells and promotes their recruitment to sites of inflammation [42], [43], was significantly increased (∼3 fold) in day 3-infected mice (Fig. 6D); however, expression of this critical chemokine receptor was significantly down-modulated by day 5 of infection, highlighting the change in the inflammatory status of the gut upon GI anthrax progression.
Discussion
Contact with B. anthracis-infected animals or consumption of tainted, undercooked meat may result in GI anthrax in humans [13], [44]. In recent years, advances have been made toward the development of experimental models to study the pathogenesis of GI B. anthracis infection and the effects of its toxins on GI health [11]–[13], [45]. Unlike direct intragastric infection with B. anthracis spores [11], [12], oral gavage with Sterne spores did not induce severe pathology in the small intestine, and the Peyer’s Patch did not appear to be the primary site of B. anthracis growth. Moreover, intragastric infection with vegetative bacilli resulted in rapid morbidity and mortality of the mice [13]. Such morbidity leads to significant nutritional deficiencies in the animals, which have known negative immunologic consequences [46]. Therefore, our model of GI anthrax provides a unique window of moderate disease, whereby the immunologic status of the animals better indicates responses to the pathogen itself. To gain a better understanding of GI anthrax pathogenesis, A/J mice were orally infected with 109 B. anthracis Sterne spores. Bacterial dissemination was observed in various visceral organs as early as one day post-infection (liver), which potentially contributed to further systemic bacterial pathogenesis resulting in impaired immunity, whereupon mice succumbed to infection as early as day 2 post-infection. Consequently, our findings provide in-depth immunologic studies as well as detailed investigations of the intestinal health of infected mice as it relates to its barrier function and microbial composition.
Emerging data suggest that B. anthracis toxins suppress host immunity, allowing the pathogen to significantly replicate, resulting in septic shock [47]. We posit that, as is the case with many other microbial pathogens, the first event is the ability of B. anthracis to disrupt epithelial barrier function via synergy of LT with B. anthracis S-layer protein A (BslA) [32], as this protein has been identified as an important surface adhesin of this deadly pathogen [48]. Disruption of intestinal barrier function undoubtedly contributes to bacterial dissemination locally and systemically, a mechanism that may change the composition of the gut microbiota, as significant decreases in the relative abundance of Enterobacteriaceae and Bifidobacterium in the feces of Sterne infected mice were observed. We also detected Enterobacteriaceae members in two important peripheral immune organs, the MLNs and the spleen. The observed bacterial translocation and dissemination could be seen as a potential cause for the lower numbers in the feces of infected mice; however, further studies are warranted to ascertain this possibility. On the other hand, Bifidobacterium, typically recognized as a beneficial bacterial group in the host gut, was not detected in any other organ, suggesting that this group was potentially out-competed for survival in the altered intestinal microenvironment upon B. anthracis Sterne infection. Indeed, this has been shown to be the case with many patients with colorectal inflammatory diseases, since Bifidobacteria protect the mucosae from damaging inflammation [49] and enhance barrier function [50]. When considering the concomitant intestinal barrier damage caused by B. anthracis infection, the dissemination of Enterobacteriaceae may have contributed to pathogenic inflammation, while elimination of the beneficial bacterium, Bifidobacterium, may have left the host with less regulatory mucosal responses involved in healthy gut homeostasis.
PCoA demonstrated a significantly altered gut microbial composition in mice after 3 days of infection, indicating a global change in the gut flora at the phylum level. Uncultured operational taxonomic units (OTU) 751 and OTU 1058 were shown to be dramatically increased, along with Anaetruncus, while uncultured OTU 339 and OTU 2186 were decreased with infection. Both unculturable OTU 751 and OTU 1058 are classified as part of the Bacteroidales S24-7 family, which has been found to be a dominant uncultured family in the gut whose function is likely the predominant contributor to the Bacteroidetes phylum expansion. Unculturable OTU 339 and OTU 2186 can be classified into the Ruminococcaceae family and Lachnospiraceae family, respectively. Depletion of both of these two families has consistently been associated with colonic inflammation [51]. Most of these two families’ members belong to Clostridium cluster IV and XIVa, which have the potential to induce butyrate production [52], Tregs [53], maintenance of barrier function [52], and competition with other pathogens in the colonization of the gut. Therefore, the collective depletion of these commensal bacteria in the gut may have negatively affected the local immunity by promoting pathogenic inflammation and disrupting mucosal homeostasis.
Controlled inflammatory responses are necessary to promote protective immunity and overcome pathogen challenge after infection. However, certain pathogens have evolved to evade immune recognition and clearance through NF-κB and MAPK signaling inhibition [54]. Accordingly, one day post-infection we found a robust increase in the transcription of genes involved in innate immune recognition (Data Not Shown); however, expression of these genes rapidly decreased to basal levels, and in certain cases, was even lower than those of the controls, suggesting suppression of critical sensing molecules to mobilize protective immunity against pathogens. Most of the genes whose expression were upregulated are MAPK-dependent, indicating that within one day endospores germinate into the vegetative form to secrete LT, which likely causes immune suppression. Indeed, infected mice exhibited global immune suppression, as infection did not elicit protective immune activation in either colonic or splenic DCs. This is consistent with previous reports showing that LT suppresses IL-6 and TNF-α in DCs by disrupting MAPK signaling [28]. Suppression is thought to occur early in infection, while in later stages, the toxin exerts inflammatory effects contributing to toxic shock and bacteremia [28]. Our studies clearly demonstrate the inhibition of MAPK signaling in intestinal immune cells (e.g., DCs) upon GI anthrax infection.
The gut microenvironment is tightly controlled by innate cells that potentially impact T cell function by regulating PD-1/B7-H1 interactions, which may result in T cell “exhaustion” as a consequence of infection [55]. Importantly, it has been shown that the MAPK-dependent transcription factor, T-bet [56], is a negative regulator of PD-1 [57]. Thus, inhibition of MAPK-activity by LT may have downregulated T-bet activation, resulting in the increased expression of PD-1 on T cells. While the expression of B7-H1 was reduced on DCs [58], B7-H1 transcription was globally upregulated in colonic cells other than DCs. Furthermore, MAPK-dependent cytokines (e.g., IL-1β, TNF-α, and IL-6) were downregulated in DCs and macrophages as a result of Sterne infection. In the colon, expression of Cxcr3, which is a signature chemokine receptor for IFNγ+ Th1 cell recruitment, was transiently increased with a significant reduction by day 5, indicating a potential lack of supporting immune mechanisms to induce microbial protective immunity in the GI tract. Conversely, in the spleen, the immune environment was shifted toward a mixed Th1/Th17 response, with increased IFNγ and IL-17A-producing CD4+ T cells, a hallmark of bacterial infection [59].
In summary, GI infection with B. anthracis is quite different and more complex than infection via the respiratory route. Respiratory surfaces maintain an anti-inflammatory environment [60], as does the GI tract [61]; however, the microbial load of the GI tract is considerably larger and more complex. Respiratory infection with B. anthracis has a higher fatality rate and requires smaller inoculums than GI infection, perhaps due to the presence of mitigating factors in the latter, including proteolytic enzyme activity, the extensive gut microbiota, and peristaltic expulsion of spores from the GI tract. Additionally, the breach in gut barrier function caused by B. anthracis and its gene products leads to translocation of B. anthracis and other gut microbes, which may engage complex signaling events, locally and systemically. Translocation of the gut microbiota could be beneficial to the host by mounting a Th1/Th17 response to clear the bacteria; conversely, it may cause bacteremia and septic shock. The precise contribution of this shift in the composition of the gut microbiome to the pathobiology of GI anthrax has yet to be examined. An early distinction between mice that survive and those that succumb to infection and a better understanding of early innate immune cell activation that bypasses suppressive attempts by B. anthracis will be critical in the prevention of fatal systemic disease in animals and in humans.
Supporting Information
Morbidity and Mortality in GI Anthrax is Dependent on Active Infection and Involves Hematogenous Spread of Infection. A. A/J mice were orally gavaged with 109 spores of the Sterne strain of B. anthracis. One day post-infection, B. anthracis Sterne bacilli could be found within the liver of some mice Bar = 50 µm. B. A/J mice were orally gavaged with 125 µg LT (PA+LF) or injected intraperitoneally (i.p.) and monitored for morbidity and death. C. IL-1β expression profile of the distal colons of A/J mice that were given LT (125 µg) by the oral route versus i.p. injection.
(TIFF)
Ex vivo Inhibition of MAPKs in Immune Cells by Sterne. Colonic LP cells were isolated from uninfected A/J mice and incubated with 1 MOI of B. anthracis spores for 1, 3, or 6 hours. Activity of p38 and Erk1/2 was subsequently analyzed with phosphorylation-specific antibodies by Western blot.
(TIFF)
Splenic Innate Immune Responses in Sterne-infected A/J Mice. A/J mice were orally gavaged with 109 spores of the Sterne strain of B. anthracis and innate immune responses analyzed at various time points by flow cytometry. Splenic DC (A) and macrophage (B) functions were inhibited after infection as measured by IL-1β and TNF-α production. n = 10 mice/group. Data represent observations from four independent experiments and are shown as mean +/− SEM. **P<0.01, ***P<0.001 compared with PBS.
(TIFF)
Sera Cytokine Levels of Sterne-infected A/J Mice. A/J mice were orally gavaged with 109 spores of the Sterne strain of B. anthracis and sera collected. Cytokines in the sera of Sterne-infected and uninfected A/J mice were measured using the Bio-Plex Pro Mouse Cytokine 23-plex immunoassay kit. Data are shown as mean +/− SEM; each symbol represents one mouse. *P<0.05, ***P<0.001 compared with PBS.
(TIFF)
Splenic T Cell Responses in Sterne-infected A/J Mice. A/J mice were orally gavaged with 109 spores of the Sterne strain of B. anthracis and Th1 (A) and Th17 (B) responses analyzed at various time points by flow cytometry. n = 10 mice/group. Data represent observations from four independent experiments and are shown as mean +/− SEM. *P<0.05 compared with PBS.
(TIFF)
List of primer sequences for Real-Time PCR analyses.
(DOCX)
List of primer sequences for 16S rDNA analyses.
(DOCX)
Acknowledgments
We would like to extend our gratitude to Dr. Arthur Friedlander and Dr. Timothy Hoover for fruitful discussions, as well as to Dr. Lieqi Tang and Eric Li for technical support.
Funding Statement
This work was supported by the National Institute of Allergy and Infectious Diseases RO1 AI093370 to MM. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Beatty ME, Ashford DA, Griffin PM, Tauxe RV, Sobel J (2003) Gastrointestinal anthrax: review of the literature. Arch Intern Med 163: 2527–2531. [DOI] [PubMed] [Google Scholar]
- 2. Mock M, Fouet A (2001) Anthrax. Annu Rev Microbiol 55: 647–671. [DOI] [PubMed] [Google Scholar]
- 3. Turnbull PC (1991) Anthrax vaccines: past, present and future. Vaccine 9: 533–539. [DOI] [PubMed] [Google Scholar]
- 4. Mikesell P, Ivins BE, Ristroph JD, Dreier TM (1983) Evidence for plasmid-mediated toxin production in Bacillus anthracis. Infect Immun 39: 371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Green BD, Battisti L, Koehler TM, Thorne CB, Ivins BE (1985) Demonstration of a capsule plasmid in Bacillus anthracis. Infect Immun 49: 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tonello F, Zornetta I (2012) Bacillus anthracis factors for phagosomal escape. Toxins (Basel) 4: 536–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Makino S, Watarai M, Cheun HI, Shirahata T, Uchida I (2002) Effect of the lower molecular capsule released from the cell surface of Bacillus anthracis on the pathogenesis of anthrax. J Infect Dis 186: 227–233. [DOI] [PubMed] [Google Scholar]
- 8. Scorpio A, Chabot DJ, Day WA, O'Brien D K, Vietri NJ, et al. (2007) Poly-gamma-glutamate capsule-degrading enzyme treatment enhances phagocytosis and killing of encapsulated Bacillus anthracis. Antimicrobial agents and chemotherapy 51: 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mohamadzadeh M, Duong T, Sandwick SJ, Hoover T, Klaenhammer TR (2009) Dendritic cell targeting of Bacillus anthracis protective antigen expressed by Lactobacillus acidophilus protects mice from lethal challenge. Proc Natl Acad Sci U S A 106: 4331–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tournier JN, Ulrich RG, Quesnel-Hellmann A, Mohamadzadeh M, Stiles BG (2009) Anthrax, toxins and vaccines: a 125-year journey targeting Bacillus anthracis. Expert Rev Anti Infect Ther 7: 219–236. [DOI] [PubMed] [Google Scholar]
- 11. Glomski IJ, Piris-Gimenez A, Huerre M, Mock M, Goossens PL (2007) Primary involvement of pharynx and peyer's patch in inhalational and intestinal anthrax. PLoS Pathog 3: e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tonry JH, Popov SG, Narayanan A, Kashanchi F, Hakami RM, et al. (2013) In vivo murine and in vitro M-like cell models of gastrointestinal anthrax. Microbes Infect 15: 37–44. [DOI] [PubMed] [Google Scholar]
- 13. Xie T, Sun C, Uslu K, Auth RD, Fang H, et al. (2013) A New Murine Model for Gastrointestinal Anthrax Infection. PLoS One 8: e66943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baldari CT, Tonello F, Paccani SR, Montecucco C (2006) Anthrax toxins: A paradigm of bacterial immune suppression. Trends in immunology 27: 434–440. [DOI] [PubMed] [Google Scholar]
- 15. Welkos SL, Keener TJ, Gibbs PH (1986) Differences in susceptibility of inbred mice to Bacillus anthracis. Infect Immun 51: 795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cheng SX (2012) Calcium-sensing receptor inhibits secretagogue-induced electrolyte secretion by intestine via the enteric nervous system. Am J Physiol Gastrointest Liver Physiol 303: G60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barman M, Unold D, Shifley K, Amir E, Hung K, et al. (2008) Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect Immun 76: 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Antharam VC, Li EC, Ishmael A, Sharma A, Mai V, et al. (2013) Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J Clin Microbiol 51: 2884–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goossens PL (2009) Animal models of human anthrax: the Quest for the Holy Grail. Mol Aspects Med 30: 467–480. [DOI] [PubMed] [Google Scholar]
- 20. Loving CL, Kennett M, Lee GM, Grippe VK, Merkel TJ (2007) Murine aerosol challenge model of anthrax. Infect Immun 75: 2689–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Welkos SL, Keener TJ, Gibbs PH (1986) Differences in susceptibility of inbred mice to Bacillus anthracis. Infection and immunity 51: 795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjoberg J, et al. (2010) Enteric defensins are essential regulators of intestinal microbial ecology. Nature immunology 11: 76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R (2011) UniFrac: an effective distance metric for microbial community comparison. ISME J 5: 169–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, et al. (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12: R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lau SK, Woo PC, Woo GK, Fung AM, Ngan AH, et al. (2006) Bacteraemia caused by Anaerotruncus colihominis and emended description of the species. J Clin Pathol 59: 748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hill JE, Fernando WM, Zello GA, Tyler RT, Dahl WJ, et al. (2010) Improvement of the representation of bifidobacteria in fecal microbiota metagenomic libraries by application of the cpn60 universal primer cocktail. Appl Environ Microbiol 76: 4550–4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hu H, Leppla SH (2009) Anthrax toxin uptake by primary immune cells as determined with a lethal factor-beta-lactamase fusion protein. PloS one 4: e7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Agrawal A, Lingappa J, Leppla SH, Agrawal S, Jabbar A, et al. (2003) Impairment of dendritic cells and adaptive immunity by anthrax lethal toxin. Nature 424: 329–334. [DOI] [PubMed] [Google Scholar]
- 29. Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, et al. (1998) Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 280: 734–737. [DOI] [PubMed] [Google Scholar]
- 30. Vitale G, Bernardi L, Napolitani G, Mock M, Montecucco C (2000) Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. The Biochemical journal 352 Pt 3: 739–745. [PMC free article] [PubMed] [Google Scholar]
- 31. Ebrahimi CM, Sheen TR, Renken CW, Gottlieb RA, Doran KS (2011) Contribution of lethal toxin and edema toxin to the pathogenesis of anthrax meningitis. Infection and immunity 79: 2510–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xie T, Auth RD, Frucht DM (2011) The effects of anthrax lethal toxin on host barrier function. Toxins 3: 591–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu L, Frucht DM (2007) Bacillus anthracis: a multi-faceted role for anthrax lethal toxin in thwarting host immune defenses. The international journal of biochemistry & cell biology 39: 20–24. [DOI] [PubMed] [Google Scholar]
- 34. Park JM, Greten FR, Li ZW, Karin M (2002) Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science 297: 2048–2051. [DOI] [PubMed] [Google Scholar]
- 35. During RL, Li W, Hao B, Koenig JM, Stephens DS, et al. (2005) Anthrax lethal toxin paralyzes neutrophil actin-based motility. The Journal of infectious diseases 192: 837–845. [DOI] [PubMed] [Google Scholar]
- 36. Chou PJ, Newton CA, Perkins I, Friedman H, Klein TW (2008) Suppression of dendritic cell activation by anthrax lethal toxin and edema toxin depends on multiple factors including cell source, stimulus used, and function tested. DNA Cell Biol 27: 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Karakhanova S, Meisel S, Ring S, Mahnke K, Enk AH (2010) ERK/p38 MAP-kinases and PI3K are involved in the differential regulation of B7-H1 expression in DC subsets. Eur J Immunol 40: 254–266. [DOI] [PubMed] [Google Scholar]
- 38. Chopra AP, Boone SA, Liang X, Duesbery NS (2003) Anthrax lethal factor proteolysis and inactivation of MAPK kinase. J Biol Chem 278: 9402–9406. [DOI] [PubMed] [Google Scholar]
- 39. Paccani SR, Benagiano M, Savino MT, Finetti F, Tonello F, et al. (2011) The adenylate cyclase toxin of Bacillus anthracis is a potent promoter of T(H)17 cell development. The Journal of allergy and clinical immunology 127: 1635–1637. [DOI] [PubMed] [Google Scholar]
- 40. Datta SK, Sabet M, Nguyen KP, Valdez PA, Gonzalez-Navajas JM, et al. (2010) Mucosal adjuvant activity of cholera toxin requires Th17 cells and protects against inhalation anthrax. Proc Natl Acad Sci U S A 107: 10638–10643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, et al. (2007) Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27: 670–684. [DOI] [PubMed] [Google Scholar]
- 42. Bonecchi R, Bianchi G, Bordignon PP, D'Ambrosio D, Lang R, et al. (1998) Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med 187: 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lacotte S, Brun S, Muller S, Dumortier H (2009) CXCR3, inflammation, and autoimmune diseases. Ann N Y Acad Sci 1173: 310–317. [DOI] [PubMed] [Google Scholar]
- 44. Swartz MN (2001) Recognition and management of anthrax–an update. N Engl J Med 345: 1621–1626. [DOI] [PubMed] [Google Scholar]
- 45. Sun C, Fang H, Xie T, Auth RD, Patel N, et al. (2012) Anthrax lethal toxin disrupts intestinal barrier function and causes systemic infections with enteric bacteria. PLoS One 7: e33583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chandra RK (1996) Nutrition, immunity and infection: from basic knowledge of dietary manipulation of immune responses to practical application of ameliorating suffering and improving survival. Proc Natl Acad Sci U S A 93: 14304–14307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Coggeshall KM, Lupu F, Ballard J, Metcalf JP, James JA, et al. (2013) The sepsis model: an emerging hypothesis for the lethality of inhalation anthrax. Journal of cellular and molecular medicine 17: 914–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kern J, Schneewind O (2010) BslA, the S-layer adhesin of B. anthracis, is a virulence factor for anthrax pathogenesis. Mol Microbiol 75: 324–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Manichanh C, Borruel N, Casellas F, Guarner F (2012) The gut microbiota in IBD. Nature reviews Gastroenterology & hepatology 9: 599–608. [DOI] [PubMed] [Google Scholar]
- 50. Ewaschuk JB, Diaz H, Meddings L, Diederichs B, Dmytrash A, et al. (2008) Secreted bioactive factors from Bifidobacterium infantis enhance epithelial cell barrier function. American journal of physiology Gastrointestinal and liver physiology 295: G1025–1034. [DOI] [PubMed] [Google Scholar]
- 51. Biddle A SL, Blanchard J, Leschine S (2013) Untangling the Genetic Basis of Fibrolytic Specialization by Lachnospiraceae and Ruminococcaceae in Diverse Gut Communities. Diversity 5: 627–640. [Google Scholar]
- 52. Thibault R, Blachier F, Darcy-Vrillon B, de Coppet P, Bourreille A, et al. (2010) Butyrate utilization by the colonic mucosa in inflammatory bowel diseases: a transport deficiency. Inflammatory bowel diseases 16: 684–695. [DOI] [PubMed] [Google Scholar]
- 53. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, et al. (2011) Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331: 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mohamadzadeh M, Chen L, Schmaljohn AL (2007) How Ebola and Marburg viruses battle the immune system. Nat Rev Immunol 7: 556–567. [DOI] [PubMed] [Google Scholar]
- 55.Gianchecchi E, Delfino DV, Fierabracci A (2013) Recent insights into the role of the PD-1/PD-L1 pathway in immunological tolerance and autoimmunity. Autoimmunity reviews. [DOI] [PubMed]
- 56. Bachmann M, Dragoi C, Poleganov MA, Pfeilschifter J (2007) M (2007) Interleukin-18 directly activates T-bet expression and function via p38 mitogen-activated protein kinase and nuclear factor-Œ∫B in acute myeloid leukemia, Äìderived predendritic KG-1 cells. Molecular Cancer Therapeutics 6: 723–731. [DOI] [PubMed] [Google Scholar]
- 57. Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, et al. (2011) Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nature immunology 12: 663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Qian Y, Deng J, Geng L, Xie H, Jiang G, et al. (2008) TLR4 signaling induces B7-H1 expression through MAPK pathways in bladder cancer cells. Cancer investigation 26: 816–821. [DOI] [PubMed] [Google Scholar]
- 59. Pepper M, Linehan JL, Pagan AJ, Zell T, Dileepan T, et al. (2010) Different routes of bacterial infection induce long-lived TH1 memory cells and short-lived TH17 cells. Nature immunology 11: 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Balhara J, Gounni AS (2012) The alveolar macrophages in asthma: a double-edged sword. Mucosal immunology 5: 605–609. [DOI] [PubMed] [Google Scholar]
- 61. Tsuji NM, Kosaka A (2008) Oral tolerance: intestinal homeostasis and antigen-specific regulatory T cells. Trends in immunology 29: 532–540. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Morbidity and Mortality in GI Anthrax is Dependent on Active Infection and Involves Hematogenous Spread of Infection. A. A/J mice were orally gavaged with 109 spores of the Sterne strain of B. anthracis. One day post-infection, B. anthracis Sterne bacilli could be found within the liver of some mice Bar = 50 µm. B. A/J mice were orally gavaged with 125 µg LT (PA+LF) or injected intraperitoneally (i.p.) and monitored for morbidity and death. C. IL-1β expression profile of the distal colons of A/J mice that were given LT (125 µg) by the oral route versus i.p. injection.
(TIFF)
Ex vivo Inhibition of MAPKs in Immune Cells by Sterne. Colonic LP cells were isolated from uninfected A/J mice and incubated with 1 MOI of B. anthracis spores for 1, 3, or 6 hours. Activity of p38 and Erk1/2 was subsequently analyzed with phosphorylation-specific antibodies by Western blot.
(TIFF)
Splenic Innate Immune Responses in Sterne-infected A/J Mice. A/J mice were orally gavaged with 109 spores of the Sterne strain of B. anthracis and innate immune responses analyzed at various time points by flow cytometry. Splenic DC (A) and macrophage (B) functions were inhibited after infection as measured by IL-1β and TNF-α production. n = 10 mice/group. Data represent observations from four independent experiments and are shown as mean +/− SEM. **P<0.01, ***P<0.001 compared with PBS.
(TIFF)
Sera Cytokine Levels of Sterne-infected A/J Mice. A/J mice were orally gavaged with 109 spores of the Sterne strain of B. anthracis and sera collected. Cytokines in the sera of Sterne-infected and uninfected A/J mice were measured using the Bio-Plex Pro Mouse Cytokine 23-plex immunoassay kit. Data are shown as mean +/− SEM; each symbol represents one mouse. *P<0.05, ***P<0.001 compared with PBS.
(TIFF)
Splenic T Cell Responses in Sterne-infected A/J Mice. A/J mice were orally gavaged with 109 spores of the Sterne strain of B. anthracis and Th1 (A) and Th17 (B) responses analyzed at various time points by flow cytometry. n = 10 mice/group. Data represent observations from four independent experiments and are shown as mean +/− SEM. *P<0.05 compared with PBS.
(TIFF)
List of primer sequences for Real-Time PCR analyses.
(DOCX)
List of primer sequences for 16S rDNA analyses.
(DOCX)