

Abstract
The nuclear lamina is an extensive protein network that contributes to nuclear structure and function. LEM domain (LAP2, emerin, MAN1 domain, LEM-D) proteins are components of the nuclear lamina, identified by a shared ∼45-amino-acid motif that binds Barrier-to-autointegration factor (BAF), a chromatin-interacting protein. Drosophila melanogaster has three nuclear lamina LEM-D proteins, named Otefin (Ote), Bocksbeutel (Bocks), and dMAN1. Although these LEM-D proteins are globally expressed, loss of either Ote or dMAN1 causes tissue-specific defects in adult flies that differ from each other. The reason for such distinct tissue-restricted defects is unknown. Here, we generated null alleles of bocks, finding that loss of Bocks causes no overt adult phenotypes. Next, we defined phenotypes associated with lem-d double mutants. Although the absence of individual LEM-D proteins does not affect viability, loss of any two proteins causes lethality. Mutant phenotypes displayed by lem-d double mutants differ from baf mutants, suggesting that BAF function is retained in animals with a single nuclear lamina LEM-D protein. Interestingly, lem-d double mutants displayed distinct developmental and cellular mutant phenotypes, suggesting that Drosophila LEM-D proteins have developmental functions that are differentially shared with other LEM-D family members. This conclusion is supported by studies showing that ectopically produced LEM-D proteins have distinct capacities to rescue the tissue-specific phenotypes found in single lem-d mutants. Our findings predict that cell-specific mutant phenotypes caused by loss of LEM-D proteins reflect both the constellation of LEM-D proteins within the nuclear lamina and the capacity of functional compensation of the remaining LEM-D proteins.
Keywords: LEM domain, laminopathies, nuclear lamina, Drosophila, germline stem cells
THE nuclear lamina is an extensive protein network underlying the nuclear envelope. This network is composed of the nucleus-specific intermediate filament proteins, the A- and B-type lamins, which form a structural platform for association of >200 proteins (Schirmer et al. 2003; Korfali et al. 2010; Malik et al. 2010). LEM domain (LAP2, emerin, MAN1 domain, LEM-D) proteins represent one family of lamin interacting proteins. This family shares an ∼45-residue bihelical domain that was first identified in LAP2, emerin, and MAN1 (Lin et al. 2000; Mansharamani and Wilson 2005; Wagner and Krohne 2007). LEM-D proteins interact with the small, conserved protein called Barrier-to-autointegration factor (BAF), a protein that binds double-strand DNA and histones (Zheng et al. 2000; Cai et al. 2001; Laguri et al. 2001; Furukawa et al. 2003; Liu et al. 2003; Montes de Oca et al. 2005). Through interactions with BAF, LEM-D proteins connect interphase chromosomes to the nuclear lamina, thereby contributing to global nuclear organization.
Metazoan genomes encode several LEM-D proteins (Lee and Wilson 2004; Berk et al. 2013). Most show enriched localization within the nuclear lamina, wherein the LEM-D proteins direct shared protein associations. For example, emerin and MAN1 interact with each other, as well as with the A- and B-type lamins and transcriptional regulators, such as the Germ-cell-less repressor (Mansharamani and Wilson 2005). In addition, LEM-D proteins direct unique interactions. For example, MAN1 associates with the receptor regulated Smads (Osada et al. 2003; Raju et al. 2003; Pinto et al. 2008; Wagner et al. 2010), the downstream effectors of the TGF-β signaling pathways, while emerin associates with β-catenin (Markiewicz et al. 2006), a mediator of Wnt signaling. Recent evidence suggests that some LEM-D proteins are not enriched in the nuclear lamina, but shuttle between the cytoplasm and nucleus. These proteins, exemplified by LEM-3/Ankle1, are distinguished by the presence of a C-terminal GIY–YIG motif associated with endonuclease activity (Brachner et al. 2012) and hypersensitivity to DNA damage (Dittrich et al. 2012). Together, these findings emphasize that LEM-D proteins have the capacity to make diverse contributions to many nuclear processes, ranging from transcriptional regulation to maintenance of genome stability.
Mutations in genes encoding the nuclear lamina LEM-D proteins cause several human diseases. The loss of LEM-D proteins is associated with bone density disorders, cardiomyopathies, and muscular dystrophies (Worman et al. 2010). These diseases display age-enhanced, tissue-restricted pathology, even though LEM-D proteins are globally expressed. The basis for tissue-restricted defects is unknown. One possibility is that the affected tissues require a function unique to one member of the LEM-D family. Alternatively, cell types may have differences in the abundance of LEM-D proteins (Solovei et al. 2013), which may predispose cells to loss of a disease-associated protein. Distinguishing between these possibilities requires an improved understanding of the functions of LEM-D proteins and their capacity for regulatory compensation.
Drosophila melanogaster serves as an excellent model to study how nuclear lamina components contribute to tissue-specific regulation during development. This model has been powerful in defining effects of novel disease-causing mutations in the human A-type lamin (Dialynas et al. 2012), demonstrating that some disease-causing mutations cause a loss of nuclear compartmentalization of lamins and associated proteins, which may contribute to muscle disease. Drosophila encodes a three-member nuclear lamina LEM-D family (Figure 1A). These LEM-D proteins include dMAN1, Otefin (Ote), and Bocksbeutel (Bocks) (Ashery-Padan et al. 1997a,b; Wagner et al. 2004, 2006; Pinto et al. 2008). dMAN1 represents the Drosophila homolog of LEM2 and MAN1, whereas Ote and Bocks are predicted to be homologs of emerin (Wagner et al. 2006; Wagner and Krohne 2007). Genetic analyses have demonstrated that both dMAN1 and Ote are required for Drosophila development. Loss of dMAN1 causes a moderate decrease in adult viability, with surviving adults showing male sterility, decreased female fertility due to egg retention, defects in neuromuscular junctions, flightlessness associated with altered wing patterning and positioning, and locomotion difficulties (Pinto et al. 2008; Wagner et al. 2010). In contrast, phenotypes associated with ote−/− mutants are restricted to the ovary, where germline stem cells (GSCs) show age-enhanced loss (Jiang et al. 2008; Barton et al. 2013). The nonoverlapping defects associated with Ote and dMAN1 loss imply that these proteins make unique regulatory contributions during development.
Figure 1.
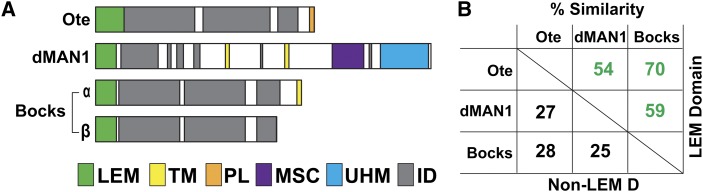
Drosophila nuclear lamina LEM-D proteins. (A) Three genes encode nuclear lamina-enriched LEM-D proteins, otefin (ote), dMAN1, and bocksbeutel (bocks). All of the encoded proteins carry an amino-terminal LEM-D (green box). The bocks gene encodes two isoforms. The Bocks α- and β-isoforms are distinguished by a unique 50-amino-acid carboxyl-terminal domain in Bocks α-isoform that carries a transmembrane domain (yellow boxes). Outside of the LEM-D, the three proteins carry regions predicted to be intrinsically disordered (ID, gray boxes) and additional domains, including the MAN1–Src1-p C-terminal domain (MSC, purple box), the U2AF homology motif (UHM, blue box), a transmembrane domain (TM, yellow box) or a peripheral localization domain (PL, orange box). (B) Shown is a chart of the percentage similarity derived from pairwise alignments of amino acid sequences of LEM-D proteins, including either the LEM-D alone or the non-LEM-D regions of the proteins. In these comparisons, the larger Bocks α-isoform was used.
The Drosophila nuclear lamina LEM-D proteins share extensive homology within the LEM-D (Figure 1B). However, outside of this domain, homology is minimal. Non-LEM-D regions in Ote and Bocks are predicted to be largely disordered, a feature shared with emerin (Wolff et al. 2001), whereas dMAN1 contains several defined domains (Wagner et al. 2006; Pinto et al. 2008). Even though homology is limited, non-LEM-D regions of the Drosophila LEM-D proteins direct common protein associations, such as interactions with the A- and B-type lamins, Lamin C and lamin Dm0, respectively (Pinto et al. 2008; Schulze et al. 2009). These shared protein partners imply that Drosophila LEM-D proteins possess overlapping functions, as found for LEM-D proteins in other organisms (Gruenbaum et al. 2002; Liu et al. 2003; Huber et al. 2009; Barkan et al. 2012; Gonzalez et al. 2012; Reil and Dabauvalle 2013). However, the extent of any regulatory compensation among the three Drosophila nuclear lamina LEM-D proteins is unknown. To begin to address this question, we generated mutations in the bocks gene. Analyses of animals carrying bocks null alleles demonstrated that complete loss of Bocks causes no overt developmental defects. Next, we generated animals carrying null alleles of two lem-d genes. We found that all lem-d double mutants fail to survive to adulthood, demonstrating that Drosophila LEM-D proteins are functionally redundant. Analyses of the developmental and cellular phenotypes in lem-d double mutants revealed distinct mutant phenotypes, implying that pairs of LEM-D proteins share different functions. Further, lem-d double mutants display phenotypes distinct from baf mutants, indicating that essential functions of LEM-D proteins extend beyond BAF regulation. Finally, we determined whether ectopic production of individual LEM-D proteins rescued tissue-restricted phenotypes found in the ote−/− and dMAN1−/− mutants. We found that production of Bocks, but not dMAN1, rescued ote−/− sterility, demonstrating that Ote and Bocks are functional homologs. In contrast, neither Bocks nor Ote rescued dMAN1−/− mutant phenotypes, implying that dMAN1 makes unique contributions to wing development. Based on our studies, we predict that cell-specific mutant phenotypes resulting from loss of individual LEM-D proteins depend upon two factors. These include the presence of other LEM-D proteins within the nuclear lamina and the capacity of remaining LEM-D proteins to functionally compensate for the lost protein.
Materials and Methods
Drosophila stocks and culture conditions
Drosophila stocks were raised at 25° at 70% humidity on standard cornmeal/agar medium, with p-hydroxybenzoic acid methyl ester as a mold inhibitor. Crosses were carried out in vials at 25°, unless otherwise noted. Stocks used in this study include y1w67c23 (the wild-type reference control), two dMAN1 alleles [dMAN1Δ81 and dMAN1Δ26 (Pinto et al. 2008)], and two ote alleles [oteB279-G and otehalPk (Barton et al. 2013)].
Generation of bocks deletion alleles
The parental line used to generate bocks mutants was bocksCB03586, a line that carries a white marked P element [P{PTT-GB}] inserted into the 5′-untranslated region of the bocks gene (Buszczak et al. 2007). P mobilization was achieved using a chromosomal source of transposase, P[ry+ Δ2-3] (99B) (Robertson et al. 1988). Homozygous y1w67c23; bocksCB03586 females were crossed to males y+w+/Y; CyO/Sp; Δ2-3 Sb/TM6, Ubx males. Single y1 w67c23; bocksCB03586/ Δ2-3 Sb red-eyed males (indicating the presence of the white marked P element) were mated to females that were y+ w67c23; bocks+/TM6, Tb. Excisions were identified as white-eyed, Tubby (TM6), non-Sb flies (y+ w67c23/Y; bocks*/TM6, Tb). DNA was isolated from these stocks and Southern analysis was completed to determine whether P excision resulted in deletions of DNA sequences containing the bocks gene. Two bocks deletion alleles were identified, bocksΔ10 and bocksΔ66 (Figure 2A). The endpoints of each deletion was determined using PCR to isolate an appropriately sized fragment from genomic DNA. These fragments were directly sequenced.
Figure 2.

Generation and analysis of bocks deletion alleles. (A) Structure of the bocks locus, including a portion of the uncharacterized CG8312 gene and the overlapping P581PK gene. The bocks coding region is shown as a large rectangle (black), depicting the location of the LEM-D (green) and the transmembrane domain (yellow). Deletion alleles of bocks were generated by imprecise deletion of a P element inserted at +11 in the bocksCB03586 allele (inverted red triangle). The bocksΔ10 has a 344-bp and bocksΔ66 has a 728-bp deletion. (B)Western blot of protein extracts isolated from bocks+/+ (+/+), bocksCB03586 (CB) adults, and adults homozygous for one of the two bocks deletion alleles. The Western blot was probed with antibodies against Bocks and reprobed with antibodies against α-tubulin, which serves as a loading control. Note that our separation conditions detect only a single band, which likely represents both Bocks isoforms. (C) Shown are Western blots of protein extracts from bocks+/+, bocks Δ10/Δ10, bocksΔ66/Δ66 adults. These blots were probed with antibodies against lamin Dm0, Lamin C, dMAN1, or Ote and reprobed with antibodies against α-tubulin. (*) A breakdown product of dMAN1, which is primarily detected in bocks+/+ extracts. (D) Top: confocal images of wild-type (wt) and bocks mutant third-instar larval salivary gland nuclei stained for DNA (DAPI), Lamin C (green), HP1 (red), and DAPI (gray), with the merged image shown on the right. Bottom: confocal images of wild-type (wt) and bocks mutant third-instar larval salivary gland nuclei stained for DNA (DAPI), lamin Dm0 (green), FG-repeat containing nuclear porins (FG-Nups, red), and the merged image. The lamin Dm0 staining shows O-shaped deformities in the bocks−/− nuclear lamina, evident in the higher magnification inset in the bottom left corner. The prevalence of any deformities, including solid lamin Dm0 accumulations in bocks +/+, is noted in the bottom right corner of lamin Dm0 images. The total number of nuclei analyzed from a minimum of five animals is in parentheses. (E) Confocal images of ventral nerve cord cells from wild-type (wt) and bocks−/− mutant third-instar larvae stained for lamin Dm0 (green) and DAPI (gray). These diploid cells show rare deformities (arrowhead) in the nuclear lamina, with a prevalence of ∼1%. Genotypes are noted at the top. Scale bars, 10 µm.
qPCR analysis of gene expression
Ten wandering, late third-instar larvae were collected, and frozen at −80° and RNA was isolated using TRIzol (Invitrogen). Total RNA was DNAse I treated using DNA-free (Ambion) and reverse transcribed using high-capacity cDNA kit with random hexamer primers (Applied Biosystems). Cycle threshold values were normalized to the housekeeping gene, RpL32. Fold-enrichment was calculated using the ΔΔCT method (Livak and Schmittgen 2001). Primer pairs for RNA quantification are listed 5′ to 3′ for RpL32 (forward AAGATGACCATCCGCCCAGCATAC and reverse ACGCACTCTGTTGTCGATACCCTTG), heat shock factor (forward TTCTCAGCGCCACTGTATTT and reverse GATGTGCCCACCAACAGTA), CG8312 Up (forward CAACACCTACAATGCCAAGAAG and reverse TCAGGGTTATGACGTTCGTG), and CG8312 Dn (forward AGCCTCCTGCCCACTATTA and reverse CCGCATCTTTCTCATCGAACT).
Western and immunohistochemical analyses
Polyclonal goat anti-Otefin antibodies were generated against the amino-terminal 187 amino acids of Otefin. The coding sequence for these amino acids was amplified from y1w67c23 genomic DNA and cloned into the pET21-a (Novagen) bacterial expression vector. The purified His-tagged Ote fragment was used for immunization in goats (Elmira Biologicals). The resulting serum was affinity purified using bacterially expressed, full length, His-tagged Otefin (Actigel, Sterogen).
Western analyses were completed on adult protein extracts. These proteins were separated on 10% SDS polyacrylamide gels and transferred to 0.2-µm nitrocellulose membrane at 70 V for 1 hr. After blocking in PBS with 5% milk, 0.1% Tween, membranes were incubated overnight in one of the following primary antibodies: guinea pig anti-Bocks [1:2,000; (Wagner et al. 2004)], mouse anti-lamin Dm0 (DSHB ADL84.12, 1:1,000), mouse anti-Lamin C (DSHB LC28.26, 1:1,000), sheep anti-dMAN1 [1:50; (Pinto et al. 2008)], and goat anti-Otefin (1:1000). Primary antibodies were detected with HRP-conjugated secondary antibodies (1:20,000, Sigma) and detected by chemiluminescence (SuperSignal West Pico, Pierce). Blots were stripped by 30 min of agitation at 40° in 10% SDS, 1% β-mercaptoethanol in PBS and reblotted for anti-α-tubulin (Sigma, T5168, 1:20,000) to serve as a loading control.
Immunohistochemical staining of adult ovaries was performed as described in Baxley et al. (2011). Nonmuscle larval tissues were fixed in 4% EM-grade paraformaldehyde, followed by a 1-hr wash in 1% TritonX-100 in PBS. Larval body wall muscles were prepared for staining as described in Budnik et al. (1990). The following primary antibodies were used for immunohistochemical analyses: mouse anti-Lamin C (DSHB LC28.26, 1:200), rabbit anti-heterochromatic protein 1 (HP1, Covance PRB291C, 1:400), rabbit anti-lamin Dm0 (Fisher, 1:200), mouse MAb414, which recognizes FG-repeat-containing nucleoporins (FG-Nups, Covance, 1:100 for nonmuscle tissue and 1:3000 for muscles), mouse anti-phospho-histone 3 (pHH3, Millipore 06-570, 1:200), rabbit anti-vasa (Santa Cruz sc-30210, 1:300), and mouse anti-spectrin (DSHB 3A9, 1:50).
Lethal phase analysis of lem-d double mutants
Heterozygous balanced lem-d double mutant stocks were generated. The genotypes of these stocks were (1) y1w67c23; otePK/CyO-y+; bocksΔ66/TM6B-Tb, Hu; (2) y1w67c23; oteB279G/CyO-y+; bocksΔ10/TM6B-Tb, Hu; (3) y1w67c23; otePK, dMAN1Δ26/CyO-y+; (4) y1w67c23; oteB279G, dMAN1Δ81/CyO-y+; (5) y1w67c23; dMAN1Δ81/CyO-y+; bocksΔ10/TM6B-Tb, Hu; (6) y1w67c23; dMAN1Δ26/CyO-y+; bocksΔ66/TM6B-Tb, Hu. Crosses between males and females from different balanced heterozygous stocks were set up in bottles. After an acclimation period, eggs were collected on yeasted orange juice plates for 24 hr and incubated at 25° for 24 hr. Hatched second-instar larval progeny were collected from these plates and genotyped, using the yellow marker to identify the second chromosome CyO, y+ balancer, and the Tubby marker (Tb) to identify the third chromosome TM6b, Tb, Hu balancer. Heterozygous and homozygous lem-d mutants of distinct genotypes were placed into different vials and allowed to develop at 25°. After 2 weeks, the number of pupae and adults produced in each vial were scored, leading to determination of the percentage survival of lem-d mutants during larval and pupal stages of development.
Complementation by ectopic expression
For each nuclear lamina LEM-D protein, a heat-inducible expression transgene was constructed. The lem-d cDNAs were cloned downstream of the heat shock 70 promoter present within a white-marked P transposon. In total, four expression transposons were made, including (1) P[hsp70:ote], (2) P[hsp70:dMAN1], (3) P[hsp70:bocks α], and (4) P[hsp70: bocks β]. Lines carrying each P[hsp70:lem-d] transposon were generated by P-element-mediated germline transformation. To assess the capacity of ectopically produced LEM-D proteins to rescue mutant phenotypes associated with dMAN1 and Ote loss, we established stocks in which each P[hsp70:lem-d] transgene was crossed into ote−/− or dMAN1−/− mutant background. The resulting y1w67c23; ote-/CyO, y+; P[hsp70:lem-d] or y1w67c23; dMAN1-/ CyO, y+; P[hsp70:lem-d] flies were crossed to y1w67c23; ote-/CyO or y1w67c23; dMAN1-/CyO flies to generate lem-d−/−, P[hsp70:lem-d] progeny. After 2 days at 25°, progeny received heat shock daily in a 37° water bath for 1 hr and returned to incubation at 25° until eclosion. The emerged lem-d−/−, P[hsp70:lem-d] adults were assessed for complementation of dMAN1 mutant phenotypes by examination of wing patterning and complementation of ote mutant phenotypes by quantification of egg production and immunohistochemical analysis of ovaries.
Results
Generation of bocks mutants
To understand the functional relationship between the Drosophila nuclear lamina LEM-D proteins, we needed to generate mutations in the bocks gene, as none existed. This gene encodes two isoforms that differ in the amino acid composition of their carboxyl-terminal domains. The Bocks α-isoform is the larger isoform, carrying a transmembrane domain in its unique C terminus that targets this protein to the nuclear envelope. In contrast, the smaller Bocks β-isoform lacks a transmembrane domain and localizes primarily to the nucleoplasm (Wagner et al. 2004). The bocks gene resides in a gene dense region, located downstream of the divergently transcribed CG8312 gene and overlapping with the P58IPK gene (Figure 2A). We generated mutations in the bocks gene through mobilization of a P transposon inserted within the 5′-untranslated region of the parental bocksCB03586 gene. This strategy produced two bocks deletion alleles, bocksΔ10 and bocksΔ66. Deletions in both alleles extend 5′ from +11, with bocksΔ10 carrying a 344-bp deletion and bocksΔ66 carrying a 728-bp deletion (Figure 2A). Western analysis revealed that both alleles fail to accumulate Bocks protein (Figure 2B), demonstrating that two null bocks alleles were generated.
The upstream CG8312 gene resides close to bocks. Three promoters regulate CG8312 transcription, positioned ∼0.3, ∼0.9, and ∼11.8 kb upstream of the bocks transcription start site. Both bocks deletion alleles remove the first CG8312 promoter. In bocksΔ10/Δ10, 609 bp of 5′ sequences remain upstream of the second CG8312 promoter, while in bocksΔ66/Δ66, 225 bp remain. To address whether these deletions alter CG8312 transcription, we isolated RNA from bocks+/+, bocksΔ10/Δ10, and bocksΔ66/Δ66 late third-instar larvae and measured levels of CG8312 RNA using quantitative real-time PCR (Supporting Information, Figure S1). Two primers sets quantified CG8312 RNA, evaluating levels of RNA produced from all promoters or only the first two promoters. In bocksΔ10/Δ10 larvae, CG8312 RNA levels were unchanged, implying that the first promoter is not used during this stage of development. In bocksΔ66/Δ66 larvae, CG8312 RNA levels were reduced by 30%, due to decreased transcription from the second CG8312 promoter. Taken together, these data suggest that phenotypes observed in bocksΔ10/Δ10 or bocksΔ10/Δ66 mutants are due to loss of Bocks function.
Once bocks mutations were available, we defined effects of Bocks loss on development. To this end, we crossed y1w67c23; bocksΔ10/Sb and y1w67c23; bocksΔ66/Sb females and males and determined the number of bocks+/− (Sb) and bocks−/− (non-Sb) progeny. We found that bocks−/− adults were obtained at the expected number, with these adults displaying normal morphology and fertility (data not shown). These experiments demonstrate that Bocks is not essential for Drosophila development. Previous studies showed that depletion of Bocks in cultured Drosophila cells had no effect on the formation of the nuclear lamina (Wagner et al. 2004). Similarly, we found that in vivo loss of Bocks did not alter the accumulation or localization of many nuclear lamina components (Figure 2, C and D). We found that localization of Lamin C, the FG-repeat nuclear pore proteins (FG-Nups), and Heterochromatin Protein 1 (HP1) was normal in bocks−/− third-instar larval tissues (Figure 2D), implying that Bocks is not required for general maintenance of the nuclear lamina structure. We did detect an abnormal lamin Dm0 distribution in many bocks−/− salivary gland polytene nuclei (43% relative to 7% in bocks+/+ salivary gland nuclei), but at a much lower level in diploid nuclei (1%). In the abnormal bocks−/− salivary gland nuclei, the lamin Dm0 was found in one to seven O-shaped lamin Dm0-containing structures, which lacked lamin C and DNA (Figure 2D). In contrast, the abnormal bocks+/+ salivary gland nuclei contained solid structures. Similarly, O-shaped structures were observed in salivary gland nuclei that express mutant forms of Lamin C, although the number of lamin aggregates in bocks−/− nuclei is lower (Schulze et al. 2005, 2009). These observations suggest that loss of Bocks may weaken the nuclear lamina in large nuclei, causing the irregularities found in salivary gland nuclei.
At least two LEM-D proteins are required for development
To understand redundancy among Drosophila nuclear lamina LEM-D proteins, we studied developmental and cellular changes associated with the simultaneous loss of pairs of LEM-D proteins. In all cases, homozygous lem-d double mutants were produced from crosses of balanced heterozygous lem-d double mutant males and females, generating offspring that carried heteroallelic combinations of mutant alleles of each lem-d gene. In this way, we eliminated confounding recessive effects of second-site mutations on homozygous mutant chromosomes.
To define the developmental potential of lem-d double mutants, we collected and genotyped second-instar larvae resulting from crosses of balanced heterozygous lem-d double mutants. These larvae were placed into separate vials and the percentage of larvae developing into pupae and adults was determined. As a control, we conducted the same experiment with second-instar larvae collected from crosses of balanced heterozygous single lem-d mutants. In each case, individual lem-d mutant second-instar larvae survived at levels similar to those of the wild-type control (Figure 3). In contrast, lem-d double mutants showed decreased survival. Several observations were made from these experiments. First, heterozygous combinations of some lem-d mutant alleles reduced adult survival (Figure S2A). For example, ote+/−; bocks+/− and dMAN1+/−; bocks+/− heterozygotes had reduced viability, while ote+/−, dMAN1+/− heterozygotes had wild-type survival (Figure S2A). Further, dMAN1−/−; bocks+/− adults were never recovered, even though dMAN1−/−, ote+/− adults were recovered at nearly the same level as dMAN1−/− single mutants (Figure S2B). These observations suggest that Bocks has a critical role in buffering the developmental consequences resulting from loss of dMAN1 or Ote. Second, no homozygous lem-d double-mutant adults were recovered (Figure 3). These data demonstrate that at least two nuclear lamina LEM-D proteins are needed for development. Interestingly, pairs of homozygous lem-d double mutants displayed distinct stages of lethality. While only ∼10% of the ote−/−; bocks−/− second-instar larvae formed pupae, ∼60% of dMAN1−/−, ote−/− and ∼55% of dMAN1−/−; bocks−/− second-instar larvae formed pupae (Figure 3). Together, these experiments reveal that pairs LEM-D proteins share distinct developmental functions, which are essential for viability. We suggest that the shared function(s) of Ote and Bocks are needed during larval stages of development, whereas the critical function(s) that Ote and Bocks share with dMAN1 are needed during pupal development.
Figure 3.

At least two nuclear Drosophila LEM-D proteins are required for adult survival. Shown is a graph of the percent survival of collected second-instar larvae (L) to pupae (P) and adults (A) of the indicated genotypes, including the following combinations of lem-d alleles: oteB279G/PK, dMAN1Δ81/Δ26, and bocks Δ10/Δ66. The total number of second-instar larvae analyzed is listed above each set of bars. At least three independent experiments were completed, with error bars corresponding to standard deviation. Student’s t-test compared the percentage survival of lem-d double mutants and the wild-type control at the matching developmental stages (* P < 0.05, ** P < 0.01).
All LEM-D proteins interact with BAF (Segura-Totten and Wilson 2004; Pinto et al. 2008). We were interested in understanding whether the lethality associated with lem-d double mutants was linked to altered BAF function. Previous studies showed that baf−/− mutants fail to develop beyond the larval–pupal transition (Furukawa et al. 2003). In baf−/− larvae, endocyclic larval tissues that undergo DNA replication without cell division were of a normal size, but mitotically active tissues, such as brains and imaginal discs were either small or absent, respectively. Further, nuclei in baf−/− tissues were misshapen, showing a convoluted nuclear lamina and intranuclear lamin Dm0 accumulation (Furukawa et al. 2003). We reasoned that if lem-d double-mutant lethality were related to loss of BAF function, then lem-d double mutants would show baf mutant phenotypes. First, we compared development of lem-d double mutants with baf mutants. Our studies revealed that ote−/−; bocks−/− mutants die earlier than baf−/− mutants, while survival of dMAN1−/−, ote−/− and dMAN1−/−; bocks−/− mutants were similar (Furukawa et al. 2003). The earlier lethality of ote−/−; bocks−/− mutants implies that the critical function(s) shared by Ote and Bocks extend beyond BAF regulation. Second, we assessed cell proliferation of lem-d double-mutant brain and in imaginal discs that ultimately form adult tissues. Proliferation was assessed using an antibody against phosphorylated serine 10 of histone H3 (pHH3), a selective marker for mitosis (Hendzel et al. 1997). Unlike baf−/− mutants, the ote−/−, dMAN1−/− and the dMAN1−/−; bocks−/− larvae had normal sized brains and imaginal discs with extensive pHH3 staining (Figure 4). However, similar to baf−/− (Furukawa et al. 2003), ote−/−; bocks−/− larval brains were small with little pHH3 staining, and imaginal disc tissues were small or absent (Figure 4). These experiments indicate that cells require either Ote or Bocks for mitotic growth. Third, we stained lem-d double-mutant tissues with antibodies against lamins, HP1, and the FG-Nups to examine nuclear organization. In most lem-d double-mutant tissues, nuclei had a uniform nuclear lamina and normal HP1 localization (Figure 5). Again, salivary gland nuclei were exceptional, wherein bocks−/− double mutants had nuclei with an abnormal localization of lamin Dm0 (Figure 5A). Additionally, many ote−/−, dMAN1−/− nuclei (∼20%) had abnormal lamin Dm0 structures (Figure 5A), although these were solid structures instead of the O-shaped structures found in bocks−/− mutants. These observations reinforce the proposal that giant polyploid nuclei are sensitive to loss of nuclear lamina components. Even so, no cytoplasmic or intranuclear accumulation of lamins or FG-Nups was observed, implying that the nuclear lamina is largely maintained in lem-d double mutants. Taken together, our data suggest that mutant phenotypes associated with loss of two LEM-D proteins do not match those found with loss of BAF. Based on the lack of aligned phenotypes, we conclude that loss of BAF function may not be a primary cause of lethality in lem-d double mutants.
Figure 4.

Cellular phenotypes associated with loss of two LEM-D proteins. Left: Schematic of the larval central nervous system and imaginal discs tissues, showing the antennal disc (AD), the eye disc (ED), the optic lobe (OL), the central brain (CB), the ventral nerve cord (VNC), and imaginal discs (IDs). Right: Confocal images of larval tissues stained with DAPI (gray) and antibodies against the mitotic marker phospho-histone H3 (pHH3, red). Tissues were obtained from wild-type (wt) and lem-d double mutants (oteB279G/PK, dMAN1Δ81/Δ26; oteB279G/PK; bocks Δ10/Δ66; dMAN1Δ81/Δ26; bocks Δ10/Δ66) third-instar larvae. Scale bars, 100 µm.
Figure 5.

The nuclear lamina remains intact in lem-d double mutants. (A) Shown are confocal images of wild-type (wt) and lem-d double mutant nuclei of third-instar larval salivary glands. Nuclei were stained for DNA (DAPI, gray), Lamin C, or lamin Dm0 (green), HP1 (red), or FG-Nups (red). The lamin Dm0 staining of salivary glands isolated from bocks−/− double mutants showed O-shaped lamin Dm0 deformities, while the wild-type and ote−/−, dMAN1−/− nuclei have solid aggregates of lamin Dm0 (boxed regions). Numbers in the bottom right corner indicate the prevalence of these deformities. The number of nuclei analyzed from a minimum of five animals is in parentheses. (B) Confocal images of diploid imaginal disc tissues isolated from wild-type (wt) and lem-d double-mutant larvae, stained for lamin Dm0 (green) and DAPI (gray). (C) Confocal images of third-instar larvae body wall muscle nuclei isolated from wt and lem-d double mutants. Muscles were stained with DAPI (blue), antibodies against FG-Nups (green), and the F-actin stain phalloidin (red). Genotypes are noted above all image sets. Scale bars, 10 µm.
LEM-D proteins directly interact with Lamin C and lamin Dm0 (Pinto et al. 2008; Schulze et al. 2009). Previous studies revealed that muscle-specific production of mutant Lamin C isoforms causes semilethality, with body wall muscles in larvae showing the formation of cytoplasmic aggregates of nuclear lamina components, such as the FG-Nups (Dialynas et al. 2012). We wondered whether lethality of lem-d double mutants might be similarly related to muscle-specific dysfunction. For this reason, we stained body wall muscles in lem-d double mutants with antibodies against the FG-Nups. We found that mutant nuclei had uniform nuclear lamina, with no evidence of cytoplasmic aggregates (Figure 5C). These studies suggest that LEM-D proteins are not required for maintenance of the nuclear lamina in muscle nuclei. Larval body wall muscles form during embryogenesis when maternally contributed LEM-D proteins are present. Thus, it remains possible that LEM-D proteins contribute to the formation of nuclear lamina in muscle nuclei.
Increased expression of Bocks rescues GSC loss in ote−/− ovaries
Loss of dMAN1 or Ote causes tissue-restricted adult phenotypes (Jiang et al. 2008; Pinto et al. 2008; Wagner et al. 2010; Barton et al. 2013). We reasoned that such phenotypes might result because of cell-specific differences in the nuclear lamina, wherein affected cells may have insufficient amounts of a compensating LEM-D protein. To test this prediction, we determined whether the dMAN1−/− or ote−/− mutant phenotypes were rescued by increased production of a different LEM-D protein. For these experiments, we generated dMAN1−/− and ote−/− flies with a single copy of a P[hsp70:lem-d] transgene. Four trangenes were generated, with each encoding one nuclear lamina LEM-D protein (dMAN1, Ote, the Bocks α-isoform or the Bocks β-isoform) under the control of the heat shock 70 promoter (hsp70). For each lem-d−/−, P[hsp70:lem-d] line, animals were subjected to a daily heat treatment beginning at the second-instar larval stage of development and the degree of the tissue-specific rescue was assessed in adult progeny (Figure 6).
Figure 6.

Expression of bocks rescues the ovary requirement for Otefin. (A) Top: Schematic of the strategy to assess rescue of the wing-patterning defect in dMAN1−/− mutants. Daily heat shocks were given to dMAN1−/−, P[hsp70:lem-d] beginning at second-instar larvae and continued until eclosion. Wing phenotypes of adults were assessed. Bottom: Quantification of the percentage of wild type (wt) wing patterning in non-heat-shocked (gray bars) and heat shocked (black bars) adults carrying either no (none) or the indicated P[hsp70:lem-d] transgene. (B) Top: Schematic of the strategy to assess rescue of the egg-production defect in ote−/− mutants. Daily heat shocks were given to ote−/−, P[hsp70:lem-d] beginning at second-instar larvae and continued to eclosion. Bottom: Quantification of eggs produced per female per day by wild type and ote−/− mutants that were not heat shocked (gray bars) or heat shocked (black bars). The number of adults analyzed is indicated above each bar. Error bars represent standard deviation from a minimum of three independent experiments. Student’s t-test P-values were obtained by comparing data obtained from comparisons between adults of the indicated genotype to dMAN1−/− (A) or ote−/− (B) adults raised under the same conditions. (* P < 0.05, ** P < 0.01).
Rescue of the wing patterning defect in dMAN1−/− mutants was used to assess the ability of individual Bocks isoforms or Ote to compensate for dMAN1 loss. All dMAN1−/− adults have wings with thick longitudinal veins, a variable number of anterior cross veins, branching of posterior cross veins, and folds in the blade, a phenotype not shared with the other lem-d mutants. As a control, we tested whether wing patterning defects were rescued by heat treatment of dMAN1−/−, P[hsp70:dMAN1] animals. We found that all dMAN1−/−, P[hsp70:dMAN1] adults had a normal wing phenotype, indicating that a functional level of dMAN1 protein was produced (Figure 6A). Low levels of rescue of the wing patterning defects (∼15%) were also observed in non-heat-treated dMAN1−/−, P[hsp70:dMAN1] adults, implying that basal expression of the P[hsp70:dMAN1] transgene is sufficient for phenotypic rescue. Next, we examined wing phenotypes of heat-treated and dMAN1−/−, P[hsp70:ote], dMAN1−/−, P[hsp70:bocks α] and dMAN1−/−, P[hsp70:bocks β] adults. In all cases, wing-patterning defects remained, implying that ectopic production of Ote or Bocks cannot rescue dMAN1 loss (Figure 6A). As wing defects in dMAN1 mutants result from upregulation of BMP signaling (Wagner et al. 2006; Pinto et al. 2008), we conclude that increased expression of neither Ote nor Bocks can restore this signaling in dMAN1 mutants.
Rescue of the egg laying defect in ote−/− mutants was used to assess the ability of individual Bocks isoforms or dMAN1 to compensate for Ote loss. All ote−/− females are sterile due to defects in GSC survival (Jiang et al. 2008; Barton et al. 2013), a phenotype not shared with the other lem-d mutants. As a control, we tested whether egg production was restored by heat treatment of ote−/−; P[hsp70:ote] animals. We found that all ote−/−; P[hsp70:ote] females laid eggs (Figure 6B; Figure S3), a rescue that required heat treatment. Next, we tested whether egg production was restored by heat treatment of other ote−/−, P[hsp70:lem-d] animals. Strikingly, we found that increased production of both Bocks isoforms, but not dMAN1, rescued egg laying (Figure 6B; Figure S3), demonstrating that Ote and Bocks share tissue-specific functions that are distinct from dMAN1.
We extended our ote−/− rescue studies to investigate the relationship between egg laying and GSC phenotypes. We reasoned that if Ote made multiple contributions during oogenesis, then increased production of other LEM-D proteins might rescue GSC phenotypes, even though no or few eggs were produced. In Drosophila ovaries, GSCs reside within the germarium, a structure comprised of somatic cells niche cells and two to three GSCs (Chen et al. 2011; Harris and Ashe 2011; Losick et al. 2011). In young ote−/− females, ovaries display a complex germarial phenotype, wherein some germaria carry no germ cells, termed GSC loss, and some germaria carry more than five GSC-like cells, termed GSC expansion (Barton et al. 2013). To determine the ability of other LEM-D proteins to rescue these phenotypes, ovaries obtained from ote−/−, P[hsp70:lem-d] heat-treated females were stained with antibodies against Vasa, a germline-specific helicase, and Spectrin, a cytoskeletal protein that localizes to the cytoplasmic periphery in somatic cells and forms a spherical structure called the spectrosome in GSCs (Lin et al. 1994). As a control for temperature effects, we stained heat-treated ote−/− ovaries with Vasa and Spectrin. Surprisingly, these studies revealed that heat treatment alone significantly reduced GSC loss in ote−/− ovaries (Figure S4). Even so, differentiation of the ote−/− germ cells remained blocked (Figure S4). The reason for the rescue of GSC loss is unknown. In ovaries obtained from heat-treated ote−/−, P[hsp70:ote] females, both GSC loss and germ-cell differentiation were rescued (Figure S3 and Figure S4), consistent with the heat-dependent rescue of egg production.
Next, we examined the germarial phenotypes in ovaries obtained from heat-treated ote−/− females carrying the Bocks α-isoform, the Bocks β-isoform, or dMAN1 expression transgene. Increased expression of the Bocks α and Bocks β-isoforms, but not dMAN1, restored GSC numbers and germ-cell differentiation in half of the analyzed germaria (Figure S4), reinforcing the finding that only Bocks is compensating for mutant GSC phenotypes associated with Ote loss. These data imply that the unique requirement for Ote in GSCs reflects insufficient levels of Bocks within the nuclear lamina to buffer against Ote loss. To determine whether Bocks contributes to nuclear lamina function in early stages of germ-cell development, we examined larval gonads. Previous studies have shown that loss of Ote reduces the number of primordial germ cells (PGCs) in the third-instar larval gonads (Barton et al. 2013). To understand whether Bocks protects PGCs from Ote loss, we stained lem-d double-mutant larval gonads with antibodies against Vasa and Spectrin (Figure 7). We found that ote−/−; bocks−/− gonads had a significantly lower number of PGCs relative to ote−/− or other lem-d double-mutant gonads (Figure 7B). Further, DAPI staining revealed pyknotic nuclei, indicating that PGC loss is associated with increased cell death in ote−/− and in the ote−/−; bocks−/− gonads (Figure 7A insets). Taken together, our data provide strong evidence that Ote and Bocks are functional homologs, with both proteins contributing to germ-cell survival during development.
Figure 7.

Bocks contributes to survival of primordial germ cells in the developing gonad. (A) Top left shows a schematic of a third-instar larval female gonad, indicating somatic cells (green and blue) and primordial germ cells (PGCs, red). Rest of A shows larval female gonads obtained from wild-type (wt) and lem-d double mutants stained for DNA (DAPI, blue), the germline specific helicase Vasa (red) and the cytoskeletal protein Spectrin (green). Boxed areas are magnified below to show pyknotic nuclei indicative of apoptosis. Genotypes are noted above each larval ovary image. Scale bars, 25 µm. (B) Box plots of the number of PGCs per lem-d wild-type (none) and double-mutant gonad, with the total number of gonads analyzed for each genotype is indicated above each box plot. For each box, the white line indicates the median PCG number, while boxes and whiskers represent the 25th to 75th percentile interval and the nonoutlier range, respectively. Student’s t-tests compared differences in PGC numbers between lem-d double mutants and controls, with P-values between compared pairs shown.
Discussion
Metazoan genomes encode multiple LEM-D proteins that show enriched localization within the nuclear lamina (Lee and Wilson 2004; Berk et al. 2013). Drosophila has three nuclear lamina LEM-D proteins, corresponding to dMAN1, Ote, and Bocks. Genetic studies demonstrated that neither dMAN1 nor ote are essential genes, with mutations in these genes causing nonoverlapping tissue-restricted developmental defects, some of which worsen with age (Jiang et al. 2008; Pinto et al. 2008; Wagner et al. 2010; Barton et al. 2013). Here, we investigated the developmental requirement of the remaining nuclear lamina lem-d gene, bocks. We demonstrate that bocks is a nonessential gene, with bocks−/− adults showing normal morphology, viability, and fertility (Figure 2; data not shown). The absence of mutant phenotypes in bocks−/− adults was surprising, given that expression of bocks is highest of all lem-d genes during development (Chintapalli et al. 2007) and that bocks is the only lem-d gene that displays alternative splicing (Wagner et al. 2004). Our data imply that the developmental functions of the nuclear lamina can be maintained, even when total levels of LEM-D proteins are significantly reduced.
Shared LEM-D protein functions extend beyond BAF regulation
We investigated the extent of such regulatory compensation among the three Drosophila nuclear lamina LEM-D proteins, using genetic and phenotypic analyses of lem-d double mutants. Our studies revealed that loss of any two LEM-D proteins causes lethality before adulthood (Figure 3). These data show that functional redundancy exists among the Drosophila LEM-D proteins, as seen for LEM-D proteins in other organisms (Gruenbaum et al. 2002; Liu et al. 2003; Huber et al. 2009; Barkan et al. 2012; Gonzalez et al. 2012; Reil and Dabauvalle 2013). Interestingly, these studies revealed that although loss of Bocks alone did not affect development, Bocks has multiple roles within the nuclear lamina. We identified dominant genetic interactions only between bocks and the other lem-d mutants. For example, ote+/−; bocks+/− and dMAN1+/−; bocks+/− double heterozygous adults showed reduced viability, while ote+/−, dMAN1+/− double heterozygous adults did not. Further, no dMAN1−/−; bocks+/− adults survived, whereas dMAN1−/−, ote+/− adults survived at wild-type levels (Figure S2). The decreased survival of both dMAN1 and ote mutants when Bocks levels are reduced suggests that Bocks compensates for the loss of both proteins. The absence of dominant interactions between dMAN1 and ote mutants suggests that Bocks compensates for different regulatory functions lost in the ote and dMAN1 mutants. The dominant interactions observed with bocks−/− mutants suggests that Bocks provides the strongest compensation for loss of other LEM-D proteins, which may be linked to the higher levels of Bocks in the nuclear lamina (Chintapalli et al. 2007).
The hallmark feature of the LEM-D protein family is BAF binding. For this reason, we postulated that the lethality caused by loss of two LEM-D proteins was linked to changes in BAF function. Loss of BAF causes a typical mitotic mutant phenotype (Furukawa et al. 2003). Lethality of baf mutants occurs at the larval–pupal transition, with larvae carrying small brains and no imaginal discs. These defects are linked to an altered nuclear lamina. Our studies uncovered striking differences between baf and lem-d double mutants. First, ote−/−, dMAN1−/− and dMAN1−/−; bocks−/− larvae had large brains and imaginal discs and showed high levels of mitosis (Figure 4A). Second, although ote−/−; bocks−/− tissues showed low levels of mitosis as found in baf−/− tissues, the ote−/−; bocks−/− mutant phenotypes are different because these mutants die earlier and have nuclei with a normal nuclear lamina structure (Figure 4A and Figure 5). Taken together, these observations suggest that some BAF function is retained in animals with only a single nuclear lamina LEM-D protein and predict that lethality found in the lem-d double mutants extends beyond loss of BAF regulation.
How the nuclear lamina LEM-D proteins compensate for loss of a different family member remains unclear. Outside of the LEM-D, none of these proteins show extensive homology (Figure 1B). Nonetheless, all three proteins carry small regions of 8–12 amino acids that show homology to each other (data not shown). Further experimentation is needed to establish the role of these regions in the function of the LEM-D proteins.
Bocks and Ote are functional homologs
As a second approach toward understanding the extent of regulatory compensation among the Drosophila nuclear lamina proteins, we determined whether increased expression of one LEM-D protein rescued the mutant phenotypes associated with loss of a different LEM-D family member. These experiments revealed that increased amounts of Bocks, but not dMAN1, rescued ote−/− phenotypes, whereas neither Bocks nor Ote could rescue a dMAN1−/− phenotype (Figure 6). We draw several conclusions from these studies. First, we infer that levels of Bocks are limiting in GSCs, thereby sensitizing these cells to Ote loss. Second, we surmise that the Ote function in GSCs is not linked to BAF regulation, because increased dMAN1 expression failed to rescue Ote loss. Third, we infer that dMAN1 contributes regulatory functions distinct from other LEM-D family members, because wing-patterning defects were not rescued by increased expression of Ote or Bocks. Based on these experiments, we suggest that Bocks and Ote are functional homologs. These findings imply that the Drosophila nuclear lamina LEM-D family is composed of one MAN1 and two emerin homologs.
We were surprised to discover that the Bocks β-isoform provided partial rescue of ote−/− fertility (Figure 6B). Previous studies have shown that Ote function in GSCs requires the C-terminal peripheral localization domain (Jiang et al. 2008), indicating a requirement for nuclear lamina localization. Yet, the Bocks β-isoform lacks the transmembrane domain required for nuclear envelope targeting, which should be incompatible with rescue of ote−/− phenotypes. However, as the Bocks β-isoform directly interacts with lamins (Pinto et al. 2008), we reason that these protein associations may be sufficient to target this protein to the nuclear lamina for the rescue of the ote−/− phenotypes.
Multiple factors contribute to tissue-specific defects caused by loss of LEM-D proteins
Loss of LEM-D proteins cause several age-enhanced, tissue-restricted human diseases (Worman et al. 2010). Our genetic analyses provide insight into mechanisms that may limit effects of individual LEM-D protein loss to specific tissues. First, our studies support previous findings that LEM-D proteins possess overlapping developmental functions. Importantly, our results suggest that these shared requirements extend beyond BAF regulation. These findings imply that functional motifs outside of the LEM-D share regulatory interactions, even though these regions display limited sequence homology. Second, we find that some LEM-D proteins may possess cell-type-specific functions that are not redundant with other family members. Third, we uncovered evidence that certain cell types may express limiting levels of individual LEM-D proteins, which sensitize the cell toward loss of other LEM-D family members. Taken together, our studies indicate that multiple factors contribute to the restricted nature of LEM-D diseases.
Acknowledgments
We thank Isabel Algaze-Gonzalez, William Barry, Emma Hornick, Jessica Ponce, and Eric Schultz for technical assistance. We thank Georg Krohne and Paul Fisher for the generous gift of antibodies. We thank members of the labs for helpful discussions and comments on the manuscript. We thank the University of Iowa Central Microscopy Research Facility for help in imaging and the use of the Zeiss LSM710, acquired via National Institutes of Health (NIH) (S10 RR025439-01). This work was supported by NIH R01 grant (GM087341) to P. Geyer.
Footnotes
Communicating editor: J. A Birchler
Literature Cited
- Ashery-Padan R., Ulitzur N., Arbel A., Goldberg M., Weiss A. M., et al. , 1997a Localization and posttranslational modifications of otefin, a protein required for vesicle attachment to chromatin, during Drosophila melanogaster development. Mol. Cell. Biol. 17: 4114–4123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashery-Padan R., Weiss A. M., Feinstein N., Gruenbaum Y., 1997b Distinct regions specify the targeting of otefin to the nucleoplasmic side of the nuclear envelope. J. Biol. Chem. 272: 2493–2499 [DOI] [PubMed] [Google Scholar]
- Barkan R., Zahand A. J., Sharabi K., Lamm A. T., Feinstein N., et al. , 2012. Ce-emerin and LEM-2: essential roles in Caenorhabditis elegans development, muscle function, and mitosis. Mol. Biol. Cell 23: 543–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton L. J., Pinto B. S., Wallrath L. L., Geyer P. K., 2013. The Drosophila nuclear lamina protein otefin is required for germline stem cell survival. Dev. Cell 25: 645–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxley R. M., Soshnev A. A., Koryakov D. E., Zhimulev I. F., Geyer P. K., 2011. The role of the Suppressor of Hairy-wing insulator protein in Drosophila oogenesis. Dev. Biol. 356: 398–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk J. M., Tifft K. E., Wilson K. L., 2013. The nuclear envelope LEM-domain protein emerin. Nucleus 4: 298–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachner A., Braun J., Ghodgaonkar M., Castor D., Zlopasa L., et al. , 2012. The endonuclease Ankle1 requires its LEM and GIY-YIG motifs for DNA cleavage in vivo. J. Cell Sci. 125: 1048–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik V., Zhong Y., Wu C. F., 1990. Morphological plasticity of motor axons in Drosophila mutants with altered excitability. J. Neurosci. 10: 3754–3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buszczak M., Paterno S., Lighthouse D., Bachman J., Planck J., et al. , 2007. The carnegie protein trap library: a versatile tool for Drosophila developmental studies. Genetics 175: 1505–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai M., Huang Y., Ghirlando R., Wilson K. L., Craigie R., et al. , 2001. Solution structure of the constant region of nuclear envelope protein LAP2 reveals two LEM-domain structures: one binds BAF and the other binds DNA. EMBO J. 20: 4399–4407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Wang S., Xie T., 2011. Restricting self-renewal signals within the stem cell niche: multiple levels of control. Curr. Opin. Genet. Dev. 21: 684–689 [DOI] [PubMed] [Google Scholar]
- Chintapalli V. R., Wang J., Dow J. A., 2007. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat. Genet. 39: 715–720 [DOI] [PubMed] [Google Scholar]
- Dialynas G., Flannery K. M., Zirbel L. N., Nagy P. L., Mathews K. D., et al. , 2012. LMNA variants cause cytoplasmic distribution of nuclear pore proteins in Drosophila and human muscle. Hum. Mol. Genet. 21: 1544–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittrich C. M., Kratz K., Sendoel A., Gruenbaum Y., Jiricny J., et al. , 2012. LEM-3: a LEM domain containing nuclease involved in the DNA damage response in C. elegans. PLoS ONE 7: e24555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa K., Sugiyama S., Osouda S., Goto H., Inagaki M., et al. , 2003. Barrier-to-autointegration factor plays crucial roles in cell cycle progression and nuclear organization in Drosophila. J. Cell Sci. 116: 3811–3823 [DOI] [PubMed] [Google Scholar]
- Gonzalez Y., Saito A., Sazer S., 2012. Fission yeast Lem2 and Man1 perform fundamental functions of the animal cell nuclear lamina. Nucleus 3: 60–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenbaum Y., Lee K. K., Liu J., Cohen M., Wilson K. L., 2002. The expression, lamin-dependent localization and RNAi depletion phenotype for emerin in C. elegans. J. Cell Sci. 115: 923–929 [DOI] [PubMed] [Google Scholar]
- Harris R. E., Ashe H. L., 2011. Cease and desist: modulating short-range Dpp signalling in the stem-cell niche. EMBO Rep. 12: 519–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendzel M. J., Wei Y., Mancini M. A., Van Hooser A., Ranalli T., et al. , 1997. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma 106: 348–360 [DOI] [PubMed] [Google Scholar]
- Huber M. D., Guan T., Gerace L., 2009. Overlapping functions of nuclear envelope proteins NET25 (Lem2) and emerin in regulation of extracellular signal-regulated kinase signaling in myoblast differentiation. Mol. Cell. Biol. 29: 5718–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X., Xia L., Chen D., Yang Y., Huang H., et al. , 2008. Otefin, a nuclear membrane protein, determines the fate of germline stem cells in Drosophila via interaction with Smad complexes. Dev. Cell 14: 494–506 [DOI] [PubMed] [Google Scholar]
- Korfali N., Wilkie G. S., Swanson S. K., Srsen V., Batrakou D. G., et al. , 2010. The leukocyte nuclear envelope proteome varies with cell activation and contains novel transmembrane proteins that affect genome architecture. Mol. Cell. Proteomics 9: 2571–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laguri C., Gilquin B., Wolff N., Romi-Lebrun R., Courchay K., et al. , 2001. Structural characterization of the LEM motif common to three human inner nuclear membrane proteins. Structure 9: 503–511 [DOI] [PubMed] [Google Scholar]
- Lee K. K., Wilson K. L., 2004. All in the family: evidence for four new LEM-domain proteins Lem2 (NET-25), Lem3, Lem4 and Lem5 in the human genome. Symp. Soc. Exp. Biol. 56: 329–339 [PubMed] [Google Scholar]
- Lin F., Blake D. L., Callebaut I., Skerjanc I. S., Holmer L., et al. , 2000. MAN1, an inner nuclear membrane protein that shares the LEM domain with lamina-associated polypeptide 2 and emerin. J. Biol. Chem. 275: 4840–4847 [DOI] [PubMed] [Google Scholar]
- Lin H., Yue L., Spradling A. C., 1994. The Drosophila fusome, a germline-specific organelle, contains membrane skeletal proteins and functions in cyst formation. Development 120: 947–956 [DOI] [PubMed] [Google Scholar]
- Liu J., Lee K. K., Segura-Totten M., Neufeld E., Wilson K. L., et al. , 2003. MAN1 and emerin have overlapping function(s) essential for chromosome segregation and cell division in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 100: 4598–4603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K. J., Schmittgen T. D., 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)). Method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- Losick V. P., Morris L. X., Fox D. T., Spradling A., 2011. Drosophila stem cell niches: a decade of discovery suggests a unified view of stem cell regulation. Dev. Cell 21: 159–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik P., Korfali N., Srsen V., Lazou V., Batrakou D. G., et al. , 2010. Cell-specific and lamin-dependent targeting of novel transmembrane proteins in the nuclear envelope. Cell. Mol. Life Sci. 67: 1353–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansharamani M., Wilson K. L., 2005. Direct binding of nuclear membrane protein MAN1 to emerin in vitro and two modes of binding to barrier-to-autointegration factor. J. Biol. Chem. 280: 13863–13870 [DOI] [PubMed] [Google Scholar]
- Markiewicz E., Tilgner K., Barker N., van de Wetering M., Clevers H., et al. , 2006. The inner nuclear membrane protein emerin regulates beta-catenin activity by restricting its accumulation in the nucleus. EMBO J. 25: 3275–3285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montes de Oca R., Lee K. K., Wilson K. L., 2005. Binding of barrier to autointegration factor (BAF) to histone H3 and selected linker histones including H1.1. J. Biol. Chem. 280: 42252–42262 [DOI] [PubMed] [Google Scholar]
- Osada S., Ohmori S. Y., Taira M., 2003. XMAN1, an inner nuclear membrane protein, antagonizes BMP signaling by interacting with Smad1 in Xenopus embryos. Development 130: 1783–1794 [DOI] [PubMed] [Google Scholar]
- Pinto B. S., Wilmington S. R., Hornick E. E., Wallrath L. L., Geyer P. K., 2008. Tissue-specific defects are caused by loss of the Drosophila MAN1 LEM domain protein. Genetics 180: 133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju G. P., Dimova N., Klein P. S., Huang H. C., 2003. SANE, a novel LEM domain protein, regulates bone morphogenetic protein signaling through interaction with Smad1. J. Biol. Chem. 278: 428–437 [DOI] [PubMed] [Google Scholar]
- Reil M., Dabauvalle M. C., 2013. Essential roles of LEM-domain protein MAN1 during organogenesis in Xenopus laevis and overlapping functions of emerin. Eur. J. Cell Biol. 92: 280–294 [DOI] [PubMed] [Google Scholar]
- Robertson H. M., Preston C. R., Phillis R. W., Johnson-Schlitz D. M., Benz W. K., et al. , 1988. A stable genomic source of P element transposase in Drosophila melanogaster. Genetics 118: 461–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer E. C., Florens L., Guan T., Yates J. R., 3rd, Gerace L., 2003. Nuclear membrane proteins with potential disease links found by subtractive proteomics. Science 301: 1380–1382 [DOI] [PubMed] [Google Scholar]
- Schulze S. R., Curio-Penny B., Li Y., Imani R. A., Rydberg L., et al. , 2005. Molecular genetic analysis of the nested Drosophila melanogaster lamin C gene. Genetics 171: 185–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze S. R., Curio-Penny B., Speese S., Dialynas G., Cryderman D. E., et al. , 2009. A comparative study of Drosophila and human A-type lamins. PLoS ONE 4: e7564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura-Totten M., Wilson K. L., 2004. BAF: roles in chromatin, nuclear structure and retrovirus integration. Trends Cell Biol. 14: 261–266 [DOI] [PubMed] [Google Scholar]
- Solovei I., Wang A. S., Thanisch K., Schmidt C. S., Krebs S., et al. , 2013. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 152: 584–598 [DOI] [PubMed] [Google Scholar]
- Wagner N., Krohne G., 2007. LEM-Domain proteins: new insights into lamin-interacting proteins. Int. Rev. Cytol. 261: 1–46 [DOI] [PubMed] [Google Scholar]
- Wagner N., Schmitt J., Krohne G., 2004. Two novel LEM-domain proteins are splice products of the annotated Drosophila melanogaster gene CG9424 (Bocksbeutel). Eur. J. Cell Biol. 82: 605–616 [DOI] [PubMed] [Google Scholar]
- Wagner N., Kagermeier B., Loserth S., Krohne G., 2006. The Drosophila melanogaster LEM-domain protein MAN1. Eur. J. Cell Biol. 85: 91–105 [DOI] [PubMed] [Google Scholar]
- Wagner N., Weyhersmuller A., Blauth A., Schuhmann T., Heckmann M., et al. , 2010. The Drosophila LEM-domain protein MAN1 antagonizes BMP signaling at the neuromuscular junction and the wing crossveins. Dev. Biol. 339: 1–13 [DOI] [PubMed] [Google Scholar]
- Wolff N., Gilquin B., Courchay K., Callebaut I., Worman H. J., et al. , 2001. Structural analysis of emerin, an inner nuclear membrane protein mutated in X-linked Emery-Dreifuss muscular dystrophy. FEBS Lett. 501: 171–176 [DOI] [PubMed] [Google Scholar]
- Worman H. J., Ostlund C., Wang Y., 2010. Diseases of the nuclear envelope. Cold Spring Harb. Perspect. Biol. 2: a000760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng R., Ghirlando R., Lee M. S., Mizuuchi K., Krause M., et al. , 2000. Barrier-to-autointegration factor (BAF) bridges DNA in a discrete, higher-order nucleoprotein complex. Proc. Natl. Acad. Sci. USA 97: 8997–9002 [DOI] [PMC free article] [PubMed] [Google Scholar]