

Abstract
Dominant mutations in GJB2 may lead to various degrees of sensorineural hearing impairment and/or hyperproliferative epidermal disorders. So far studies of dominant GJB2 mutations were mostly limited to case reports of individual patients and families. In this study, we identified 7 families, 11 subjects with dominant GJB2 mutations by sequencing of GJB2 in 2168 Chinese Han probands with sensorineural hearing impairment and characterized the associated spectrum, de novo rate and genotype-phenotype correlation. We identified p.R75Q, p.R75W and p.R184Q as the most frequent dominant GJB2 mutations among Chinese Hans, which had a very high de novo rate (71% of probands). A majority (10/11) of subjects carrying dominant GJB2 mutations exhibited palmoplantar keratoderma in addition to hearing impairment. In two families segregated with additional c.235delC or p.V37I mutations of GJB2, family members with the compound heterozygous mutations exhibited more severe phenotype than those with single dominant GJB2 mutation. Our study suggested that the high de novo mutation rate gives rise to a significant portion of dominant GJB2 mutations. The severity of the hearing and epidermal phenotypes associated with dominant GJB2 mutations may be modified by additional recessive mutations of GJB2.
Introduction
Numerous gap junction proteins, termed as connexins (Cx), are expressed in inner ear and epidermis. Formed by hexameric connexin hemichannels (connexon) between adjacent cells, those gap junctions play an important role in the potassium homeostasis of the inner ear as well as in the keratinocyte growth and differentiation of the epidermis [1].
Mutations in the GJB2 gene, encoding the gap-junction protein connexin 26 (Cx26), may lead to sensorineural hearing loss (HL) and hyperproliferative epidermal disorder. Recessive GJB2 mutations are predominantly associated with non-syndromic HL (DFNB1A, MIM220290) and are the most common cause of hereditary HL [2]. In contrast, dominant GJB2 mutations may cause both non-syndromic (DFNA3A, MIM601544) and syndromic HL [3], [4]. Differing mostly in various types of epidermal disorders, the syndromic HL associated with dominant GJB2 mutations includes keratitis-ichthyosis-deafness (KID, MIM148210) syndrome, hystrix-like ichthyosis with deafness (HID, MIM602540), palmoplantar keratoderma (PPK, MIM148350) with deafness, Vohwinkel syndrome (MIM124500) and Bart-Pumphrey syndrome (MIM149200). Major epidermal phenotypes associated with dominant GJB2 mutations include ichthyosis, pseudoainhum, palmoplantar hyperkeratosis, knuckle pads and nail abnormalities [5].
De novo mutation occurs in the germline during early embryogenesis or somatically in some overgrowth syndromes [6]. With the advent of whole-exome and whole-genome sequencing approaches, its etiologic contribution to both rare and common genetic diseases has been increasingly recognized [7], [8]. Though on average more deleterious than their inherited counterparts due to less stringent selective pressure [9], de novo mutations have nevertheless been reported in some disorders with lesser impact on fitness, including a number of de novo GJB2 mutations detected in sporadic patients with sensorineural HL and hyperproliferative epidermal disorder [10].
Unlike recessive GJB2 mutations, for which the mutation spectrum and the genotype-phenotype correlation have been well characterized, previous studies of dominant GJB2 mutations were mostly limited to case reports of individual patients and families. In the present study, we aimed to systematically characterize the spectrum, de novo rate and genotype-phenotype correlation of dominant GJB2 mutations in Chinese Han patients with syndromic or non-syndromic HL.
Materials and Methods
Subject recruitment
A total of 2168 Chinese Han probands with sensorineural HL were recruited from patients seeking genetic testing and counseling in Department of Otolaryngology - Head and Neck Surgery, Xinhua Hospital, Shanghai, China. Among them, 1904 (87.8%) probands had severe or profound HL, while the rest 264 (12.2%) had mild or moderate HL. Additional family members were recruited for those carrying known dominant GJB2 mutations. All subjects or their parents gave written, informed consent to participate in this study. This study was approved by the Ethics Committee of Xinhua Hospital, Shanghai Jiao tong University School of Medicine.
Mutation screening of GJB2
Genomic DNA was extracted from the whole blood samples using the Blood DNA kit (TIANGEN BIOTECH, Beijing, China). Mutation screening of all exons and flanking splicing sites of GJB2 was performed by PCR amplification and bidirectional sequencing as previously described [11]. The dominant mutations identified in this study were also screened in 100 ethnically-matched normal hearing controls.
Auditory and epidermal evaluations
For subjects and family members carrying known dominant GJB2 mutations, physical examination was performed with special attention to hearing and epidermal abnormalities. Comprehensive auditory evaluations were performed including otoscope examination, tympanometry and pure-tone audiometry (PTA). Hearing threshold was calculated as the average of the hearing levels at 0.5, 1.0, 2.0, 4.0 KHz for the better ear. The severity of hearing impairment was defined as mild (20–40 dB), moderate (41–70 dB), severe (71–95 dB) and profound (>95 dB). Histological examination was performed in skin biopsies of patient C209-1.
Short tandem repeat analysis
For subjects carrying potential de novo mutations of GJB2, short tandem repeat (STR) analysis was performed in the affected individuals and their unaffected parents to confirm the parenthood using informative STRs D5S435, D5S629, D5S610, D5S351, D6S1714, D6S1573, D6S1344, D6S469, D3S1597, D3S3611, D3S3601 and D3S3589.
Results
Spectrum and phenotypes of dominant GJB2 mutations
By mutation screening of GJB2 in 2168 Chinese Han probands with sensorineural HL, we identified 7 (0.32%) probands carrying known dominant GJB2 mutations p.R75Q (n = 3), p.R75W (n = 2) and p.R184Q (n = 2). Four additional family members carrying p.R75Q (n = 2) and p.R75W (n = 2) mutations were also recruited in this study. All other GJB2 variants detected in this study were either previously reported recessive mutations or non-pathogenic polymorphisms. None of the three dominant mutations in GJB2 was seen in 100 ethnically-matched normal hearing controls.
Table 1 summarized the genotypes and phenotypes observed in the 11 subjects carrying dominant GJB2 mutations. Except for subject D439-2, who had mild HL with delayed-onset, the majority of the subjects (10/11) had congenital, severe-to-profound HL. Ten of the 11 subjects had additional PPK-associated epidermal abnormalities in their hands and feet, including thickening and peeling of the skin, keratoderma, erythema, callus, deep fissures, brittle, thickened or spoon nails, knuckle pads and circular keratotic constriction bands (Figure 1).
Table 1. Genotypes and phenotypes of subjects with dominant GJB2 mutations.
| Family | Patient | Relation to the proband | Age (years) | GJB2 genotype | Hearing impairment | Epidermal abnormalities | Origin of mutation |
| D439 | D439-1* | - | 17 | p.R75Q/p.V37I | Congenital, profound | Thickening (palms and soles) and peeling (soles) of the skin, keratoderma (palms), erythema (hands and feet), knuckle pads and circular keratotic constriction band (fingers) | Parental inherited |
| D439-2 | Half sibling | 26 | p.R75Q/+ | Delayed-onset (∼10 years), mild | Minor thickening of the skin (palms only) and keratoderma (palms) | Parental inherited | |
| D439-4 | Mother | 49 | p.R75Q/p.V37I | Congenital, profound | Thickening (palms and soles) of the skin, keratoderma (palms), knuckle pads and circular keratotic constriction band (fingers), callus (soles) | Unknown | |
| D898 | D898-1* | - | 3 | p.R75Q/+ | Congenital, profound | Thickening and peeling of the skin (palms and soles), erythema (hands and feet) | De novo |
| D1057 | D1057-1* | - | 31 | p.R75Q/+ | Congenital, severe | Thickening of the skin (palms), keratoderma (palms), knuckle pads (fingers) | De novo |
| D441 | D441-1* | - | 17 | p.R75W/c.235delC | Congenital, profound | Severe peeling (soles) and thickening (palms and soles) of the skin, keratoderma (palms and feet), erythema (hands and feet), knuckle pads (fingers), deep fissures (hand and feet); the epidermal abnormalities were extended to wrists, ankles and the dorsal area of hands and feet | Parental inherited |
| D441-2 | Mother | 44 | p.R75W/+ | Congenital, profound | Thickening of the skin (palms and soles), keratoderma (feet), erythema (hands and feet), knuckle pads (fingers), callus (soles) | De novo | |
| D493 | D493-1* | - | 5 | p.R75W/+ | Congenital, profound | Thickening of the skin (palms), keratoderma (palms), thickened (toes) or brittle nails (fingers) | De novo |
| D493-2 | Sibling | 3 | p.R75W/+ | Congenital, profound | Thickening of the skin (palms), minor peeling and erythema (palms), brittle nails (fingers) | De novo | |
| C209 | C209-1* | - | 33 | p.R184Q/+ | Congenital, severe | Thickening and peeling of the skin (soles), keratoderma (soles), callus (soles), thickened nails (fingers), spoon nails (toes) | De novo |
| D40 | D40-1* | - | 6 | p.R184Q/+ | Congenital, profound | None | De novo |
*Probands.
Figure 1. Representative epidermal abnormalities of the subjects carrying dominant GJB2 mutations.
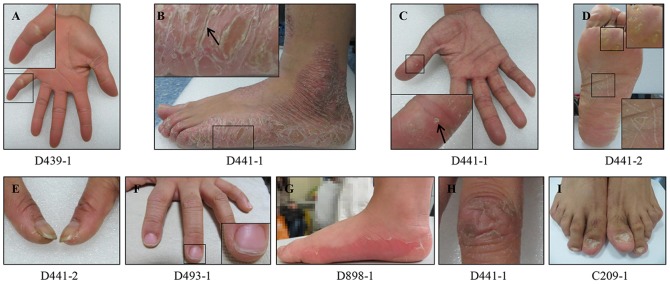
(A) Thickening of the skin and the circular keratotic constriction band (box); (B) Peeling (box) and deep fissures (arrow in the box); (C) Punctiform keratoderma; (D) Callus (the upper box) and striped keratoderma (the lower box); (E) Thickened nails; (F) Brittle nails; (G) Erythema; (H) knuckle pad; (I) spooned nails.
All subjects with the p.R75Q (n = 5) and p.R75W (n = 4) mutations had syndromic HL with PPK. C209-1 and D40-1, two subjects with the p.R184Q mutations, had syndromic and non-syndromic HL, respectively. Since the p.R184Q mutation has not been reported to be associated with epidermal abnormalities previously (Table S1), we further characterized the PPK phenotype of subject C209-1 (Figure 2). In addition to a variety of epidermal abnormalities, skin biopsy of his soles confirmed the hyperkeratosis, thickening of the granular layer, acanthosis, and sparse lymphocytes infiltration in the superficial layer of dermis.
Figure 2. Genotype and phenotype characterization of Fmaily 209.

(A) Pedigree and the genotypes of family members. Proband C209-1 with de novo p.R184Q mutation of GJB2 was pointed by the arrow. (B) HL phenotypes of C209-1. (C) PPK phenotypes of C209-1 including the thickening and peeling of the skin, keratoderma, callus (the left panel), spoon nails of the toes (the middle panel) and thickened nails of the fingers (the right panel). (D) Skin biopsy of the left sole showing hyperkeratosis, thickening of the granular layer, acanthosis, and sparse lymphocytes infiltration in the superficial layer of dermis. (Hematoxylin-eosin stain; original magnification ×200.).
High de novo rate of dominant GJB2 mutations
In 6 of the 7 families detected with dominant GJB2 mutations, the probands (D898-1, D1057-1, D493-1, C209-1 and D40-1) and/or their first-degree relatives (D441-2 and D493-2) had unaffected parents. STR analysis of the trios confirmed the true parenthood (data not shown). Parental genotyping showed that the dominant GJB2 mutations detected in those 7 subjects were de novo (Figure 2A, 4A and Figure S1). One particular pair of siblings, D493-1 and D493-2, both had de novo mutations of p.R75W, possibly through parental germline mosaicism.
Figure 4. Genotype and phenotype characterization of Family D441.

(A) Pedigree and the genotypes of family members. Subject D441-2 with de novo p.R75W mutation of GJB2 was pointed by the arrow. (B) HL phenotypes of D441-1 and D441-2. (C) PPK phenotypes of D441-1 and D441-2. Note that D441-1 had more severe keratoderma (the third panels) and erythema (the second, fourth and fifth panels) than D441-2, and that the PPK phenotype of D441-1 was extended to the wrist (the second panels), the ankle (the fourth and fifth panels) and the back of the hands and feet (the second and fourth panels).
Phenotypic modification by additional GJB2 mutations
The p.R75Q and p.R75W mutations were identified in multiple family members in Family D439 (D439-1, D439-2 and D439-4) and Family D441 (D441-1 and D441-2), respectively. Interestingly, intrafamilial variations of the degree of HL and PPK were observed in both families and the severity of phenotype was apparently modified by the presence of additional GJB2 mutations.
In Family D439, both proband D439-1 and his mother D439-4 had the p.R75Q/p.V37I compound heterozygous mutations of GJB2, while the proband's half-sibling D439-2 had the p.R75Q heterozygous mutation only (Figure 3A). Consistent with the genotypes, the HL of both D439-1 and D439-4 was congenital and profound, while that of D439-2 was delayed-onset and mild (Figure 3B). In addition, D439-1 and D439-4 had more severe PPK than D439-2, including symptoms of knuckle pad and circular keratotic constriction band of the fingers that were absent in D439-2.
Figure 3. Genotype and phenotype characterization of Fmaily D439.

(A) Pedigree and the genotypes of family members. (B) HL phenotypes of D439-1, D439-2 and D439-4. (C) PPK phenotypes of D439-1, D439-2 and D439-4. Note that D439-1 and D439-4 had more severe keratoderma than D439-2 (the left and right panels), and that a number of PPK-associated epidermal abnormalities observed in D439-1 and D439-4 were absent in D439-2, including peeling of the skin (arrows in the right panel), knuckle pad (arrows, the middle panel) and circular keratotic constriction band (arrows, the left panel).
In Family 441, proband D441-1 had the p.R75W/c.235delC compound heterozygous mutations of GJB2, while the proband's mother D441-2 had the p.R75W heterozygous mutation only (Figure 4A). Though the degree of HL was similar (Figure 4B), D441-1 exhibited more severe PPK phenotypes than D441-2, and the affected area was extended to his wrists, ankles and the back of hands and feet (Figure 4C), areas that were typically unaffected in other PPK patients.
Discussion
In this study, we characterized the spectrum, de novo rate and genotype-phenotype correlation of dominant GJB2 mutations associated with hearing and epidermal disorders in Chinese Hans. Out of 2168 probands with sensorineural HL, 7 (0.32%) was detected with known dominant GJB2 mutations. Though not as common as its recessive counterpart, dominant mutations in GJB2 still represent one of the most frequent causes for dominantly inherited HL. To date, more than 20 dominant mutations of GJB2 has been reported worldwide (The Human Gene Mutation Database, http://www.hgmd.cf.ac.uk). The mutation spectrum revealed by the current study of Chinese Hans, however, evolved only three of them: p.R75Q (n = 3), p.R75W (n = 2) and p.R184Q (n = 2). Considering that all these three mutations have also been reported in sporadic cases with HL in Taiwan or mainland China (Table S1), they are likely among the most common dominant GJB2 mutations in Chinese Hans.
As summarized in Table S1, the p.R75Q and p.R75W mutations were associated with both non-syndromic and syndromic HL in previous reports, while the p.R184Q mutation was exclusively associated with non-syndromic HL. Interestingly however, in our study all 5 families, 9 patients with the p.R75Q or p.R75W mutations and one patient with the p.R184Q mutation exhibited syndromic HL with PPK (Table 1). Our results suggested that the genotype-phenotype correlation of dominant GJB2 mutations may be variable among different ethnic groups.
One of the most interesting findings of our study was the very high de novo rate of dominant GJB2 mutations in Chinese Hans, which occurred in 71% (5/7) of the probands and two additional family members with the p.R75Q (n = 2), p.R75W (n = 3) and p.R184Q (n = 2) mutations (Table 1, Figure 2A, 4A and Figure S1). It has been shown that most de novo mutations constitute transitions (purine↔purine or pyrimidine↔pyrimidine) rather than transversions (purine↔pyrimidine), and the mutation rate can be elevated at the CpG-rich region [12]. The p.R75Q (CGG>CAG), p.R75W (CGG>TGG) and p.R184Q (CGG>CAG) mutations all fit in with this characteristics of de novo mutations, which may partly explain the relatively high frequency of these three dominant GJB2 mutations in Chinese Hans.
In two families segregated with one dominant and one recessive GJB2 mutations, family members with both mutations exhibited more severe HL and PPK phenotypes than those with the dominant GJB2 mutation only (Figure 3 and 4). This intrafamilial variation of the phenotypes leads us to hypothesize that the additional heterozygous recessive mutation of GJB2 may modify the HL and PPK phenotypes caused by the dominant GJB2 mutations. Consistent with our observation, the speculated phenotypic modification has also been supported in several other families with dominant GJB2 mutations [13], [14]. Functional studies are needed to investigate this hypothesized modification effect.
In summary, our study provided a preliminary overview of the spectrum, de novo rate and genotype-phenotype correlation of dominant GJB2 mutations in Chinese Hans. Characterization of these features may result in more accurate genetic testing and counseling of the associated disorders.
Supporting Information
Four additional families with de novo mutations of GJB2 . The affected individuals with de novo mutations were pointed by the arrows.
(TIF)
Genotype-phenotype correlation of the p.R75Q, p.R75W and p.R184Q mutation in previous reports and the current study.
(DOC)
Acknowledgments
We thank all the research staff involved in this study from the Xinhua Hospital. We also thank the study patients for their participation in this study.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. The majority of data can be found in the manuscript and the remained data has uploaded as Supporting Information files.
Funding Statement
This research was supported by grants from National Science Foundation of China (81222010 and 30971596 to TY, 81170924 to HW), grant from National Basic Research Program of China (2011CB504501 to HW), and the Oriental Scholar project from Shanghai Municipal Education Commission (to TY). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Kelsell DP, Di WL, Houseman MJ (2001) Connexin mutations in skin disease and hearing loss. Am J Hum Genet 68: 559–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Snoeckx RL, Huygen PL, Feldmann D, Marlin S, Denoyelle F, et al. (2005) GJB2 mutations and degree of hearing loss: a multicenter study. Am J Hum Genet 77: 945–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maestrini E, Korge BP, Ocana-Sierra J, Calzolari E, Cambiaghi S, et al. (1999) A missense mutation in connexin26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel's syndrome) in three unrelated families. Hum Mol Genet 8: 1237–1243. [DOI] [PubMed] [Google Scholar]
- 4. Morle L, Bozon M, Alloisio N, Latour P, Vandenberghe A, et al. (2000) A novel C202F mutation in the connexin26 gene (GJB2) associated with autosomal dominant isolated hearing loss. J Med Genet 37: 368–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Iossa S, Marciano E, Franze A (2011) GJB2 Gene Mutations in Syndromic Skin Diseases with Sensorineural Hearing Loss. Curr Genomics 12: 475–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lindhurst MJ, Sapp JC, Teer JK, Johnston JJ, Finn EM, et al. (2011) A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med 365: 611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hoischen A, van Bon BW, Gilissen C, Arts P, van Lier B, et al. (2010) De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat Genet 42: 483–485. [DOI] [PubMed] [Google Scholar]
- 8. Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, et al. (2007) Strong association of de novo copy number mutations with autism. Science 316: 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crow JF (2000) The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet 1: 40–47. [DOI] [PubMed] [Google Scholar]
- 10. Heathcote K, Syrris P, Carter ND, Patton MA (2000) A connexin 26 mutation causes a syndrome of sensorineural hearing loss and palmoplantar hyperkeratosis (MIM 148350). J Med Genet 37: 50–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dai P, Yu F, Han B, Liu X, Wang G, et al. (2009) GJB2 mutation spectrum in 2,063 Chinese patients with nonsyndromic hearing impairment. J Transl Med 7: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Piazza V, Beltramello M, Menniti M, Colao E, Malatesta P, et al. (2005) Functional analysis of R75Q mutation in the gene coding for Connexin 26 identified in a family with nonsyndromic hearing loss. Clin Genet 68: 161–166. [DOI] [PubMed] [Google Scholar]
- 13. Iossa S, Chinetti V, Auletta G, Laria C, De Luca M, et al. (2009) New evidence for the correlation of the p.G130V mutation in the GJB2 gene and syndromic hearing loss with palmoplantar keratoderma. Am J Med Genet A 149A: 685–688. [DOI] [PubMed] [Google Scholar]
- 14. Welch KO, Marin RS, Pandya A, Arnos KS (2007) Compound heterozygosity for dominant and recessive GJB2 mutations: effect on phenotype and review of the literature. Am J Med Genet A 143A: 1567–1573. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Four additional families with de novo mutations of GJB2 . The affected individuals with de novo mutations were pointed by the arrows.
(TIF)
Genotype-phenotype correlation of the p.R75Q, p.R75W and p.R184Q mutation in previous reports and the current study.
(DOC)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. The majority of data can be found in the manuscript and the remained data has uploaded as Supporting Information files.