

Abstract
We present an extensible interface between the AMBER molecular dynamics (MD) software package and electronic structure software packages for quantum mechanical (QM) and mixed QM and classical molecular mechanical (MM) MD simulations within both mechanical and electronic embedding schemes. With this interface, ab initio wave function theory and density functional theory methods, as available in the supported electronic structure software packages, become available for QM/MM MD simulations with AMBER. The interface has been written in a modular fashion that allows straight forward extensions to support additional QM software packages and can easily be ported to other MD software. Data exchange between the MD and QM software is implemented by means of files and system calls or the message passing interface standard. Based on extensive tests, default settings for the supported QM packages are provided such that energy is conserved for typical QM/MM MD simulations in the microcanonical ensemble. Results for the free energy of binding of calcium ions to aspartate in aqueous solution comparing semiempirical and density functional Hamiltonians are shown to demonstrate features of this interface.
Keywords: AMBER, QM/MM, ab initio, density functional theory, molecular dynamics, calcium binding
1 Introduction
Hybrid quantum mechanical and molecular mechanical (QM/MM) approaches are used extensively to study local electronic events in large molecular systems with a diverse area of applications ranging from enzymatic catalysis to properties of materials systems1–13. In QM/MM schemes, part of the system that includes the chemically relevant region is treated quantum mechanically while the remainder, often referred to as environment, is treated at the classical level using MM force fields. This multiscale approach reduces the computational cost significantly as compared to a QM treatment of the entire system and makes simulations possible that otherwise would not be feasible. At the same time the numerical results obtained from QM/MM simulations should converge to full QM results if the QM region is sufficiently large such that the effect of artifacts at the QM/MM boundary is minimized and if the MM force field affords an adequate representation of the environment.
The AMBER14,15 software package for biomolecular simulations supports QM/MM approaches that use semiempirical neglect of diatomic differential overlap (NDDO) type Hamiltonians16 as well as density functional tight binding (DFTB) Hamiltonians17. These QM methods have the advantage of being computationally efficient, facilitating sampling sufficient phase space during molecular dynamics (MD) simulations. On the downside, the approximate nature of semiempirical Hamiltonians limits their accuracy and transferability, often requiring specific parameterizations for a given problem18,19. In addition, most semiempirical Hamiltonians are only available for selected elements of the periodic table. It thus is frequently desirable to employ more accurate and generally applicable ab initio wave function theory or density functional theory (DFT) methods in the QM region.
Combining existing software packages for classical MD simulations with electronic structure programs is an effective approach to enable ab initio wave function theory and DFT based QM/MM MD simulations. It avoids duplication of programming effort and exploits the functionality and performance that are offered by the interfaced programs which frequently are the result of many years of software development. It also immediately benefits the existing user base of the simulation package who can continue to use their software infrastructure such as automated workflow schemes that rely on established input and output syntax. Consequently, several such interfaces have been developed and described in the literature20–32. With the exception of PUPIL33 and the scripting environment ChemShell34,35, however, these are mostly limited to support only one specific electronic structure program. In addition, some interfaces are either not well maintained or have not entered the main release branch of the simulation software package and are thus not available to the end user.
In this work we present a versatile and easily extensible interface for QM/MM simulations within mechanical and electronic embedding schemes that supports a wide range of electronic structure software packages. This interface has been integrated into the MD engine SANDER of the AMBER14,15 software package and has been made available with release version 12 in April 2012. The interface is written in Fortran90 using a modular fashion, which makes it easily extensible to include support for additional electronic structure software as well as portable to be included into MD software engines other than SANDER. The AMBER implementation supports the link atom approach that is available for semiempirical QM/MM simulations16 as well as the full range of advanced sampling and free energy methods that are available in SANDER. This manuscript serves as a reference for the new interface and begins with a review of the QM/MM theory before describing features and technical details of the implementation and integration with AMBER. The numerical accuracy of the implementation is then shown by analyzing geometry optimizations of the water dimer and the energy conservation during constant energy QM/MM MD simulations of N-methylacetamide and alanine dipeptide in explicit solvent followed by a short discussion of typical time scales that are accessible with ab initio or DFT based QM/MM MD simulations. We finally demonstrate features of the new QM/MM interface in AMBER using the problem of calcium binding by proteins as example. We compare results for the free energy of binding of calcium ions to aspartate in aqueous solution obtained from MD simulations using both a classical MM potential as well as QM/MM potentials using the semiempirical PM636 Hamiltonian and DFT before summarizing with concluding remarks.
2 QM/MM theory
The total energy in a QM/MM system can be written in an additive way as
| (1) |
where the three terms represent the QM energy EQM of the QM region in absence of perturbations due to the MM environment, the classical MM energy EMM of the MM region, and the QM/MM interaction energy EQM/MM between the QM and the MM region. In addition to the QM and MM methods employed, a QM/MM calculation thus requires also a choice for the form of the interaction energy EQM/MM.
The simplest approach is to neglect any electronic coupling between the QM and the MM system and treat all non-bonded interactions, that is, van der Waals (vdW) and electrostatic, at the level of the classical MM force field. This is useful to impose steric constraints on the embedded QM system and commonly referred to as mechanical embedding. It can become problematic if reactive events are studied that involve significant charge transfer within the QM region since the atom types and thus both the vdW parameters and the charges remain constant during the course of the simulation. As a consequence the interaction between the QM and MM region of the transition and product states is typically not properly described. To improve this situation, some mechanical embedding implementations use point charges for the QM region atoms that are derived from the electronic structure calculation at each step of a simulation.
In many cases it is also important to allow for polarization of the embedded QM region due to the electric field of the surrounding MM environment which is referred to as electronic embedding. In this case the QM/MM interaction energy for a system consisting of NQM atoms in the QM region and NMM atoms in the MM region is given as
| (2) |
Here, the first term is the Coulomb interaction between the total charge density ρQm of the QM region (which consists of the electron density and in general nuclear point charges) and the fixed MM point charges Qk. The second term is the classical vdW interaction between the atoms in the QM and MM region as given by the underlying MM force field in terms of an empirical Lennard-Jones (LJ) potential. In mechanical embedding, ρQM in the first term of Equation (2) is replaced with fixed point charges that are typically taken from the corresponding MM force field or derived from the electronic structure calculations on the fly. It is worth mentioning that electronic embedding is not always superior to mechanical embedding and an extensive study comparing different QM/MM approaches can be found for example in the work by Hu et al.37.
If the QM/MM boundary crosses covalent bonds, the QM/MM interaction energy EQM/MM additionally includes bonded terms from the classical MM force field accounting for corresponding bond stretch, angle and dihedral forces between the QM and MM subsystems.
The forces acting on the atoms in a QM/MM calculation are given in terms of derivatives of the total energy expression Equation (1) with respect to the Cartesian coordinates of the atoms,
| (3) |
The first two terms are the standard gradient expression for the QM method and the classical MM force field that are employed in the QM and MM regions, respectively. What remains, are the forces acting on the QM atoms A and the MM atoms k due to the QM/MM interaction term for which we obtain
| (4) |
where we have introduced for the LJ potential between QM atom A and MM atom k, and
| (5) |
where EQM is the electric field due to the QM charge density ρQM. The forces thus obtained can then be used for geometry optimizations or to propagate the system coordinates during MD simulations within the QM/MM framework.
3 Implementation
We have developed a self-contained, easily extensible interface for QM/MM calculations in which the geometry optimization or MD simulation is driven by a classical MM program. As such, the purpose of the interface is to provide the driving MM program with the QM contribution to the energy and the forces, Equations (1) to (5), as obtained from an external electronic structure program. The interface is written in Fortran 90 with a simple application programming interface (API) that makes it easy to be linked with the MM program at the source code level while communication between the interface and the electronic structure programs is implemented via either file based data exchange or alternatively, as initially implemented within TeraChem38–40, via a client/server model for data exchange based on version 2 of the message passing interface41 (MPI-2) standard. The interface has been integrated into the MD engine SANDER of the AMBER14,15 software package for biomolecular simulations and was released with version 12 of AMBER in April 2012. The usefulness of this interface has already been demonstrated for DFT based QM/MM MD simulations of aqueous systems42 and the simulation of electronic absorption spectra of the photoactive yellow protein40. An overview of the capabilities of the interface and details of its implementation are given in the remainder of this section.
3.1 Features
The interface supports both mechanical and electronic embedding. For the latter the electronic structure program has to support QM calculations in an external electric field of point charges including the ability to calculate either the electric field due to the QM charge density at the position of the MM point charges or directly the forces exterted on the MM atoms that arise due to electrostatic interaction with the QM charge density, see Equation (5). At the time of writing the following electronic structure programs are supported for mechanical embedding:
and the following programs are supported for mechanical and electronic embedding:
This represents a set of widely used programs, both commercial and freely available, each with its own strengths for different electronic structure methods and computing platforms ranging from desktop workstations to supercomputers. In the case of TeraChem this also includes accelerator hardware in the form of graphics processing units (GPUs). Most researchers, both academic and industrial, will have access to one or the other of these software packages. The development version of AMBER also includes support for Q-Chem51 and it is our intention to add support for additional electronic structure software with future releases, including plane wave DFT codes for materials science related QM/MM simulations.
The implementation within AMBER’s MD engine SANDER builds upon the existing QM/MM functionality for semiempirical QM methods16 and thus inherits all of its features with the exception of approaches that are either not available for ab initio wave function theory and DFT methods or that would require changes to the electronic structure software. For example, the automatic link atom setup16 for simulations in which the QM/MM boundary crosses covalent bonds is available in exactly the same fashion as for the built-in semiempirical methods. However, both generalized Born (GB) solvent models52,53 and, in the case of simulations with periodic boundary conditions and electronic embedding, the treatment of long-range electrostatic interactions between QM and MM regions and electrostatic interactions of the QM region with its own periodic images via the particle mesh Ewald (PME) approach16,54,55 is not available. Instead, electrostatic interactions, the first term in Equation (2), are truncated beyond a cutoff that is defined as the minimum distance between an MM point charge and any atom of the QM region. For non-periodic simulations this is generally not problematic as they can be run without truncation by increasing the cutoff beyond the system size, thus enabling energy-conserving MD. However, under periodic boundary conditions this is not possible and it is current practice to use a cutoff that is as large as possible while employing a thermostat to dissipate the heat that is introduced in MD simulations due to discontinuities in the potential resulting from the truncation of the electrostatic interactions.
The AMBER implementation supports QM/MM geometry optimizations, standard MD simulations, as well as the advanced sampling and free energy methods such as umbrella sampling56,57 that are available in the SANDER MD program. Parallelization of replica exchange58,59 MD (REMD) simulations and the various quantum dynamics approaches such as path integral60,61 MD (PIMD) simulations are supported with each replica or bead running concurrently via the MPI implementation of SANDER. Parallelization of the individual QM calculations is available as provided by the corresponding electronic structure software.
A comprehensive set of regression tests covering all supported external QM software packages and different simulation options, including geometry optimizations, standard MD simulations and advanced sampling methods like REMD and PIMD has been included with the release of AMBER 12. These tests not only help to maintain code integrity and functionality with future releases of AMBER and the interfaced electronic structure programs but also serve as examples for a user on how to use the interface.
3.2 User interface
One of the driving forces when integrating the interface with AMBER was that it should be easy to use for anybody who is familiar with the AMBER MD software package. No expert knowledge with the external QM software package is required as long as the most commonly used electronic structure methods such as DFT and second order Møller–Plesset perturbation theory (MP2) are used. For a user it is thus irrelevant which of the supported QM software packages is installed as long as it supports the electronic structure method the user wishes to employ. Only minor modifications to an input file that would be used for semiempirical QM/MM simulations with AMBER are required.
3.2.1 Simulation setup and QM region selection
The setup of QM/MM simulations using the new interface follows the same scheme as for the built-in semiempirical models16. For completeness we summarize the main steps involved, details can be found in the AMBER user manual. A QM/MM simulation with AMBER requires initially setting up input files for an MM simulation, including parameters, topology and coordinates. This can be useful in itself for example to equilibrate a system at the MM level, however, requires providing MM parameters for non-standard residues if these are not available in the AMBER force field library. In particular it is important to realize that vdW parameters and in the case of mechanical embedding also charges of atoms in the QM region will be used during the QM/MM simulations, see Equation (2). This also holds for covalent force field terms that cross the QM/MM boundary if the QM and MM regions are covalently linked.
Once an MM simulation has been set up, the required modification to the input file are minimal. The user only needs to specify the atoms that make up the QM region, the QM region charge and spin multiplicity, as well as the QM method to be employed. Settings of details of the QM/MM embedding scheme that deviate from the default electronic embedding and cutoff for the real-space electrostatic interactions between QM and MM region are optional.
From this point on the QM/MM implementation takes care of everything else automatically, making sure that the QM and MM codes calculate the required contributions to the energy and forces, Equations (1) to (5). This is straightforward if the QM region is not covalently linked to the MM region. In this case the MM force field is modified by deleting the covalent force field terms (bond, angle, dihedral terms) and, in the case of electronic embedding, by deleting the atom point charges for all atoms in the QM region. A link atom scheme16 is used if the QM/MM boundary crosses a covalent bond to saturate the dangling bond of the QM region, the important point of which is that it does not introduce any additional degrees of freedom into the simulation. The link atom is added automatically for the QM calculations without user intervention but details can be controlled by the user. The point charge on the MM region atom whose bond crosses the QM/MM boundary is set to zero in the case of electronic embedding to avoid overpolarization of the QM region and any residual charge due to this procedure is evenly distributed among all remaining atoms in the MM region to maintain charge neutrality. This behavior can also be modified by the user. Finally, bonded force field terms crossing the QM/MM boundary are retained if at least one atom is part of the MM region.
3.2.2 QM program and method selection
Figure 1 shows the relevant parts of an AMBER control input file mdin that uses the QM program Orca50 for a QM/MM simulation with the B3LYP/6–31G* method in the QM region. To specify that an external software package shall be used for the QM/MM calculation, instead of the built-in semiempirical QM methods of AMBER, it is sufficient to set the qmmm namelist variable qm theory to ‘EXTERN’. The settings for the QM approach to be employed by Orca, in this case the B3LYP density functional method with the 6–31G* basis set, need to be provided in the orc namelist. If instead, for example, the Gaussian program is to be used, then orc needs to be replaced with gau and similar for other electronic structure software packages.
Figure 1.

Example of minimal modifications (bold face) required to an AMBER input file to perform QM/MM MD simulations with B3LYP/6-31G* using the Orca program via the new interface instead of semiempirical methods implemented in AMBER.
Default parameters for Hartree-Fock and DFT calculations are provided for all supported QM programs such that the forces are computed with sufficient accuracy for good energy conservation during MD simulations in the microcanonical ensemble. Specifically these are the self-consistent field (SCF) convergence and associated integral neglect thresholds as well as grid size parameters for the numerical quadrature of the exchange-correlation (XC) potential and energy in DFT calculations. If possible, the interface instructs the QM program to use the converged wave function from the previous geometry optimization or MD step as the initial guess for the present step. This extrapolation results in substantial computational savings but leads to an energy drift for MD simulations in the microcanonical ensemble62,63. However, the resulting energy drift is typically small enough to be acceptable for many applications, in particular if tight SCF convergence thresholds are used and a simulation is coupled to a thermostat. The user can modify these settings for a given program via its corresponding namelist in the AMBER control input file mdin.
It is not possible for a simple interface to account for all input options that are available in the supported QM software packages. Furthermore, new options may be added to the external software package which cannot be anticipated. Therefore, the control options have been restricted to the most likely usage scenarios, that is DFT and MP2 since most other electronic structure methods are computationally too expensive to run routine QM/MM MD simulations. An expert user may wish to use advanced input options for the supported external QM software packages that go beyond what is supported with the present version of the interface. To this end the interface also supports input for the external software packages via user-provided template files. Such a template file needs to contain all information that is required to fully specify the QM method that shall be used for a simulation (such as density functional and basis set), including accuracy settings (such as SCF convergence thresholds), that deviate from default settings of the electronic structure program. The AMBER interface will then use the information provided in the template file and supplement it with the missing data: coordinates for atoms in the QM region; coordinates of point charges within the specified cutoff (in the case of electronic embedding); instructions to perform a single point energy or gradient calculation as required for post-processing snapshots of MD trajectories or performing geometry optimizations and MD simulations.
3.3 Technical details
One of the key design goals of the interface has been modularity and extensibility. To achieve this, the interface was written entirely in Fortran 90 with data types, subroutines and functions for each of the supported electronic structure packages collected into separate modules. Only the driver subroutine for exchanging relevant information with the MD program (QM region atom types and coordinates, charge, spin multiplicity, MM region point charges and coordinates, QM contribution to the energy and forces) is exposed, all other functionality that handles communication with the QM program is private to the modules. A separate module collects utility routines that are common to all QM program-specific modules, such as debug and printing functions.
Two different communication methods are implemented for data exchange between the interface and the QM programs (see Figure 2). Communication via files and system calls is available for all supported software packages. In this case, upon each call of its driver routine, the interface proceeds by
writing input files for the QM program that contain the current QM region atomic numbers and coordinates and MM point charges and coordinates,
executing the QM program via a system call, and
parsing the output files of the QM program to retrieve the energy and forces
If requested by the user the interface will also store the dipole moment and atomic partial charges of the QM region along an MD trajectory. The interface stores the in- and output files for the QM calculation of the present and the last geometry optimization or MD step. This simplifies debugging in case of program crashes.
Figure 2.
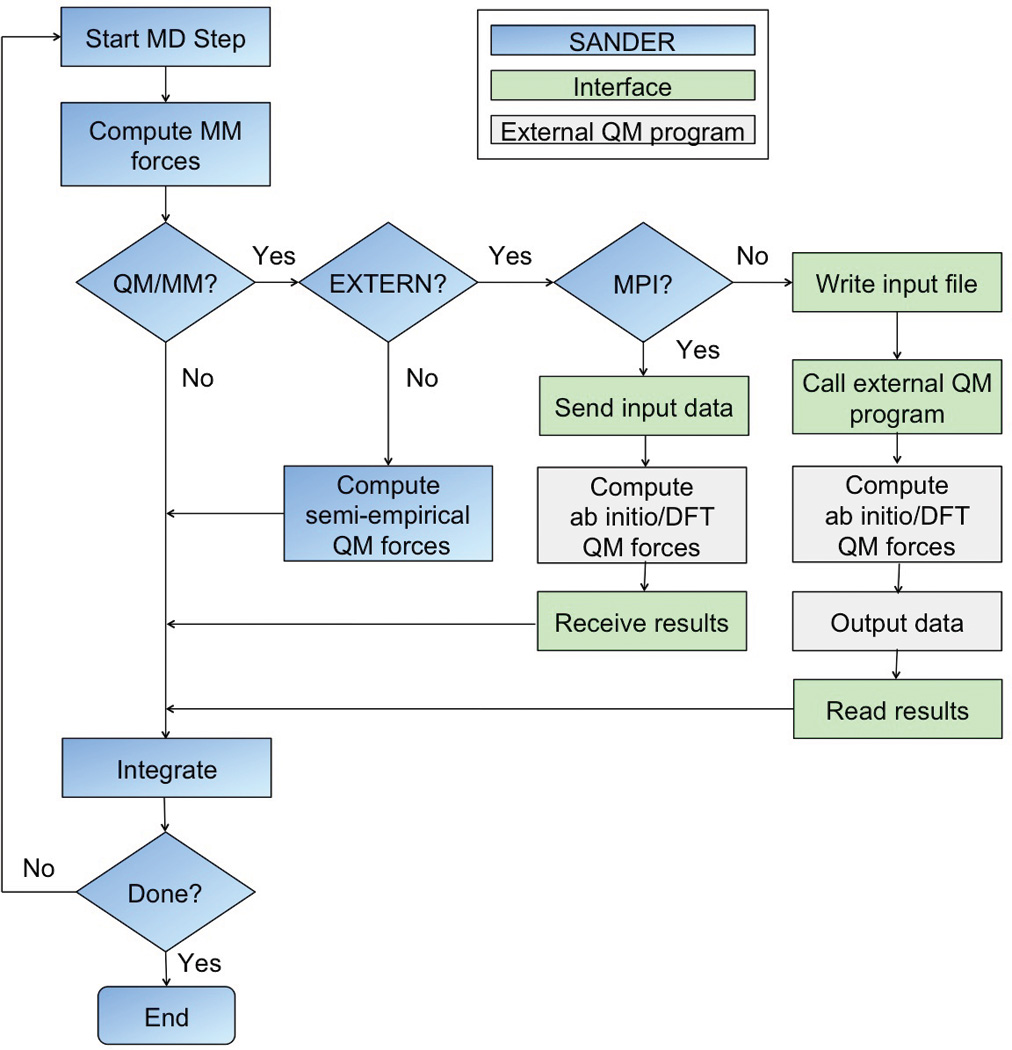
Flow chart for a QM/MM simulation with the AMBER MD program SANDER using the interface to external QM programs. Data exchange is either based on files and system calls or, in the client/server model, proceeds by sending and receiving the required data between SANDER and the QM program using the MPI-2 standard. The client/server model requires corresponding changes to the QM software package and is currently supported by TeraChem.
Data exchange is also implemented via a client/server model based on MPI-2. This is currently supported only by TeraChem40 since it requires source code changes to the QM program. However, the interface is very generic and support could be easily added to other electronic structure software. For use with the MPI-2 interface, the QM program is started in server mode at the beginning of a simulation. The interface then connects as a client to the QM program and all subsequent data exchange proceeds via standard MPI send and receive calls. At the end of the simulation the interface sends a signal for the QM program to quit and disconnects.
The client/server model of data exchange has several advantages over communication that proceeds through files and system calls. It avoids the initialization of the QM program that is otherwise required during each geometry optimization or MD step and thus reduces computational overhead that can be significant for small QM regions. More importantly this avoids any loss of precision during the data transfer that typically occurs when formatted in- and output files are used. In addition, data exchange between the MM and QM program is possible during the optimization of the wave function or electron density in the SCF procedure. With corresponding modifications in the QM program and the interface this could be useful for more advanced QM/MM coupling schemes including generalized Born (GB) solvation models or the treatment of long-range QM/MM eletrostatics under periodic boundary conditions via PME approaches that are available for semiempirical QM meth-ods16. In comparison to the file based interface, the MPI-2 based client/server interface is easier to maintain since it is robust to changes in the format of either in- or output files. Alternative inter-process communication methods relying on different protocols, for example using sockets, could also be envisioned.
For the implementation into the MD engine SANDER, the existing QM/MM code in AMBER has been refactored and if an external electronic structure program is used for a QM/MM calculation an AMBER-specific driver routine for the new interface is called instead of the built-in semi-empirical code. Additional details about the implementation including the API of the interface are available in the Supporting Information.
4 Computational details
The software base used for all simulations in this work was a development version of AMBER 13. The executables were built under the Rocks Cluster Distribution 5.4.3 (based on CentOS 5.6) with the Intel compiler and MKL library version 12.1.1.256 and the MVAPICH2 MPI-2 implementation version 1.8a1p1. QM/MM calculations were either performed with the PM636 semiempirical model as implemented by us in AMBER or using the Gaussian 0949 electronic structure program. Standard MNDO64-type expressions are used in AMBER with semiempirical Hamiltonians for QM/MM interactions between point-charges and electrons and between point-charges and QM cores (nuclei plus core electrons). The exponential damping function of the point-charge core interaction uses a value of 5.0 for the exponent corresponding to the point charge and the value that has been optimized for the corresponding Hamiltonian for the exponent corresponding to the QM atom. The PM6 Hamiltonian uses the PM365 exponents where available. No PM3 parameters are available for Ca2+ and thus we used exponents of 1.3 and 2.0 in this work, denoted as PM6/a and PM6/b, respectively. The SCF was considered converged when the energy difference between two consecutive SCF cycles dropped below 10−10 kcal/mol for the PM6 implementation in AMBER or when the root-mean-square of the difference density matrix elements between two SCF steps dropped below 10−8 in the Gaussian calculations. Other than this, default Gaussian 09 settings were used. DFT calculations were performed using the BP8666,67, BLYP66,68 and B3LYP69 XC functionals and the 6–31G*70,71 or 6–311G**72, TZVP73 and aug-cc-pVQZ74,75 basis sets and MP2 calculations were performed using the cc-pVDZ74 basis set. All simulations have been set up with the tleap program of AmberTools.
Geometry optimizations of the water dimer were performed with AMBER using either the TIP3P76 or the SPC77 rigid three-site point-charge water models and a combination of different QM/MM methods with electronic embedding and a truncated Newton conjugate gradient (TNCG) algorithm with a termination threshold of 10–2 kcal/mol/ Å for the root-mean-square of the gradient and a maximum of 100 optimization steps.
N-methylacetamide (NMA) and alanine dipeptide (ADP) were solvated in a droplet of SPC/Fw78 flexible three-site point-charge water molecules of 15 Å radius (408 and 403 water molecules, respectively). A soft half-harmonic restraining potential was employed beyond this radius. The ff99SB force field79 was used for NMA and ADP in MM calculations. QM/MM calculations used electronic embedding with NMA in the QM region. A QM/MM boundary crossing covalent bonds was tested with ADP, selecting the QM region such that peptide bonds were not cut: the acetyl capping group and its adjacent nitrogen atom as well as the methyl group on the N-methyl capping group were kept in the MM region, leaving a total of 12 atoms in the QM region including 2 hydrogen link atoms that are automatically placed along the broken bonds between carbon and nitrogen. A time step of 0.5fs was used for all NMA and ADP simulations. Nonbonded interactions were not truncated. The system was equilibrated for 20 ps at the MM level with Langevin dynamics80 at 300K using a collision frequency of 5 ps−1 before switching to constant energy QM/MM simulations.
Geometry optimizations of the Ace–Asp–NMe peptide (acetyl and N-methyl capped aspartate) and Ca2+ ion were performed with AMBER using the ff99SB force field and a limited-memory Broyden-Fletcher-Goldfarb-Shanno algorithm with a termination threshold of 10–2 kcal/mol/Å for the root-mean-square of the gradient. In order to keep the Ca2+ ion at a fixed distance from the carboxylate group, two strong restraints were used: an angle restraint was added to keep the Ca2+ ion aligned with the bond between the carboxyl atom and the β-carbon atom of aspartate, that is, to keep the angle Cβ-Ccarboxyl–Ca2+ at 180 degrees; the distance RC,Ca2+ between the Ca2+ ion and the carboxyl carbon atom was restrained to values ranging from 2Å to 6Å with a spacing of 0.1 Å. QM single point calculations were performed at the geometries obtained from the MM geometry optimizations.
For the MD simulations, the Ace-Asp-NMe peptide and Ca2+ ion were solvated with TIP3P76 rigid three-site point-charge water molecules. A rectangular box of approximately 63.3 × 63.9 × 58.9 Å3 (6,384 water molecules) was used for MM simulations and a water droplet with soft half-harmonic potential beyond 20 Å radius (1,006 water molecules) was used for both MM and QM/MM simulations. The ff99SB79 force field was used for the MM simulations and the QM/MM calculations used electronic embedding with the peptide and the Ca2+ ion in the QM region. A time step of 2.0 fs was used for all simulations with bond distances to hydrogen atoms constrained using the SHAKE81,82 algorithm. Nonbonded interactions were not truncated for the water droplet simulations. Simulations using periodic boundary conditions (MM only) were performed with a cutoff of 8 Å for the real-space nonbonded interactions and the particle mesh Ewald (PME) algorithm54 to account for long-range electrostatics beyond the cutoff. The water droplet was equilibrated for 100 ps at the MM level using Langevin dynamics at 300 K with a collision frequency of 5 ps–1. The position of the carbon atom of the carboxyl group and the position of the calcium ion were restrained with a harmonic potential with a force constant of 100 kcalmol–1 Å–2 during the equilibration to avoid diffusion towards the droplet boundaries. The water box was equilibrated using the same protocol using constant volume Langevin dynamics followed by another 100 ps equilibration using constant pressure Langevin dynamics with the Berendsen barostat83 with a target pressure of 1 bar and a pressure relaxation time of 1 ps.
All subsequent simulations to determine the potential of mean force (PMF) for calcium binding to the aspartate carboxyl group were performed using Langevin dynamics at 300K and constant volume in the case of periodic boundaries. The position restraint on the carboxyl carbon atom was retained and a harmonic angle restraint with force constant 300 kcal mol−1 Å−2 rad−2 was added to keep the Ca2+ ion aligned with the bond between the carboxyl carbon atom and the β-carbon atom of aspartate, as described above for the geometry optimizations. The reaction coordinate chosen for the biased MD simulations57 was the distance RC,Ca2+ between the Ca2+ ion and the carboxyl carbon atom, ranging from 2Å to 6 Å with a window spacing of 0.1Å. A harmonic biasing potential with a force constant of 300 kcal mol−1 Å−2 was used and initial configurations along the reaction coordinate were generated by equilibrating for 50 ps using MM. QM/MM simulations were equilibrated for another 4ps starting from the MM equilibrated configurations. The data from the biased MD simulations were collected for 20 ps and unbiased using the weighted histogram analysis method56,84,85 (WHAM) with a bin size of 0.05 Å and a stringent tolerance of 10−4 kcal/mol on every point in the PMF.
Figures were generated with VMD86 version 1.8.7 and gnuplot87 version 4.4.
5 Numerical accuracy and performance
5.1 Water dimer geometry optimization
We have chosen the water dimer to benchmark our QM/MM implementation since this is a standard test system and reference data is available for comparison. Table 1 shows results for MM, QM and QM/MM geometry optimizations using the TIP3P and SPC classical water models and DFT (BP86/TZVP, BLYP/aug-cc-pVQZ, B3LYP/TZVP) as QM method as well as experimental data. In the QM/MM calculations, either the hydrogen bond donor molecule (D) or acceptor molecule (A) is in the QM region (see Figure 3). All geometry optimizations result in CS symmetry which we have tested also starting from distorted geometries.
Table 1.
Binding energy (kcal/mol), distances (Å) and angles (degrees) of a water dimer in vacuum using different QM, MM and QM/MM (hydrogen bond donor D or acceptor A in QM region) Hamiltonians with electronic embedding. BP86 and B3LYP calculations were performed with the TZVP basis set, BLYP calculations with the aug-cc-pVQZ basis set.
| D-A | Reference | E | d(H··O) | d(O··O) | a(O-H··O) |
|---|---|---|---|---|---|
| Experiment | Refs88–90 | 5.44 | N/A | 2.98 | 174 ± 20 |
| Full QM | |||||
| BP86 | this work | 5.72 | 1.91 | 2.89 | 174.5 |
| Meier et al.†32 | N/A | 1.91 | 2.89 | 174.0 | |
| Loferer et al.‡27 | 5.68 | 1.89 | 2.86 | 165.5 | |
| B3LYP | this work | 5.99 | 1.93 | 2.90 | 175.6 |
| Meier et al.†32 | N/A | 1.93 | 2.90 | 176.0 | |
| Full MM: TIP3P water model | |||||
| TIP3P | this work | 6.55 | 1.79 | 2.75 | 174.0 |
| Jorgensen et al.76 | 6.50 | N/A | 2.74 | N/A | |
| Loferer et al.27 | 7.08 | 1.75 | 2.73 | 176.3 | |
| Lev et al.30 | 6.14 | 1.83 | 2.81 | 178.7 | |
| Full MM: SPC water model | |||||
| SPC | this work | 6.61 | 1.75 | 2.75 | 176.1 |
| Jorgensen et al.76 | 6.59 | N/A | 2.75 | N/A | |
| Meier et al.†32 | N/A | 1.93 | 2.99 | 164.9 | |
| QM/MM: TIP3P water model | |||||
| BP86-TIP3P | this work | 8.09 | 1.65 | 2.65 | 179.9 |
| Loferer et al.‡27 | 7.97 | 1.73 | 2.72 | 178.0 | |
| BLYP-TIP3P | this work | 8.18 | 1.64 | 2.63 | 178.9 |
| Lev et al.30 | 8.34 | 1.69 | 2.68 | 179.0 | |
| TIP3P–BP86 | this work | 6.62 | 1.82 | 2.78 | 177.7 |
| Loferer et al.‡27 | 6.87 | 1.78 | 2.76 | 177.9 | |
| TIP3P–BLYP | this work | 5.56 | 1.86 | 2.82 | 176.7 |
| Lev et al.30 | 6.06 | 1.84 | 2.81 | 178.3 | |
| QM/MM: SPC water model | |||||
| BP86-SPC | this work | 7.79 | 1.68 | 2.67 | 179.7 |
| Meier et al.†32 | N/A | 1.68 | 2.65 | 166.8 | |
| B3LYP-SPC | this work | 7.72 | 1.70 | 2.68 | 179.6 |
| Meier et al.†32 | N/A | 1.70 | 2.66 | 166.7 | |
| SPC-BP86 | this work | 6.66 | 1.79 | 2.79 | 179.6 |
| Meier et al.†32 | N/A | 1.98 | 2.97 | 172.2 | |
| SPC-B3LYP | this work | 6.72 | 1.79 | 2.79 | 179.9 |
| Meier et al.†32 | N/A | 1.97 | 2.97 | 171.8 | |
Steepest descent geometry optimization with energy as convergence criterium
DFT calculations employed density fitting (RI-J approximation)
Figure 3.
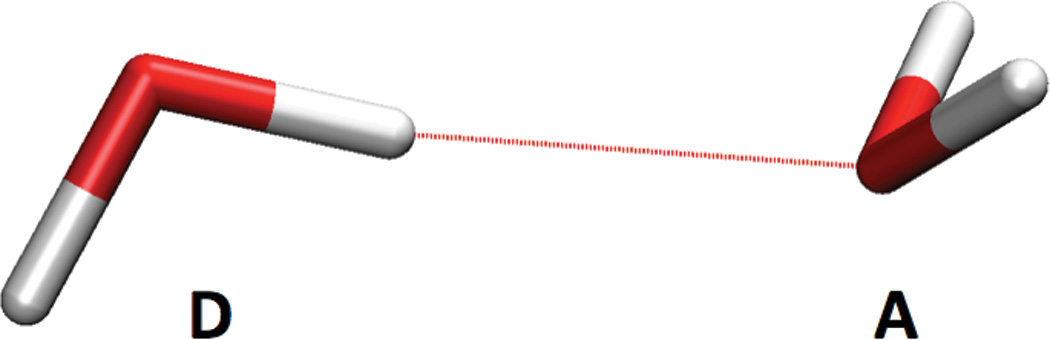
Water dimer as optimized with B3LYP/TZVP. The hydrogen bond donor (D) is on the left, the hydrogen bond acceptor (A) is on the right.
We first note that in general our results are in very good agreement with previously published data with numerical differences likely due to details of the employed DFT implementations and geometry optimization algorithms. Loferer et al.27 and Lev et al.30 employed density fitting (also called RI-J approximation) for their BP86/TZVP and BLYP/aug-ccpVQZ calculations which slightly affects energetics and geometries while Meier et al.32 used a simple steepest descent algorithm with the energy as convergence criterium for their geometry optimizations. The latter can be problematic as can be seen from the purely classical SPC results for which we obtain a binding energy and D–A distance in agreement with Jor-gensen et al.76 and the similar TIP3P model while Meier et al.32 report a much longer D–A distance and smaller bond angle a(OH··O). We confirmed that a steepest-descent geometry optimization starting from the geometry reported by Meier et al.32 remains in the vicinity of the starting point. The values reported by Meier et al.32 thus have to be interpreted with care.
The DFT calculations result in binding energies and geometries that are close to the experimental values, underestimating the hydrogen bond distance by 0.1 Å. The TIP3P and SPC water models, being parameterized to reproduce bulk water properties, overestimate the binding energy by 1 kcal/mol and underestimate the hydrogen bond distance over 0.2 Å. Compared to the reference QM calculations, the QM/MM calculations all result in increased binding energies and reduced D–A distances while the bond angle a(OH··O) remains close to linear. The hydrogen bond is shorter with a larger binding energy if the hydrogen bond donor water molecule is in the QM region. The QM/MM calculations are thus closer to the reference QM results and experimental data if the QM water is the hydrogen bond acceptor.
5.2 MD energy conservation for NMA and ADP in explicit water
The forces obtained from electronic structure software packages using default settings are in some instances not accurate enough for reasonable energy conservation during constant energy MD simulations, although the numerical accuracy may be sufficient for standard quantum chemical applications such as explorative geometry optimizations with loose convergence criteria. We thus have established default settings for Hartree–Fock and DFT calculations for all supported electronic structure programs that are employed by the QM/MM interface to reduce the numerical noise such that the energy is conserved to a high degree during MD simulations in the microcanonical ensemble. This typically involves tightening the default SCF convergence criteria and associated integral neglect thresholds as well as increasing the accuracy of the numerical quadrature grid for the XC potential and energy in the case of DFT calculations. As stated earlier, by default the converged wave function or electron density is used as the initial guess in the subsequent MD step to speed up the SCF convergence. This can lead to an energy drift62,63, however, using a tight SCF convergence threshold that minimizes numerical noise in the gradients, reduces this substantially.
Figure 4 shows the total energy during constant energy QM/MM MD simulations of Nmethylacetamide and alanine dipeptide in a droplet of SPC/Fw water molecules. The trajectories were started after an initial equilibration with MM MD at 300 K, used electronic embedding, a time step of 0.5 fs, and the default settings of the QM/MM interface. Only part of alanine dipeptide is in the QM region which is chosen such that peptide bonds do not cross the QM/MM boundary. Hydrogen link atoms are used to saturate dangling bonds in the QM region. The energy conservation is excellent both without (NMA) and with link atoms (ADP), with an energy drift1 of 2.5 × 10–3 kcal/mol/ps (which is equivalent to 1.1 × 10–6 kT/dof/ps, where dof is degrees of freedom) for the B3LYP/6–311G** simulation of NMA, an energy drift of 3.5 × 10–3 kcal/mol/ps (equivalent to 1.6 × 10–6 kT/dof/ps) for the MP2/cc-pVDZ simulation of NMA, an energy drift of 3.8×10–3 kcal/mol/ps (equivalent to 1.7 × 10–6 kT/dof/ps) for the B3LYP/6–311G** simulation of ADP, and an energy drift of –3.6 × 10–3 kcal/mol/ps (equivalent to –1.6 × 10–6 kT/dof/ps) for the MP2/cc-pVDZ simulation of ADP.
Figure 4.

Energy conservation during constant energy QM/MM MD simulations of N-methylacetamide (left) and alanine dipeptide (right) in a droplet of 408 and 403 SPC/Fw water molecules, respectively, with electronic embedding at 300 K using a time step of 0.5 fs. The QM region is highlighted and consists of the entire N-methylacetamide molecule but only part of alanine dipeptide with the QM/MM boundary crossing covalent bonds such that peptide bonds are left intact. Hydrogen link atoms are used to saturate dangling bonds in the QM region.
5.3 Typical computational throughput
The computational throughput that can be achieved with QM/MM MD simulations depends on many parameters, in particular the QM method and basis set employed, and to some extent also the electronic structure software and the available hardware. It is not our intention to benchmark the performance of the different electronic structure codes that are supported by the QM/MM interface and each of the different software packages has its own strengths and advantages, both in terms of available QM methods and in terms of computational performance on different hardware. However, it is useful to have an idea of the order of magnitude of the time scales that are currently accessible with QM/MM MD. Of particular interest is DFT since it has an excellent cost/accuracy ratio. For QM/MM MD simulations with a QM region size of 50 to 100 atoms using a time step of 0.5 fs and running on 16 stateof-the art CPU cores we have observed a computational throughput of around 0.1 ps/day to 0.5 ps/day using hybrid DFT methods with split valence basis sets and polarization functions on all atoms. If constraining the bond distances to hydrogen atoms using the SHAKE81,82 algorithm does not affect the simulation results, a time step of 2.0 fs can be used also for QM/MM MD simulations, with a corresponding increase of the computational throughput by a factor of four as compared to a time step of 0.5 fs. For some applications a larger degree of numerical noise in the forces than provided with the default settings of the QM/MM interface may be acceptable, in particular if one is not interested in dynamical quantities but average statistics and a thermostat or stochastic dynamics are employed. In this case the SCF convergence threshold and XC quadrature grid parameters may be loosened which would lead to a corresponding speedup, however, the order of magnitude of accessible time scales would remain.
6 Calcium coordination to aspartate
Binding of calcium ions to carboxylate groups is of importance for biological function, for example in cell signaling based on ion exchange proteins91, but also for technological applications such as ion exchangers based on polyelectrolytes92. Here we present studies of calcium coordination to the carboxyl group in acetyl and N-methyl capped aspartate as a model system for ion/protein interactions in aqueous solution. We compare results from MM simulations and QM/MM simulations that serve to demonstrate the functionality of the new interface in combination with the advanced sampling techniques that are available in AMBER.
6.1 Binding energy curve in vacuum
Figure 5 shows the potential energy profile for binding of a Ca2+ ion by the carboxylate group of Ace–Asp–NMe in vacuum as obtained with different MM and QM potentials at geometries optimized with the AMBER ff99SB force field. The reaction coordinate chosen is the distance between the carbon atom of the carboxylate group and the calcium ion which was restrained during the geometry optimizations. The binding curves obtained with the various methods are distinctively different. However, all QM models show an encouraging agreement around the minimum of the binding curve which is found at a reaction coordinate value of approximately 2.6 Å. The MM binding curve has its minimum at a larger distance of approximately 2.9 Å, indicating that the classical vdW potential in the AMBER ff99SB force field is too repulsive. At large separation the binding curves will be dominated by the classical 1/R behavior of the electrostatic interaction between the two ions. At intermediate distances, however, dispersion interactions are of importance. The latter are not properly accounted for in the DFT models employed here which explains the discrepancy between MP2 and DFT results with increasing ion separation. Based on the potential energy profiles presented here, one would expect results for the free energy profile of this coordination process in aqueous solution that differ between MM and QM/MM models. It is reasonable to expect that similar results should be obtained with the semiempirical PM6 model and DFT, which is not the case as shown below.
Figure 5.

Potential energy profile of calcium ion coordination to the carboxyl group in acetyl and N-methyl capped aspartate in vacuum. QM energies are evaluated at MM geometries optimized with the AMBER ff99SB force field. The angle Cβ–Ccarboxyl–Ca2+ was kept at 180 degrees.
6.2 Binding free energy curve in explicit water
Figure 6 shows the PMF for the reaction coordinate defined above as obtained from MM and QM/MM MD simulations in aqueous solution. This PMF is an upper bound to the PMF that would be obtained if the Ca2+ ion were allowed to move freely instead of restraining the angle Cβ–Ccarboxyl–Ca2+ to 180 degrees. However, this additional restraint greatly aids in converging the simulations and the effect is expected to be rather small (see also below).
Figure 6.

Free energy profile of calcium ion coordination to the carboxyl group in acetyl and N-methyl capped aspartate solvated with TIP3P water. Results are presented for periodic boundary conditions (PBC) and a water droplet. The peptide and Ca2+ ion are treated with the AMBER ff99SB force field in the MM simulations and quantum mechanically in the QM/MM simulations. PM6/a and PM6/b use different semiempirical QM/MM interaction potentials.
6.2.1 Results from MM simulations
MM simulations have been performed both with the peptide and ion solvated in a water box using periodic boundary conditions (PBCs) as well as a water droplet. MM based MD simulations often employ PBCs which is computationally very efficient because a cutoff is used for the real-space nonbonded interactions while long-range electrostatics beyond the cutoff are accounted for with the PME algorithm54. DFT based QM/MM simulations under PBCs, however, have to apply a cutoff for the electrostatic interactions between the QM and MM regions, first term on the right hand side of Equation (2). There is also no drawback in terms of computational efficiency since the QM calculation dominates the computational effort. Figure 6 clearly shows that the results obtained from the PBC and water droplet simulations are indistinguishable, thus justifying the use of a water droplet instead of PBCs for the QM/MM simulations.
In a study of calcium binding to polyacrylates92 using classical MM force fields, the free energy gain for binding to a single carboxylate group was found to be approximately 6 kcal/mol with a barrier for detachment of approximately 11 kcal/mol. This data is in good agreement with the MM results for calcium binding to Ace-Asp-NMe presented here which are 4 kcal/mol for the free energy of binding and 9 kcal/mol for the detachment barrier using the AMBER ff99SB force field and TIP3P water. This good agreement also justifies the use of the distance based reaction coordinate in conjunction with the angle restraint as discussed above. The minimum of the binding curve is found at approximately 3.1 Å, a slightly larger value than obtained from the geometry optimizations with restrained reaction coordinate (Figure 5), in agreement with the intuitive picture that the presence of a polar solvent facilitates ion dissociation.
6.2.2 Results from QM/MM simulations
In all QM/MM simulations presented here, both the Ace–Asp–NMe peptide and the Ca2+ ion are treated quantum mechanically while the TIP3P water model is retained for the surrounding water droplet. This is a rather drastic approximation since it neglects all charge transfer between the ions and the solvent, however, is a useful model to compare semiempirical and DFT methods. The semiempirical simulation setup requires some additional explanation. In AMBER, the electrostatic interaction between QM cores and MM point charges is modeled with the standard MNDO64-type core repulsion function that uses atom-specific parameters for the exponents in its damping function. The PM6 Hamiltonian36, however, uses a core repulsion function with an explicit atom pair-wise parameterization and parameters for an interaction between QM cores and MM point charges are not available. Such parameters could certainly be optimized, however, for the present work we have chosen to retain the MNDO-type expression with parameters borrowed from the PM365 Hamiltonian, where available. Since PM3 parameters are not available for calcium, we have tested two different values for the corresponding exponent to be used in conjunction with the PM6 Hamiltonian: a) a value of 1.3 Å−1 (denoted as PM6/a) which is close to the value for magnesium and b) a value of 2.0 Å−1 (denoted as PM6/b) which reduces the magnitude of the interaction between the QM core and the point charges. The choice of this parameter will thus clearly have an effect on the solvation behavior of the Ca2+ ion.
From Figure 6 we can see that the parameterization used for the electrostatic QM/MM interaction of the semiempirical QM/MM Hamiltonian has a pronounced effect on the free energy of binding and the corresponding barrier for ion dissociation. On the other hand, there is virtually no effect close to the equilibrium binding distance. We also note that the minimum for the PM6 binding curve remains approximately at the same value as obtained from the static calculations (Figure 5). The PM6/b results lead to a much lower binding energy and barrier for ion dissociation than the PM6/a results which can be understood in terms of the discussion above: the larger exponent used in the core repulsion function for QM/MM core/point charge interactions for PM6/b leads to an improved hydration of the Ca2+ ion which counterbalances the energy loss upon ion dissociation. This lowers the barrier to approximately 31 kcal/mol which is much closer to the DFT results than the PM6/a results.
Unlike the semiempirical models, DFT based QM/MM MD simulations do not depend on any additional parameters for QM/MM interactions and are uniquely defined through Equations (1) and Equation (2) and the choice of the MM force field and the QM Hamiltonian. The PMF obtained with B3LYP/6–31G* in the QM region shows large differences from the purely classical MM result (see Figure 6). In particular, the minimum of the binding curve is at a shorter distance of approximately 2.8 Å. This could be expected based on the results from the static calculations (Figure 5) which also have the DFT minimum at a shorter distance than the MM minimum. Similar to the MM results the minimum in the DFT based QM/MM free energy profile in aqueous solution is at larger distance than in gas phase which again can be rationalized in terms of the polar solvent facilitating ion dissociation. Note that this is not the case for the QM/MM calculations with the semiempirical PM6 Hamiltonian, which, compared to the DFT results, have the minimum at a distance that is too short. The free energy barrier for ion dissociation with B3LYP/6–31G* is approximately 25 kcal/mol, which is lower than the 31 kcal/mol obtained with PM6/b, but much larger than the 9 kcal/mol obtained from the MM simulations.
6.2.3 Remaining error sources
There are several potential sources of error for the QM/MM MD simulations of ion dissociation in solution as presented in this work. In the case of the PM6 simulations, the semiempirical Hamiltonian itself puts a strict limitation on the attainable numerical accuracy and, as shown above, distinctively different results are obtained with a more sophisticated DFT model such as B3LYP/6–31G*.
A major source of error for all QM/MM simulations is the QM/MM boundary. In the simulations presented here, the QM/MM boundary must have a pronounced effect on the PMF for ion dissociation in solution because it is situated right next to the atoms involved in the dissociation process and crosses coordination bonds between the calcium ion and the solvent. For one, the QM/MM van der Waals interactions93,94 can be expected to significantly affect the reaction free energies and barrier heights for ion association or dissociation processes. At least as important, however, is the fact that charge transfer between the ions and the solvent is neglected and that the TIP3P water model lacks polarizability, both of which can be expected to be important for the stabilization of highly ionic systems such as the ones studied here.
The effect of the QM/MM boundary can often be minimized by increasing the size of the QM region such that the QM/MM boundary is sufficiently far from the region of interest. This is possible for example for studies of reactive events in active sites of enzymes that are deeply buried within the protein and thus have an environment that does not change during the course of a simulation. For reactive events in solution such as the ion dissociation studied here, however, this is not possible with conventional QM/MM schemes that require a selection of atoms belonging to the QM and MM region at the beginning of a simulation. Alternative approaches, such as adaptive QM/MM42,95,96 that allow for a diffusion of solvent molecules into and out of the QM region, are thus required to include solvent surrounding the ions into the QM region to improve upon the results presented in this work.
7 Conclusions and Outlook
We have presented a versatile and easily extensible QM/MM interface that supports a wide range of electronic structure software packages. This interface has been integrated with the AMBER MD software package, enabling ab initio wave function theory and DFT based QM/MM geometry optimizations and MD simulations within both mechanical and electronic embedding schemes. The implementation supports all of AMBER’s advanced sampling techniques and has been designed to be easy to use for anybody who is familiar with classical MD simulations, requiring not much more than a straight forward selection of the QM region and the QM method in addition to the classical MD simulation setup. An automated link atom setup is used for simulations in which the QM/MM boundary crosses covalent bonds.
The interface controls the required data exchange between the MD software and the electronic structure software which is implemented in two fashions: a) traditional, file based data exchange with system calls is available for all supported QM software packages (at the time of writing ADF, GAMESS-US and NWChem for mechanical embedding and Gaussian, Orca and TeraChem for mechanical and electronic embedding) b) a client/server model based on the MPI-2 standard that increases performance and portability but requires corresponding modifications to the electronic structure code. This interface is currently supported by TeraChem. Additional electronic structure software can use the interface defined by the API of the MPI-2 client/server model without modification of the present implementation of the interface or its integration with AMBER.
The QM/MM interface defines default parameters for the electronic structure programs that lead to good energy conservation during MD simulations in the microcanonical ensemble, which we have shown with constant energy QM/MM MD simulations of Nmethylacetamide and alanine dipeptide in a water droplet using both DFT and MP2 in the QM region. Results for QM and QM/MM geometry optimizations of the water dimer are in good agreement with published data.
We have furthermore demonstrated geometry optimizations with restraints and QM/MM free energy calculations of a calcium ion binding to the carboxylate group of acetyl and Nmethyl capped aspartate in aqueous solution as a model for ion/protein interactions, comparing the semiempirical PM6 Hamiltonian to DFT with the B3LYP XC functional and the 6–31G* basis set. We have shown that the PM6 results depend strongly on the semiempirical parameters chosen for the QM/MM core/point charge interactions and that the DFT based QM/MM simulation predicts an equilibrium binding distance that lies inbetween the PM6 and MM results. The free energy of binding and corresponding dissociation barrier obtained from the QM/MM simulations is too large as compared to MM results. The QM/MM results will need to be improved with an appropriate description of the solvent in the vicinity of the ion and the carboxylate group, for example through inclusion into the QM region via adaptive QM/MM methods.
The new interface will be useful for such investigations as well as applications towards electronic events in large biomolecular systems, for example the photophysics of chromophores embedded in proteins or enzymatic reactions.
Supplementary Material
A new interface to electronic structure programs provides ab initio wave function theoryand density functional theory for mixed quantum mechanical and molecular mechanical simulations with the AMBER software package. Data exchange between the programs is implemented by means of files and system calls or the message passing interface standard. Results for the free energy of binding of calcium ions to aspartate in aqueous solution are presented to demonstrate features of this interface.
Acknowledgments
A.W.G. acknowledges funding provided by the Department of Energy, Office of Biological and Environmental Research and Office of Advanced Computing Research, SciDAC award DE-AC36-99G0-10337. This work was funded in part by the University of California through UC Lab award 09-LR-06-117792 to R.C.W., the National Science Foundation through the Scientific Software Innovations Institutes program – NSF SI2-SSE (NSF1148276) grant to R.C.W., and also by the National Institutes of Health through grant 1R01GM100934-01 to R.C.W and A.W.G. R.C.W. additionally acknowledges funding through the NSF XSEDE program and through a fellowship from NVIDIA Inc. Computer time was provided by the San Diego Supercomputer Center through National Science Foundation XSEDE awards CHE100149 to A.W.G. and CHE130010 to R.C.W and A.W.G.
Footnotes
Energy drifts are obtained from a linear regression of total energies along the trajectory.
Supporting Information Available
Details about the implementation including the API of the interface to electronic structure software.
References
- 1.Warshel A, Levitt M. J. Mol. Bio. 1976;103:227. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- 2.Singh UC, Kollman PA. J. Comput. Chem. 1986;7:718. [Google Scholar]
- 3.Field MJ, Bash PA, Karplus M. J. Comput. Chem. 1990;11:700. [Google Scholar]
- 4.Bakowies D, Thiel W. J. Phys. Chem. 1996;100:10580. [Google Scholar]
- 5.Sherwood P. In: in Modern Methods and Algorithms of Quantum Chemistry Proceedings. Grotendorst J, editor. Vol. 3. Jülich: John von Neumann Institute for Computing; 2000. pp. 285–305. >NIC series. [Google Scholar]
- 6.Gao J, Truhlar DG. Annu. Rev. Phys. Chem. 2002;53:467. doi: 10.1146/annurev.physchem.53.091301.150114. [DOI] [PubMed] [Google Scholar]
- 7.Friesner RA, Guallar V. Ann. Rev. Phys. Chem. 2005;56:389. doi: 10.1146/annurev.physchem.55.091602.094410. [DOI] [PubMed] [Google Scholar]
- 8.Lin H, Truhlar DG. Theoret. Chem. Acc. 2007;117:185. [Google Scholar]
- 9.Thiel W. In: in Multiscale Simulation Methods in Molecular Sciences. Gro-tendorst J, Attig N, Blu¨gel S, Marx D, editors. Vol. 42. Jülich: Institute for Advanced Simulation, Forschungszentrum Jülich; 2009. pp. 203–214. of NIC series. [Google Scholar]
- 10.Senn HM, Thiel W. Angew. Chem. Int. Ed. 2009;48:1198. doi: 10.1002/anie.200802019. [DOI] [PubMed] [Google Scholar]
- 11.Bernstein N, Kermode JR, Csányi G. Rep. Prog. Phys. 2009;72:1. [Google Scholar]
- 12.Zhang R, Lev B, Cuervo JE, Noskov SY, Salahub DR. In: in Advances in Quantum Chemistry. Sabin JR, Br¨andas E, editors. Vol. 59. San Diego: Elsevier Academic Press Inc.; 2010. pp. 353–400. chap. 10. [Google Scholar]
- 13.Sousa SF, Fernandes PA, Ramos MJa. Phys. Chem. Chem. Phys. 2012;14:12431. doi: 10.1039/c2cp41180f. [DOI] [PubMed] [Google Scholar]
- 14.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, et al. Amber 12. San Francisco: University of California; 2012. [Google Scholar]
- 15.Salomon-Ferrer R, Case DA, Walker RC. WIREs Comput. Mol. Sci. 2013;3:198. [Google Scholar]
- 16.Walker RC, Crowley MF, Case DA. J. Comput. Chem. 2008;29:1019. doi: 10.1002/jcc.20857. [DOI] [PubMed] [Google Scholar]
- 17.de M, Seabra G, Walker RC, Elstner M, Case DA, Roitberg AE. J. Phys. Chem. 2007;A 111:5655. doi: 10.1021/jp070071l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rossi I, Truhlar DG. Chem. Phys. Lett. 1995;233:231. [Google Scholar]
- 19.Nam K, Cui Q, Gao J, York DM. J. Chem. Theory Comput. 2007;3:486. doi: 10.1021/ct6002466. [DOI] [PubMed] [Google Scholar]
- 20.Wei D, Salahub D. Chem. Phys. Lett. 1994;224:291. [Google Scholar]
- 21.Stanton RV, Little LR, Merz KM. J. Phys. Chem. 1995;99:17344. [Google Scholar]
- 22.Stanton RV, Hartsough DS, Merz KM. J. Comp. Chem. 1995;16:113. [Google Scholar]
- 23.Ryde U. J. Comp. Aided Mol. Des. 1996;10:153. doi: 10.1007/BF00402823. [DOI] [PubMed] [Google Scholar]
- 24.Lyne PD, Hodoscek M, Karplus M. J. Phys. Chem. A. 1999;103:3462. [Google Scholar]
- 25.Eichinger M, Tavan P, Hutter J, Parinello M. J. Chem. Phys. 1999;110:10452. [Google Scholar]
- 26.VandeVondele ALJ, Rothlisberger U. J. Chem. Phys. 2002;116:69416947. [Google Scholar]
- 27.Loferer MJ, Loeffler HH, Liedl KR. J. Comput. Chem. 2003;24:1240. doi: 10.1002/jcc.10283. [DOI] [PubMed] [Google Scholar]
- 28.Biswas PK, Gogonea V. J. Chem. Phys. 2005;123:164114. doi: 10.1063/1.2064907. [DOI] [PubMed] [Google Scholar]
- 29.Woodcock HL, III, Hodoscek M, Gilbert ATB, Gill PMW, Schaefer HF, III, Brooks BR. J. Comput. Chem. 2007;28:1485. doi: 10.1002/jcc.20587. [DOI] [PubMed] [Google Scholar]
- 30.Lev B, Zhang R, de la Lande A, Salahub D, Noskov SY. J. Comp. Chem. 2010;31:1015. doi: 10.1002/jcc.21387. [DOI] [PubMed] [Google Scholar]
- 31.Okamoto T, Yamada K, Koyano Y, Asada T, Koga N, Nagaoka M. J. Com-put. Chem. 2011;32:932. doi: 10.1002/jcc.21678. [DOI] [PubMed] [Google Scholar]
- 32.Meier K, Schmid N, van Gunsteren WF. J. Comput. Chem. 2012;33:2108. doi: 10.1002/jcc.23047. [DOI] [PubMed] [Google Scholar]
- 33.Torras J, Deumens E, Trickey SB. J. Comp.-Aid. Mat. Des. 2006;13:201. [Google Scholar]
- 34.ChemShell, a Computational Chemistry Shell, see www.chemshell.org
- 35.Sherwood P, de Vries AH, Guest MF, Schreckenbach G, Cat-low CRA, French SA, Sokol AA, Bromley ST, Thiel W, Turner AJ, et al. J. Mol. Struct. (Theochem) 2003;632:1. [Google Scholar]
- 36.Stewart JJP. J. Mol. Model. 2007;13:1173. doi: 10.1007/s00894-007-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu L, Söderhjelm P, Ryde U. J. Chem. Theory Comput. 2011;7:761. doi: 10.1021/ct100530r. [DOI] [PubMed] [Google Scholar]
- 38.Ufimtsev IS, Martinez TJ. J. Chem. Theory Comput. 2009;5:1004. doi: 10.1021/ct800526s. [DOI] [PubMed] [Google Scholar]
- 39.Ufimtsev IS, Martinez TJ. J. Chem. Theory Comput. 2009;5:2619. doi: 10.1021/ct9003004. [DOI] [PubMed] [Google Scholar]
- 40.Isborn C, Götz AW, Clark MA, Walker RC, Martínez TM. J. Chem. Theory Comput. 2012;8:5092. doi: 10.1021/ct3006826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.MPI: A Message-passing Interface Standard, Version 2.2. Germany: High-Performance Computing Center Stuttgart, University of Stuttgart, No-beliusstr. 19, 70550 Stuttgart; 2009. Message Passing Interface Forum. [Google Scholar]
- 42.Park K, G¨otz AW, Walker RC, Paesani F. J. Chem. Theory Comput. 2012;8:2868. doi: 10.1021/ct300331f. [DOI] [PubMed] [Google Scholar]
- 43.te Velde G, Bickelhaupt FM, Baerends EJ, Guerra CF, van Gisbergen SJA, Snijders JG, Ziegler T. J. Comput. Chem. 2001;22:931. [Google Scholar]
- 44.Guerra CF, Snijders J, te Velde G, Baerends EJ. Theoret. Chem. Acc. 1998;99:391. [Google Scholar]
- 45.ADF2012, SCM, Theoretical Chemistry. Amsterdam, The Netherlands: Vrije Universiteit; http://www.scm.com. [Google Scholar]
- 46.Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S, et al. J. Comput. Chem. 1993;14:1347. [Google Scholar]
- 47.Gordon MS, Schmidt MW. In: Theory and Applications of Computational Chemistry, the first forty years. by edited, Dykstra CE, Frenking G, Kim KS, Scuseria GE., editors. Asterdam: Elsevier; 2005. pp. 1167–1189. chap. 41. [Google Scholar]
- 48.Valiev M, Bylaska EJ, Govind N, Kowalski K, Straatsma TP, Dam HJJV, Wang D, Nieplocha J, Apra E, Windus TL, et al. Comp. Phys. Comm. 2010;181:1477. [Google Scholar]
- 49.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, et al. Gaussian 09, Revision C.01. Wallingford CT: Gaussian, Inc.; 2010. [Google Scholar]
- 50.Neese F. WIREs Comput. Mol. Sci. 2012;2:73. [Google Scholar]
- 51.Shao Y, Fusti-Molnar L, Jung Y, Kussmann J, Ochsenfeld C, Brown ST, Gilbert ATB, Slipchenko LV, Levchenko SV, ONeill DP, et al. Phys. Chem. Chem. Phys. 2006;8:3172. [Google Scholar]
- 52.Still WC, Tempczyk A, Hawley RC, Hendrickson T. J. Am. Chem. Soc. 1990;112:6127. [Google Scholar]
- 53.Pellegrini E, Field MJ, Phys J. Chem. A. 2002;106:1316. [Google Scholar]
- 54.Darden T, York D, Pedersen L. J. Chem. Phys. 1993;98:10090. [Google Scholar]
- 55.Nam K, Gao J, York DM. J. Chem. Theory Comput. 2005;1:2. doi: 10.1021/ct049941i. [DOI] [PubMed] [Google Scholar]
- 56.Roux B. Comp. Phys. Comm. 1995;91:275. [Google Scholar]
- 57.Kästner J. WIREs Comput. Mol. Sci. 2011;1:932. [Google Scholar]
- 58.Mitsutake A, Sugita Y, Okamoto Y. 2001;60:96. doi: 10.1002/1097-0282(2001)60:2<96::AID-BIP1007>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 59.Nymeyer H, Gnanakaran S, Gárcia AE. Methods Enzymol. 2004;383:119. doi: 10.1016/S0076-6879(04)83006-4. [DOI] [PubMed] [Google Scholar]
- 60.Ceperley D. Rev. Mod. Phys. 1995;67:279. [Google Scholar]
- 61.Berne BJ, Thirumalai D. Ann. Rev. Phys. Chem. 1986;37:401. [Google Scholar]
- 62.Pulay P, Fogarasi G. Chem. Phys. Lett. 2004;386:272. [Google Scholar]
- 63.Herbert JM, Head-Gordon M. Phys. Chem. Chem. Phys. 2005;7:3269. doi: 10.1039/b509494a. [DOI] [PubMed] [Google Scholar]
- 64.Dewar M, Thiel W. J. Am. Chem. Soc. 1977;99:4899. [Google Scholar]
- 65.Stewart JJP. J. Comput. Chem. 1989;10:209. [Google Scholar]
- 66.Becke AD. Phys. Rev. A. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 67.Perdew JP. Phys. Rev. B. 1986;33:8822. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 68.Lee C, Yang W, Parr RG. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 69.Becke AD. J. Chem. Phys. 1993;98:5648. [Google Scholar]
- 70.Hehre WJ, Ditchfield R, Pople JA. J. Chem. Phys. 1972;56:2257. [Google Scholar]
- 71.Hariharan PC, Pople JA. Theoret. Chim. Acta. 1973;28:213. to get. [Google Scholar]
- 72.Krishnan R, Binkley JS, Seeger R, Pople JA. J. Chem. Phys. 1980;72:650. [Google Scholar]
- 73.Sch¨afer A, Huber C, Ahlrichs R. J. Chem. Phys. 1994;100:5829. [Google Scholar]
- 74.Dunning TH. J. Chem. Phys. 1989;90:1007. [Google Scholar]
- 75.Kendall RA, Dunning TH, Jr., Harrison RJ. J. Chem. Phys. 1992;96:6796. [Google Scholar]
- 76.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J. Chem. Phys. 1983;79:926. [Google Scholar]
- 77.Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J. Interaction models for water in relation to protein hydration. 1981:331–342. Reidel. [Google Scholar]
- 78.Wu Y, Tepper HL, Voth GA. J. Chem. Phys. 2006;124:024503. doi: 10.1063/1.2136877. [DOI] [PubMed] [Google Scholar]
- 79.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Proteins. 2006;65 doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grest GS, Kremer K. Phys. Rev. A. 1986;33:3628. doi: 10.1103/physreva.33.3628. [DOI] [PubMed] [Google Scholar]
- 81.Ryckaert J-P, Ciccotti G, Berendsen HJ. J. Comp. Phys. 1977;23:327. [Google Scholar]
- 82.Miyamoto S, Kollman PA. J. Comput. Chem. 1992;13:952. [Google Scholar]
- 83.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. J. Chem. Phys. 1984;81:3684. [Google Scholar]
- 84.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. J. Com-put. Chem. 1992;13:1011. [Google Scholar]
- 85.Grossfield A. “WHAM: the weighted histogram analysis method”, version 2.0.6. http://membrane.urmc.rochester.edu/content/wham.
- 86.Humphrey W, Dalke A, Schulten K. Journal of Molecular Graphics. 1996;14:33. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 87.Williams T, Kelley C, et al. [accessed January 27th, 2013];Gnuplot: an interactive plotting program. http://gnuplot.sourceforge.net/ [Google Scholar]
- 88.Dyke T, Muenter J. J. Chem. Phys. 1974;60:2929. [Google Scholar]
- 89.Curtiss L, Frurip D, Blander M. J. Chem. Phys. 1979;60439:2703. [Google Scholar]
- 90.Kim KS, Mhin BJ, Choi U-S, Lee K. J. Chem. Phys. 1992;97:6649. [Google Scholar]
- 91.Liao J, Li H, Zeng W, Sauer DB, Belmares R, Jiang Y. Science. 2012;335:686. doi: 10.1126/science.1215759. [DOI] [PubMed] [Google Scholar]
- 92.Bulo RE, Donadio D, Laio A, Molnar F, Rieger J, Parrinello M. Macromolecules. 2007;40:3437. [Google Scholar]
- 93.Tu Y, Laaksonen A. J. Phys. Chem. 1999;111 [Google Scholar]
- 94.Riccardi D, Li G, Cui Q. J. Phys. Chem. B. 2004;108:6467. doi: 10.1021/jp037992q. [DOI] [PubMed] [Google Scholar]
- 95.Bulo RE, Ensing B, Sikkema J, Visscher L. J. Chem. Theory Comput. 2009;5:2212. doi: 10.1021/ct900148e. [DOI] [PubMed] [Google Scholar]
- 96.Götz AW, Park K, Bulo RE, Paesani F, Walker RC. 2013 in preparation. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.