

Abstract
The presence of furan in common cooked foods along with evidence from experimental studies that lifetime exposure to furan causes liver tumors in rats and mice has caused concern to regulatory public health agencies worldwide; however, the mechanisms of the furan-induced hepatocarcinogenicity remain unclear. The goal of the present study was to investigate whether or not long-term exposure to furan causes epigenetic alterations in rat liver. Treating of male Fisher 344 rats by gavage 5 days per week with 0, 0.92, 2.0, or 4.4 mg furan/kg body weight (bw)/day resulted in dose- and time-dependent epigenetic changes consisting of alterations in DNA methylation and histone lysine methylation and acetylation, altered expression of chromatin modifying genes, and gene-specific methylation. Specifically, exposure to furan at doses 0.92, 2.0, or 4.4 mg furan/kg bw/day caused global DNA demethylation after 360 days of treatment. There was also a sustained decrease in the levels of histone H3 lysine 9 and H4 lysine 20 trimethylation after 180 and 360 days of furan exposure, and a marked reduction of histone H3 lysine 9 and H3 lysine 56 acetylation after 360 days at 4.4 mg/kg bw/day. These histone modification changes were accompanied by a reduced expression of Suv39h1, Prdm2, and Suv4-20h2 histone methyltransferases and Ep300 and Kat2a histone acetyltransferases. Additionally, furan at 2.0 and 4.4 mg/kg bw/day induced hypermethylation-dependent down-regulation of the Rassf1a gene in the livers after 180 and 360 days. These findings indicate possible involvement of dose- and time-dependent epigenetic modifications in the furan hepatotoxicity and carcinogenicity. Key words: Furan; liver; rat; epigenetic changes.
Furan is a volatile heterocyclic compound widely used in the chemical manufacturing industry and is also found in a variety of common heat-processed foods (Bakhiya and Apple, 2010; Morehouse et al., 2008; Moro et al., 2012). The average exposure of the U.S. consumer to furan is ∼0.25 μg/kg body weight (bw)/day (Morehouse et al., 2008). Importantly, relatively high furan levels have been detected in infant formulas and baby foods, and it has been estimated that the average consumption by children during their first year of life and for infants consuming only formula is 0.41 and 0.9 μg/kg bw/day, respectively (Morehouse et al., 2008). Additionally, it has been reported that furan is a constituent in tobacco smoke (Charles et al., 2008). The presence of furan in food has caused concern to regulatory public health agencies such as the U.S. Food and Drug Administration and European Food Safety Authority and led to the research and evaluation of furan by organizations, including the National Toxicology Program (NTP, 1990) and the International Agency for Research on Cancer (IARC, 1995). Based on findings from high-dose animal studies, furan is considered to be a “reasonably anticipated to be a human carcinogen” by the NTP and a “possible human carcinogen (Group 2B)” by the IARC.
Furan is metabolized in the liver by CYP2E1 to a highly reactive cis-2-butene-1,4-dial that may react with various biological macromolecules, including proteins and DNA (Peterson, 2006). A lifetime exposure of rats and mice to furan produced substantial evidence of carcinogenic activity of furan (Moser et al., 2009; NTP, 1990). Specifically, exposure to a low dose of furan (2 mg/kg bw/day) resulted mainly in the development of liver cholangiocarcinomas in male and female Fisher 344 (F344) rats, whereas higher doses of furan (4 mg/kg bw/day and greater) induced hepatocellular adenomas, hepatocellular carcinomas, and mononuclear cell leukemia in both sexes of F344 rats, and hepatocellular adenoma and carcinoma in male and female B6C3F1 mice (Cordelli et al., 2010; Moser et al., 2009; NTP, 1990).
Despite a large body of evidence for furan-induced hepatocarcinogenicity, the mechanisms leading to the liver tumor development are still unclear (Bakhiya and Apple, 2010). Although some reports have demonstrated furan genotoxicity in both in vitro and in vivo model systems (Banda et al., 2013; Cordelli et al., 2010; Johansson et al., 1997; Kellert et al., 2008; Neuwirth et al., 2012), other genotoxicity studies have been negative or produced equivocal results (Durling et al., 2007; McDaniel et al., 2012) suggesting a predominantly nongenotoxic mode of furan carcinogenicity. This is further evidenced by results of several studies indicating that furan may cause liver carcinogenesis via the induction of cytotoxicity, inflammation, oxidative stress, inactivation of protein functions through covalent binding of the furan reactive metabolites, increased cell proliferation, and liver injury (Hickling et al., 2010; Jackson et al., 2014; Moro et al., 2012; Moser et al., 2009).
Accumulating evidence indicates that some adverse biological effects caused by exposure to toxicants, regardless of their DNA reactivity, may be attributed to the ability of chemical compounds to provoke epigenetic alterations, including DNA hypomethylation, promoter region DNA hypermethylation of tumor suppressor genes, histone modifications, and changes in the function of chromatin modifying enzymes (Baccarelli and Bollati, 2009). Consequently, it has been hypothesized that chemically induced epigenetic alterations may play an important role in the carcinogenic process in some instances (Goodman and Watson, 2002; Pogribny and Beland, 2013). The results of several studies support the link between the adverse biological effects of furan and nongenotoxic epigenetic abnormalities (Chen et al., 2010, 2012; Jackson et al., 2014; Recio et al., 2013). For example, Chen et al. (2010) demonstrated that exposure of male F344 rats to low doses of furan (0.1–2 mg/kg bw/day) for 28 days altered the expression profiles of critical cancer-related genes, including those controlling cell cycle and cell death, which were independent of the DNA methylation status. More recently, Chen et al. (2012) reported that the exposure of male Sprague Dawley rats to a high concentration of furan (30 mg/kg bw/day) for 3 months resulted in noticeable changes in gene expression accompanied by hypermethylation and hypomethylation of specific gene promoter regions. Additionally, Recio et al. (2013) reported an altered expression of long noncoding RNAs in the livers of female B6C3F1 mice exposed to furan at 4 and 8 mg/kg bw/day for 3 weeks. Although these data suggest the possible involvement of epigenetic mechanisms in furan hepatotoxicity and carcinogenicity, there is a lack of conclusive information to clarify the role of epigenetic aberrations in their pathogenesis. In light of this, we investigated changes in the status of global DNA methylation and histone modifications, promoter methylation, and the expression of tumor suppressor and chromatin modifying genes in the livers of male F344 rats exposed to furan for 90, 180, and 360 days at doses 0, 0.92, 2.0, and 4.4 mg/kg bw/day.
MATERIALS AND METHODS
Animals, experimental design, and treatments
Male F344 rats (3 weeks of age) were obtained from the breeding colony at the National Center for Toxicological Research (NCTR). This strain of rats was used in a furan carcinogenicity bioassay conducted by the National Toxicology Program (NTP, 1990). Rats were housed in sterilized cages in a temperature controlled room (24°C) with a 12 h light/dark cycle, and given ad libitum access to water and a NIH-41 irradiated pelleted diet. At 7 weeks of age, rats were treated by gavage 5 days per week with 0, 0.92, 2.0, or 4.4 mg furan/kg bw/day dissolved in corn oil, a protocol identical that used in the previous furan carcinogenicity bioassay (NTP, 1990). Five experimental rats and five control rats from each treatment group were euthanized by exsanguination following deep isoflurane anesthesia after a period of 90, 180, or 360 days of treatments. The livers were excised, and a slice of the median lobe was fixed in neutral buffered formalin for 48 h for histopathological examination. The remaining median lobe was snap-frozen immediately in liquid nitrogen and stored at −80°C for subsequent analyses. All experimental procedures were reviewed and approved by the NCTR Animal Care and Use Committee.
Global DNA methylation
The methylation status of genomic DNA was evaluated by fluorometric MethylFlash Methylated DNA Quantification kits (Epigentek, Farmingdale, NY) according to the manufacturer's protocol.
Western blotting
The level of trimethylation of histone 3 lysine 4 (H3K4me3), histone 3 lysine 9 (H3K9me3), histone 3 lysine 27 (H3K27me3), and histone 4 lysine 20 (H4K20me3), and of acetylation of histone 3 lysine 9 (H3K9ac) and histone H3 lysine 56 (H3K56ac) were determined by Western blot analysis. Primary antibodies against histone H3K4me3, H3K9me3, H3K27me3, H4K20me3, and H3K56ac were purchased from Millipore Corporation (Billerica, MA); primary antibodies against H3K9ac were obtained from Abcam (Cambridge, MA). All primary antibodies were diluted to 1:1000. Horseradish peroxidase (HRP)-coupled secondary antirabbit antibodies (1:4000; Santa Cruz Biotechnology, Santa Cruz, CA) were used for visualization. Chemiluminescence was detected with the HRP Substrate (Millipore Corporation) and measured directly by a BioSpectrum AC Imaging System (UVP, Upland, CA). Signal intensities were analyzed using ImageQuant 5.1 software (Molecular Dynamics, Sunnyvale, CA). Equal protein loading was confirmed by immunostaining with antihistone H3 and antihistone H4 antibodies (Millipore Corporation).
RNA extraction and quantitative reverse transcription PCR
Total RNA was extracted from liver tissue using RNeasy Mini kits (Qiagen, Valencia, CA) according to the manufacturer's instructions. Total RNA (2 μg) was reverse transcribed using random primers and High Capacity cDNA Reverse Transcription kits (Life Technologies, Grand Island, NY) according to the manufacturer's protocol. cDNA were analyzed in a 96-well plate assay format using a 7900HT Fast Real-Time PCR System (Life Technologies). Each plate contained one experimental gene and a housekeeping gene. All primers for the gene expression analysis were obtained from Applied Biosystems and are listed in Supplementary table 1. The cycle threshold (Ct) for each sample was determined from the linear region of the amplification plot. The ΔCt values for all genes were determined relative to the endogenous control glyceraldehyde-3-phosphate dehydrogenase (Gapdh) or β-actin (Actb). The ΔΔCt values were calculated using treated group means relative to the control group means. The fold change data were calculated from the ΔΔCt values. A gene was considered to be differentially expressed if the change in expression corresponded to the following criteria: a p-value <0.05 and a fold change >1.5. All quantitative reverse transcription-PCR (qRT-PCR) reactions were conducted in triplicate and the experiments were repeated twice.
Methylation-specific PCR
The methylation status of CpG sites located in the promoter regions of Ras association (RalGDS/AF-6) domain family member 1 (Rassf1a), cyclin-dependent kinase inhibitor 2A (Cdkn2a; p16INK4A), and E-cadherin (Cdh1) was determined by methylation-specific PCR (MSP). Briefly, genomic DNA (1 μg) was modified by a standard bisulfite treatment, followed by PCR amplification using two sets of primers specific to unmethylated and methylated cytosine residues in the promoter region of the gene of interest. The sequence of gene-specific unmethylated and methylated primers used in this study are listed in Supplementary table 2. The MSP products were then visualized on a 3% high-resolution agarose gel. The presence or absence of a band after PCR amplification with the methylated primer set was indicative of methylation status. Negative control PCR amplifications were performed by using both sets of primers with unmodified DNA.
Statistical analyses
Results are presented as mean ± SD (n = 5 from each treatment group at each time interval). Data were analyzed by one-way analysis of variance (ANOVA), with pairwise comparisons being made by the Student-Newman-Keuls method. Linear regression analysis was used to determine dose-related trends. When necessary, the data were natural log transformed before conducting the analyses to maintain a more equal variance or normal data distribution. p-values <0.05 were considered significant.
RESULTS
Liver Global DNA Methylation and Expression of DNA Methyltransferases
Accumulating evidence indicates that exposure to well-known chemical carcinogens may perturb the status of DNA methylation in target organs (Baccarelli and Bollati, 2009; Goodman and Watson, 2002; Herceg et al., 2013; Pogribny and Beland, 2013). Furthermore, it has been suggested that the disruption of normal DNA methylation patterns may be responsible for the induction and promotion of tumor development (Feinberg, 2004; Jaffe, 2003). In light of this, the status of global DNA methylation in the livers of the F344 rats exposed to furan was determined. Figure 1A shows that genomic DNA methylation in the livers of furan-treated rats at 0.92, 2.0, and 4.4 mg/kg bw/day decreased in a dose-related fashion after a 360-day treatment period. At that time, the level of genomic DNA methylation in the livers of rats treated with furan at the doses 0.92, 2.0, and 4.4 mg/kg bw/day was lower by 30, 36, and 49%, respectively, as compared to the control rats (Fig. 1A).
FIG. 1.
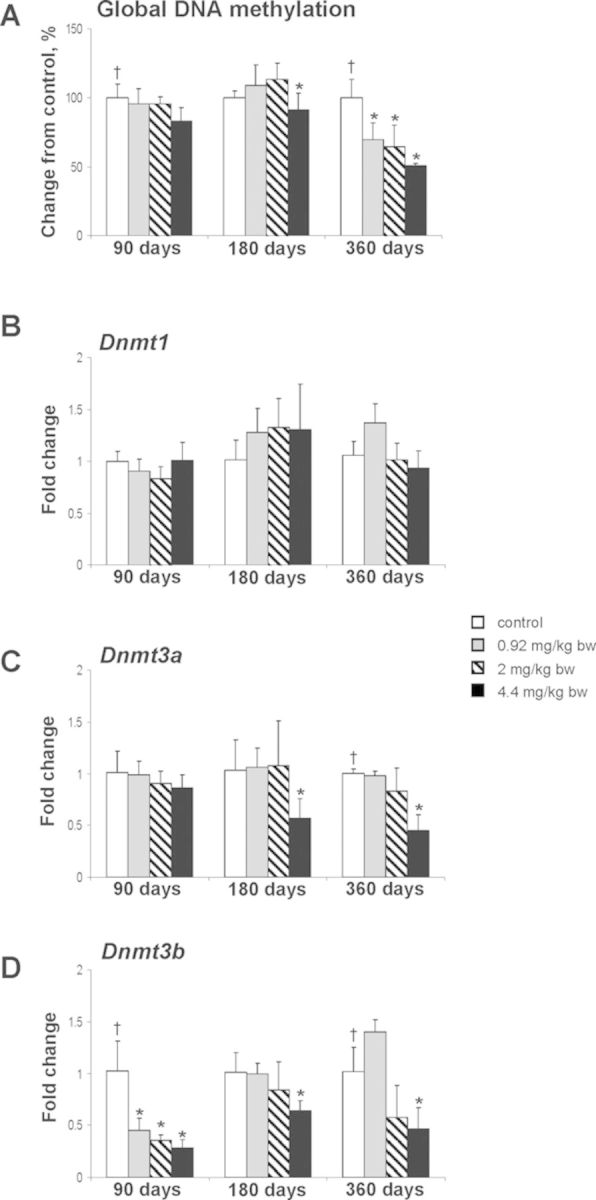
DNA methylation and the expression of DNA methyltransferases in the livers of control rats and rats treated with furan. (A) The extent of DNA methylation. The results are presented as an average percentage change in the degree of DNA hypomethylation in the livers of rats from the furan-treated group relative to that in the control group, which was assigned a value of 100%. The expression of Dnmt1 (B), Dnmt3a (C), and Dnmt3b (D) genes was determined by qRT-PCR as detailed in the Materials and Methods section. The results are presented as an average fold change in the expression of each gene in the livers of rats from each experimental group relative to that in the control group, which were assigned a value 1. Values are mean ± SD (n = 5). * denotes a significant (p < 0.05) difference from the control rats; † denotes significant (p < 0.05) trend.
The expression of Dnmt1 did not change at any time interval or dose in the livers of furan-treated rats (Fig. 1B). In contrast, the expression of Dnmt3a in the livers of rats treated with furan demonstrated a dose-related decreasing trend at 360 days, with the 4.4 mg/kg bw/day dose causing a significant reduction (Fig. 1C). Likewise, Dnmt3b was significantly down-regulated at 90, 180, and 360 days in rats treated with 4.4 mg/kg bw/day (Fig. 1D).
Status of Liver Histone Lysine Modifications
In addition to chemically induced alterations in DNA methylation, the disruption of a normal pattern of covalent histone modifications is another epigenetic change frequently observed upon chemical exposure. In order to determine whether or not furan treatment causes any alterations in histone lysine modifications, the levels of H3K4me3, H3K9me3, H3K27me3, H4K20me3, H3K9ac, and H3K56ac in the livers of control rats and rats exposed to furan were examined using Western immunoblotting (Fig. 2A). It is well established that these histone lysine modifications play a crucial role in the maintenance of genomic stability, chromatin assembly and organization, DNA damage and repair, and regulation of gene transcription (Jenuwein, 2006; Tanaka et al., 2012).
FIG. 2.

Westernblot analysis of histone H3K9, and H4K20 trimethylation and H3K9 and H3K56 acetylation in the livers of control rats and rats treated with furan. (A) Representative Western blot images. (B) Densitometric analysis of the immunostaining results, shown as percent change in histone modification level in the each experimental group relative to the corresponding values in the control group. Values are mean ± SD (n = 5). * denotes a significant (p < 0.05) difference from the control rats; † denotes significant (p < 0.05) trend.
Figure 2B shows that the levels of histone H3K9 and H4K20 trimethylation decreased in a dose-dependent manner after 180 and, especially, 360 days of exposure to furan. After 360 days, the levels of histone H3K9me3 and H4K20me3 in the livers of rats treated with furan decreased by 35 and 36%, respectively, as compared to the control rats. Treatment with furan had little to no effect on histone H3K4 or H3K27 trimethylation in the rat livers (Supplementary fig. 1).
The levels of histone H3K9ac and H3K56ac in livers of F344 rats treated with furan at 4.4 mg/kg remained unchanged after 90 and 180 days, but decreased significantly after 360 days of exposure (Fig. 2). At that time, the levels of histone H3K9ac and H3K56ac decreased by 45 and 32%, respectively.
Expression of Chromatin Modifying Genes in Liver
To investigate further the mechanisms of epigenetic alterations, the expression of histone modifying genes was determined by qRT-PCR. Exposure to furan at 4.4 mg/kg bw/day causes a significant down-regulation of Prdm2, Suv39h1, Suv4-20h2, and Ehmt2 histone methyltransferases and dose-dependent down-regulation of Ep300 and Kat2a histone acetyltransferases after a 360-day treatment period (Fig. 3).
FIG. 3.

The expression of chromatin-modifying genes in the livers of control rats and rats treated with furan. The expression of Prdm2, Suv39h1, Suv420h2, Ehmt2, Ep300, and Kat2a genes was determined by qRT-PCR as detailed in the Materials and Methods section. The results are presented as an average fold change in the expression of each gene in the livers of rats from each experimental group relative to that in the control group, which were assigned a value 1. Values shown are mean ± SD (n = 5). * denotes a significant (p < 0.05) difference from the control rats; † denotes significant (p < 0.05) trend.
Gene-Specific DNA Methylation Changes in the Liver of Fisher 344 Rats Exposed to Furan
One of the most compelling examples of the mechanistic link between epigenetic alterations and carcinogenesis is silencing of critical tumor-suppressor genes by hypermethylation of promoter CpG islands (Jones and Baylin, 2007). Hence, the promoter methylation and expression status of p16INK4a, Rassf1a, and Cdh1, tumor suppressor genes that are frequently silenced epigenetically in human and rodent liver carcinogenesis (Pogribny and Rusyn, 2014), was investigated.
Exposure to furan at a dose 4.4 mg/kg bw/day caused hypermethylation of p16INK4a and Rassf1a after 180 days, and dose-dependent hypermethylation of these genes after 360 days in the livers rats treated with furan at 2.0 and 4.4 mg/kg (Table 1, Figs. 4A and 4C), whereas no methylation changes were found at the earlier, 90-day time interval. In contrast, furan treatment did not change the methylation status of the Cdh1 gene at any time interval (Table 1, Fig. 4C). qRT-PCR analysis demonstrated that only the expression of Rassf1a (Fig. 4B) significantly decreased in association with its promoter hypermethylation, whereas the expression of hypermethylated p16INK4a (Fig. 4D) or unmethylated Cdh1 (Fig. 4E) did not change.
TABLE 1. Percentage of Methylated Samples in Liver Rats Treated with Furan.
| Gene name | 180 days | 360 days | ||||
|---|---|---|---|---|---|---|
| 0.92 mg/kg | 2 mg/kg | 4.4 mg/kg | 0.92 mg/kg | 2 mg/kg | 4.4 mg/kg | |
| Cdh1 | 0% (0/5)a | 0% (0/5) | 0% (0/6) | 0% (0/4) | 0% (0/4) | 0% (0/5) |
| p16INK4a | 0% (0/5) | 0% (0/5) | 80% (4/5) | 50% (2/4) | 75% (3/4) | 100% (4/4) |
| Rassf1a | 0% (0/5) | 0% (0/5) | 83% (5/6) | 0% (0/4) | 75% (3/4) | 100% (5/5) |
Note. Samples in control groups were unmethylated.
aMethylated/total samples ratio.
FIG. 4.
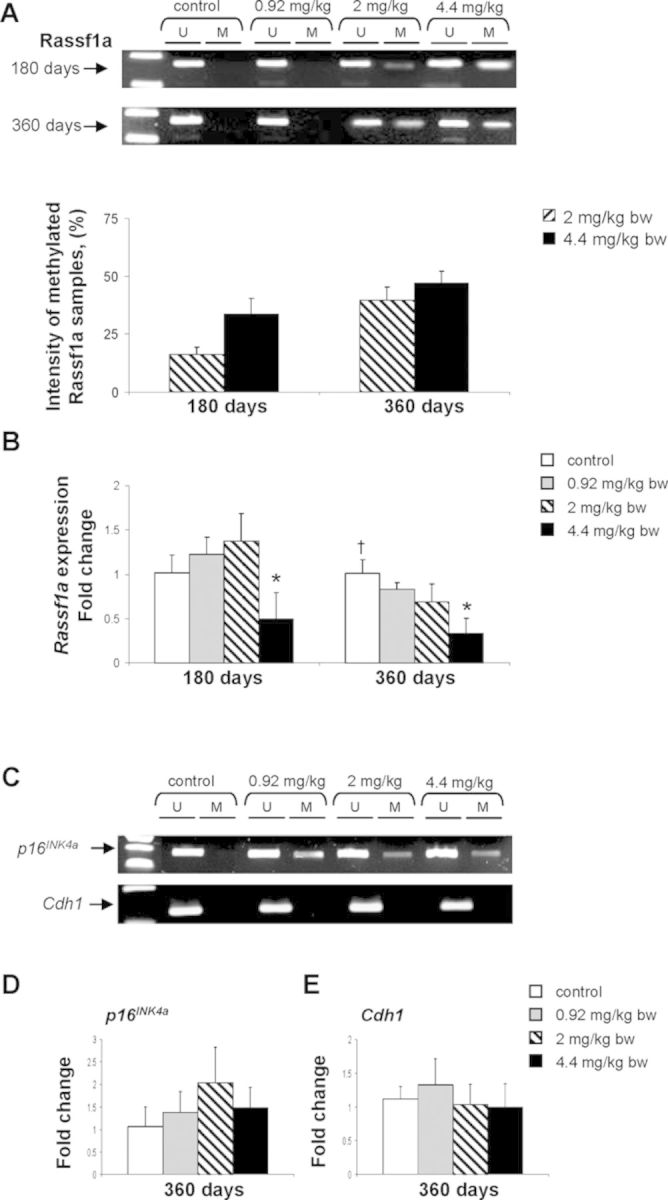
Promoter methylation and expression of Rassf1a, and Cdh1 tumor suppressor genes in the livers of control rats and rats treated with furan. (A) Methylation-specific PCR analysis of Rassf1a promoter methylation. Bisulfite-modified DNA was amplified with two sets of primers specific to unmethylated (U) and methylated (M) CpG sites in the promoter region. Presence of methylated PCR product band after amplification with methylation-specific primers indicates appearance of de novo methylation. (B) The expression of Rassf1a as determined by qRT-PCR. (C) Methylation-specific PCR analysis of p16INK4a and Cdh1 promoter methylation. (D) The expression of p16INK4a as determined by qRT-PCR. (D) The expression of Cdh1 as determined by qRT-PCR. Values are mean ± SD (n = 5). * denotes a significant (p < 0.05) difference from the control rats; † denotes significant (p < 0.05) trend.
DISCUSSION
It is widely believed that continuous exposure to certain natural and man-made chemical and physical agents plays a causative role in the development of cancer. This suggestion is based on a wealth of data showing the ability of some these agents to trigger the mutational and/or nonmutational (i.e., epigenetic) events that may affect the proper expression of genetic information leading to the accumulation of multiple cancer-specific phenotypes in target organ. Recent work on mechanisms of chemical carcinogenesis indicates that both genotoxic and nongenotoxic carcinogens affect the cellular epigenetic state via induction of various types epigenotoxic abnormalities (Baccarelli and Bollati, 2009; Herceg et al., 2013; Pogribny and Beland, 2013).
The present study demonstrated that long-term exposure of F344 rats to furan produced prominent dose- and time-dependent epigenetic changes in the liver consisting of alterations in the global pattern of DNA methylation and histone lysine methylation and acetylation, gene-specific methylation, and altered expression of chromatin modifying genes. Specifically, exposure to furan caused global demethylation of hepatic DNA after 360 days of treatment. We also observed a sustained decrease in the levels of histone H3K9 and H4K20 trimethylation in the livers after 180 and 360 days of furan exposure, and a marked reduction of histone H3K9 and H3K56 acetylation after 360 days at the highest dose of 4.4.mg/kg bw/day. These histone modification changes were accompanied by a reduced expression of Suv39h1, Prdm2, and Suv4-20h2 histone methyltransferases and Ep300 and Kat2a histone acetyltransferases. Additionally, treatment with furan induced hypermethylation-dependent down-regulation of the Rassf1a tumor suppressor gene in the livers after 180 and 360 days of treatment at the highest furan dose.
Previous reports have demonstrated that carcinogen-induced demethylation of the genome in target organs is one of the main epigenetic responses to a range of well-known chemical liver carcinogens, including arsenic, 2-acetylaminofuorene, and aflatoxin B1 (Chen et al., 2004; Wu et al., 2013). In contrast, Chen et al. (2010, 2012) previously reported that exposure of male F344 rats to furan at 0.1 and 2.0 mg/kg bw/day for 28 days, or male Sprague Dawley rats to 30 mg/kg day/bw for 3 months did not alter the status of global DNA methylation in the livers. Similarly, in the present study, the status of genomic DNA methylation remained unaffected in the livers of furan-treated rats at any dose at 90 days; however, after 360 days we found substantial decrease in genomic DNA methylation in the livers of F344 rats exposed to all three dose levels (0.92, 2.0, and 4.4 mg/kg bw/day).
It is well-established that the normal pattern of DNA methylation is closely related to the functioning of the DNA methylation machinery, including DNA methyltransferase DNMT1, DNMT3a and DNMT3b, DNA demethylases, DNA integrity, and the status of one carbon metabolism. The results of the present study indicate that furan-induced loss of genomic DNA methylation may be explained by a reduced expression of both de novo Dnmt3a and Dnmt3b after 180 and 360 days of treatment. This suggestion corresponds to previous findings demonstrating that inactivation of either Dnmt3a or Dnmt3b resulted in global DNA hypomethylation (Dodge et al., 2005; Raddatz et al., 2012).
The decrease in global DNA methylation observed upon furan treatment was accompanied by a decrease in histone H3K9me3, H4K20me3, H3K9ac, and H3K56ac. Trimethylated states of histone H3K9 and H4K20 have been shown to play a central role in the formation of heterochromatin and the maintenance of genomic stability and chromatin integrity (Jenuwein, 2006). Additionally, H3K56 acetylation is a key player in DNA replication and repair (Tanaka et al., 2012). Therefore, it is possible that a loss of any of these histone marks may compromise genomic stability and integrity and impair the viability of cells.
The mechanisms responsible for the furan exposure-related histone alterations may be attributed, in part, to an altered expression of histone-modifying genes. Specifically, decreased time- and dose-dependent levels of histone H3K9 and H4K20 trimethylation in the livers of rats exposed to furan were accompanied by a reduced expression of Suv39h1, Prdm2, and Suv4-20h2 histone methyltransferases. Likewise, the decrease in acetylation of histone H3K9 and H3K56 can be explained by down-regulation of Ep300 and Kat2a histone acetyltransferases.
An additional important finding in this study is promoter hypermethylation of p16INK4a and Rassf1a tumor suppressor genes in the livers of furan-treated rats; however, of these hypermethylated genes, only the expression of Rassf1a changed. This is unexpected but not unusual observation since any change in gene expression or even a greater expression of hypermethylated genes has been reported during hepatocarcinogenesis and in human HCC (Revil et al., 2013). These findings suggest that not all DNA methylation changes are of equal importance during carcinogenesis and that some DNA methylation changes may be purely “passenger events” as is the case with genetic mutations (Kalari and Pfeifer, 2010). The results of this study demonstrate that the expression of Rassf1a was significantly decreased in association with its promoter methylation. This indicates that hypermethylation of Rassf1a may be a significant exposure-related epigenetic event in the pathogenesis of furan-induced liver carcinogenesis. This suggestion is supported by the wealth of data that has established a role of Rassf1a down-regulation in the loss of cell cycle control, resistance to apoptosis, and enhanced genomic instability (Richter et al., 2009). Additionally, several reports have documented that hypermethylation of Rassf1a is not only one of the most common epigenetic alterations in full-fledged liver tumors, but also a frequent epigenetic abnormality found in the early stages of aflatoxin B1- and hepatitis B virus-related human hepatocarcinogenesis (Um et al., 2011; Zhang et al., 2002).
In the present study, epigenetic changes in the livers of furan-exposed rats occurred in the absence of the evident morphological neoplastic abnormalities. This suggests that these epigenetic alterations are directly related to the effect of furan exposure and are not a consequence of neoplastic morphological changes; however, we cannot exclude the possibility that nonneoplastic morphological lesions, e.g., cellular proliferation, cell death, inflammation, could have contributed, in part, to the observed epigenetic alterations. Further research is needed to determine the mechanistic role of epigenetic alterations in furan-induced liver carcinogenesis.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
Acknowledgments
This work was carried out under the auspices of the National Toxicology Program (NTP) and supported by an Interagency Agreement between Foodand Drug Administration (FDA) and National Institute of Environmental Health Sciences (NIEHS) (FDA IAG no. 224-12-0003/NIEHS IAG no. AES12013) and supported in part by appointment (A.d.C., T.K.) to the Postgraduate Research Program at the NCTR administered by the Oak Ridge Institute for Science and Education (ORISE).
REFERENCES
- Baccarelli A., Bollati V. Epigenetics and environmental chemicals. Curr. Opin. Pediatr. 2009;21:243–251. doi: 10.1097/mop.0b013e32832925cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhiya N., Apple K.E. Toxicity and carcinogenicity of furan in human diet. Arch. Toxicol. 2010;84:563–578. doi: 10.1007/s00204-010-0531-y. [DOI] [PubMed] [Google Scholar]
- Banda M., Recio L., Parsons B.L. ACB-PCR measurement of spontaneous and furan-induced H-ras codon 61 CAA to CTA and CAA to AAA mutation in B6C3F1 mouse liver. Environ. Mol. Mutagen. 2013;54:659–667. doi: 10.1002/em.21808. [DOI] [PubMed] [Google Scholar]
- Charles S.M., Jia C., Batterman S.A., Godwin C. VOC and particulate emission from commercial cigarettes: Analysis of 2,5-DMF as an ETS tracer. Environ. Sci. Technol. 2008;42:1324–1331. doi: 10.1021/es072062w. [DOI] [PubMed] [Google Scholar]
- Chen H., Li S., Liu J., Diwan B.A., Barrett J.C., Waalkes M.P. Chronic inorganic arsenic exposure induces hepatic and individual gene hypomethylation: Implications for arsenic hepatocarcinogenesis. Carcinogenesis. 2004;25:1779–1786. doi: 10.1093/carcin/bgh161. [DOI] [PubMed] [Google Scholar]
- Chen T., Mally A., Ozden S., Chipman J.K. Low doses of the carcinogen furan alter cell cycle and apoptosis gene expression in rat liver independent of DNA methylation. Environ. Health Perspect. 2010;118:1597–1602. doi: 10.1289/ehp.1002153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T., Williams T.D., Mally A., Hamberger C., Mirbahai L., Hickling K., Chipman J.K. Gene expression and epigenetic changes by furan in rat liver. Toxicology. 2012;292:63–70. doi: 10.1016/j.tox.2011.10.020. [DOI] [PubMed] [Google Scholar]
- Cordelli E., Leopardi P., Villani P., Marcon F., Macri C., Caiola S., Siniscalchi E., Conti L., Eleuteri P., Malchiodi-Albedi F., et al. Toxic and genotoxic effects of oral administration of furan in mouse liver. Mutagenesis. 2010;25:305–314. doi: 10.1093/mutage/geq007. [DOI] [PubMed] [Google Scholar]
- Dodge J.E., Okano M., Dick F., Tsujimoto N., Chen T., Wang S., Ueda Y., Dyson N., Li E. Inactivation of Dnmt3b in mouse embryonic fibroblasts results in DNA hypomethylation, chromosomal instability, and spontaneous immortalization. J. Biol. Chem. 2005;280:17986–17991. doi: 10.1074/jbc.M413246200. [DOI] [PubMed] [Google Scholar]
- Durling L.J.K., Svensson K., Abramsson-Zetterberg L. Furan is not genotoxic in the micronucleus assay in vivo or in vitro. Toxicol. Lett. 2007;169:43–50. doi: 10.1016/j.toxlet.2006.08.020. [DOI] [PubMed] [Google Scholar]
- Feinberg A.P. The epigenetic of cancer etiology. Semin. Cancer Biol. 2004;14:427–432. doi: 10.1016/j.semcancer.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Goodman J.I., Watson R.E. Altered DNA methylation: A secondary mechamism involved in carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 2002;42:501–525. doi: 10.1146/annurev.pharmtox.42.092001.141143. [DOI] [PubMed] [Google Scholar]
- Herceg Z., Lambert M.P., van Veldhoven K., Demetriou C., Vineis P., Smith M.T., Straif K., Wild C.P. Towards incorporating epigenetic mechanisms into carcinogen identification and evaluation. Carcinogenesis. 2013;34:1955–1967. doi: 10.1093/carcin/bgt212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickling K.C., Hitchcock J.M., Oreffo V., Mally A., Hammond T.G., Evans J.G., Chipman J.K. Evidence of oxidative stress and associated DNA damage, increased proliferative drive, and altered gene expression in rat liver produced by the cholangiocarcinogenic agent furan. Toxicol. Pathol. 2010;38:230–243. doi: 10.1177/0192623309357946. [DOI] [PubMed] [Google Scholar]
- IARC. Furan. IARC Monogr. Eval. Carcinog. Risks Hum. 1995;63:393–407. IARC, Lyon, France. [PMC free article] [PubMed] [Google Scholar]
- Jackson A.F., Williams A., Recio L., Waters M.D., Lambert I.B., Yauk C.L. Case study on the utility of hepatic global gene expression profiling in the risk assessment of the carcinogen furan. Toxicol. Appl. Pharmacol. 2014;274:63–77. doi: 10.1016/j.taap.2013.10.019. [DOI] [PubMed] [Google Scholar]
- Jaffe L.F. Epigenetic theories of cancer initiation. Adv. Cancer Res. 2003;90:209–230. doi: 10.1016/s0065-230x(03)90007-8. [DOI] [PubMed] [Google Scholar]
- Jenuwein T. The epigenetic magic of histone methylation. FEBS J. 2006;273:3121–3135. doi: 10.1111/j.1742-4658.2006.05343.x. [DOI] [PubMed] [Google Scholar]
- Johansson E., Reynolds S., Anderson M., Maronpot R. Frequency of Ha-ras-1 gene mutations inversely correlated with furan dose in mouse liver tumors. Mol. Carcinog. 1997;18:199–205. doi: 10.1002/(sici)1098-2744(199704)18:4<199::aid-mc3>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Jones P.A., Baylin S.B. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalari S., Pfeifer G.P. Identification of driver and passenger DNA methylation in cancer by epigenomic analysis. Adv. Genet. 2010;70:277–308. doi: 10.1016/B978-0-12-380866-0.60010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellert M., Brink A., Richter I., Schlatter J., Lutz W.K. Tests for genotoxicity and mutagenicity of furan and its metabolite cis-2-butene-1,4-dial in L5178Y tk+/− mouse lymphoma cells. Mutat. Res. 2008;657:127–132. doi: 10.1016/j.mrgentox.2008.08.014. [DOI] [PubMed] [Google Scholar]
- McDaniel L.P., Ding W., Dobrovolsky V.N., Shaddock J.G., Jr, Mittelstaedt R.A., Doerge D.R., Heflich R.H. Genotoxicity of furan in Big Blue rats. Mutat. Res. 2012;742:72–78. doi: 10.1016/j.mrgentox.2011.12.011. [DOI] [PubMed] [Google Scholar]
- Morehouse K.M., Nyman P.J., McNeal T.P., Dinovi M.J., Perfetti G.A. Survey of furan in heat processed foods by headspace gas chromatography/mass spectrometry and estimated adult exposure. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2008;25:259–264. doi: 10.1080/02652030701552949. [DOI] [PubMed] [Google Scholar]
- Moro S., Chipman J.K., Wegener J.W., Hamberger C., Dekant W., Mally A. Furan in heat-treated foods: Formation, exposure, toxicity, and aspects of risk assessment. Mol. Nutr. Food Res. 2012;56:1197–1211. doi: 10.1002/mnfr.201200093. [DOI] [PubMed] [Google Scholar]
- Moser G.J., Foley J., Burnett M., Goldsworthy T.L., Maronpot R. Furan-induced dose-response relationships for liver cytotoxicity, cell proliferation, and tumorigenicity (furan-induced liver tumorigenicity) Exp. Toxicol. Pathol. 2009;61:101–111. doi: 10.1016/j.etp.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Neuwirth C., Mosesso P., Pepe G., Fiore M., Malfatti M., Turteltaub K., Dekant W., Mally A. Furan carcinogenicity: DNA binding and genotoxicity of furan in rats in vivo. Mol. Nutr. Food Res. 2012;56:1363–1374. doi: 10.1002/mnfr.201200226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NTP. Toxicology and carcinogenesis studies of Furfural (CAS No. 98-01-1) in F344/N rats and B6C3F1 mice (gavage studies) Natl. Toxicol. Program Tech. Rep. Ser. 1990;382:1–201. [PubMed] [Google Scholar]
- Peterson L.A. Eletrophilic intermediates produced by bioactivation of furan. Drug Metab. Rev. 2006;38:615–626. doi: 10.1080/03602530600959417. [DOI] [PubMed] [Google Scholar]
- Pogribny I.P., Beland F.A. DNA methylome alterations in chemical carcinogenesis. Cancer Lett. 2013;334:215–232. doi: 10.1016/j.canlet.2012.09.010. [DOI] [PubMed] [Google Scholar]
- Pogribny I.P., Rusyn I. Role of epigenetic aberrations in the development and progression of human hepatocellular carcinoma. Cancer Lett. 2014;342:223–230. doi: 10.1016/j.canlet.2012.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raddatz G., Gao Q., Bender S., Jaenisch R., Lyko F. Dnmt3a protects active chromosome domains against cancer-associated hypomethylation. PLoS Genet. 2012;8:e1003146. doi: 10.1371/journal.pgen.1003146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recio L., Phillips S.L., Maynor T., Waters M., Jackson A.F., Yauk C.L. Differential expression of long noncoding RNAs in the livers of female B6C3F1 mice exposed to the carcinogen furan. Toxicol Sci. 2013;135:369–379. doi: 10.1093/toxsci/kft153. [DOI] [PubMed] [Google Scholar]
- Revil K., Wang T., Lachenmayer A., Kohima K., Harrington A., Li J., Hoshida Y., Llovet J.M., Powers S. Genome-wide methylation analysis and epigenetic unmasking identify tumor suppressor genes in hepatocellular carcinoma. Gastroenterology. 2013;145:1424–1425. doi: 10.1053/j.gastro.2013.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter A.M., Pfeifer G.P., Dammann R.H. The RASSF proteins in cancer; from epigenetic silencing to functional characterization. Biochim. Biophys. Acta. 2009;1796:114–128. doi: 10.1016/j.bbcan.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Tanaka A., Tanizawa H., Sriswasdi S., Iwasaki O., Chatterjee A.G., Speicher D.W., Lewin H.L., Noguchi E., Noma K. Epigenetic regulation of condensing-mediated genome organization during the cell cycle and upon DNA damage through histone H3 lysine 56. Mol. Cell. 2012;48:532–546. doi: 10.1016/j.molcel.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um T.H., Kim H., Oh B.K., Kim M.S., Kim K.S., Jung G., Park Y.N. Aberrant CpG island hypermethylation in dysplastic nodules and early HCC of hepatitis B virus-related human multistep hepatocarcinogenesis. J. Hepatol. 2011;54:939–947. doi: 10.1016/j.jhep.2010.08.021. [DOI] [PubMed] [Google Scholar]
- Wu H.C., Wang Q., Yang H.I., Tsai W.Y., Chen C.J., Santella R.M. Global DNA methylation in a population with aflatoxin B1 exposure. Epigenetics. 2013;8:962–969. doi: 10.4161/epi.25696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.J., Ahsan H., Chen Y., Lunn R.M., Wang L.Y., Chen S.Y., Lee P.H., Chen C.J., Santella R.M. High frequency of promoter hypermethylation of RASSF1A and p16 and its relationship to aflatoxin B1-DNA adduct levels in human hepatocellular carcinoma. Mol. Carcinog. 2002;35:85–92. doi: 10.1002/mc.10076. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.