

Abstract
Multiple lines of evidence indicate that Wnt/β-catenin signaling plays a fundamental role in colorectal cancer (CRC) initiation and progression. Recent genome-wide data have confirmed that in CRC this pathway is one of the most frequently modified by genetic or epigenetic alterations affecting almost 90% of Wnt/β-catenin gene members. A major challenge is thus learning how the corrupted coordination of this pathway is tied to other signalings to enhance cell growth. Peroxisome proliferator activated receptor γ (PPARγ) is emerging as a growth-limiting and differentiation-promoting factor. In tumorigenesis it exerts a tumor suppressor role and is potentially linked with the Wnt/β-catenin pathway. Based on these results, the identification of new selective PPARγ modulators with inhibitory effects on the Wnt/β-catenin pathway is becoming an interesting perspective. Should, in fact, these molecules display such properties, new research avenues would be opened aimed at developing new molecular targeted drugs. Herein, we review the basic principles and present new hypotheses underlying the crosstalk between Wnt/β-catenin and PPARγ signaling. Furthermore, we discuss the advances in our understanding as to how their altered regulation can culminate in colon cancer and the efforts aimed at designing novel PPARγ agonists endowed with Wnt/β-catenin inhibitory effects to be used as therapeutic and/or preventive agents.
Keywords: Colorectal cancer, Wnt/β-catenin pathway, Peroxisome proliferator-activated receptor γ, Genomic instability, Peroxisome proliferator activated receptor γ ligands
Core tip: Genetic and epigenetic modifications of the Wnt/β-catenin pathway play a fundamental role in the initiation and progression of colorectal cancer (CRC). The nuclear receptor peroxisome proliferator activated receptor γ (PPARγ) acts as a differentiation-promoting transcription factor with a potential link with Wnt/β-catenin. In this review, we discuss the basic principles underlying the crosstalk between Wnt/β-catenin and PPARγ signaling, present the most recent progress in understanding as to how their alterations can culminate in CRC and, finally, suggest new hypotheses and perspectives on the identification of selective PPARγ modulators endowed with Wnt/β-catenin inhibitory effects to be used as molecular targeted drugs.
INTRODUCTION
More than 1.2 million colorectal cancers (CRC) are diagnosed every year, accounting for approximately 10% of all cancers worldwide. Despite the progress made in surgical and therapeutic management, still 600000 deaths are caused every year by CRC representing over half of all gastrointestinal cancer deaths. CRC survival is highly dependent on the tumor stage at the time of diagnosis; over one-third of patients die within five years from the initial diagnosis and most of fatal outcomes result from liver metastases[1,2].
Colorectal cancer can have a hereditary (10%) or sporadic (90%) origin; in both cases, environmental factors contribute to its development. Interestingly, one third of patients have an increased risk of developing a CRC due to familial factors synthesized by yet unidentified genes. It is well established that CRC results from the cumulative effects of sequential genetic and epigenetic alterations, leading to a progressive and irreversible loss of cell growth and differentiation control[3]. In the last decades, a large number of investigations have identified several “driver genes” in CRC initiation and progression, including wingless-type (WNT) MMTV integration site family, RAS, mitogen-activated protein kinase (MAPK), phosphatidyl-inositol3-kinase (PI3K), trasforming growth factor β (TGF-β), tumor protein p53 (TP53) and DNA mismatch-repair genes. The various and sequential pathways in which these genes are involved support the theory that CRC is a heterogeneous, complex and multifactorial disease[3]. Indeed, at least three well-defined pathways, the traditional (adenoma-carcinoma sequence or Vogelstein’s model[4,5]), the alternative and the serrated pathway underlie these malignancies, leading to genomic instability that perpetuates a widespread loss of DNA integrity. Thus, genomic instability is emerging as a hallmark of the carcinogenic process and at least three distinct types have been described: chromosomal instability (CIN); microsatellite instability (MSI) and CpG island methylator phenotype (CIMP). CIN is the most common type of genomic instability, occurs in 60%-80% of CRCs and results in an imbalance of the chromosome number “manifested as aneuploidy”. MSI is an alternative pathway that accounts for 15%-20% of sporadic CRCs in which the characteristic signature is deletion of repetitive regions of DNA that in most cases generates frameshift mutations in the coding sequences of genes leading to their inactivation. CIMP is a novel instability pathway characterized by the widespread hypermethylation of CpG islands at several genomic loci[2,6,7]. These data suggest that CRC is a highly heterogeneous disease, i.e., clinicopathologically similar tumors strikingly differ as distinct biological subtypes and, consequently, in their response to treatment and patient’s survival. Extensive molecular profiling, genome-wide studies and the integrative analysis of genomic data support the notion that clinically distinct subtypes exist and provide insights into the pathways that are dysregulated in CRC[8,9]. Multiple lines of evidence indicate that Wnt signaling plays a fundamental role in CRC development as it is altered from the very early stages[10]. Consistently, almost 90% of CRCs present genetic or epigenetic alterations of Wnt players such as adenomatous polyposis coli (APC), AXIN, β-catenin, sex determining region Y (SRY)-box 9 (SOX9), regardless of the CIN or MSI signature, according to the cancer genome atlas[11]. These observations support the hypothesis that mutationally corrupted cancer (stem) cells, distributed among normal epithelial colonic cells, are the driving force of initiation and progression. Along with driver genes mutations implicated in tumor initiation, Wnt signaling alterations may influence the CRC course and prognosis and be instrumental in determining the optimal patients’ treatment.
In this context, peroxisome proliferator-activated receptor gamma (PPARγ) signaling is drawing increasing attention because of its role in CRC pathogenesis and because novel compounds identified as selective ligands could be used as pharmaceutics to improve therapies’ efficacy[12,13]. A number of studies have shown that PPARγ levels within primary tumors correlate with patients’ prognosis[14-16]. Evidence has also been provided that PPARγ tumor suppressive activity can be altered at multiple levels through aberrant phosphorylation, DNA promoter hypermethylation and microRNAs modulation. Thus, modifications of PPARγ appropriate levels, subcellular localization and activity presumably play a key role in colorectal tumorigenesis[13-15].
In this review, we present recent evidence and new hypotheses that underscore the growing impact of dysregulated Wnt and PPARγ signaling in CRC initiation and progression. Furthermore, we discuss the advances in our understanding as to how these pathways crosstalk and impact colon cancer biology and response to the therapy. Finally, we discuss new therapeutic perspectives of molecular target drugs represented by selective PPARγ modulators endowed with Wnt/β-catenin inhibitory effects.
WNT/β-CATENIN PATHWAY AND CRC INITIATION AND PROGRESSION
The epithelial cells of the intestine have a relatively short life span compared to cells of other epithelial tissues. They, in fact, orchestrate a unique mechanism of constant cell migration starting from the bottom of the crypts and going upwards to the luminal surface. It is well established that long-lived multipotent intestinal stem cells (ISCs) reside at the bottom of the intestinal crypts and give rise to transit amplifying progenitors that, upon constant upward migration, undergo cell cycle arrest and terminally differentiate into the diverse intestinal cell lineages. Once a differentiated cell has reached the villus tip or the colonic surface, it undergoes apoptosis and is shed into the lumen. The intestinal renewal system is tightly controlled and depends on the spatial organization of signals that emanate from supportive mesenchymal as well as from differentiated epithelial cells[17]. Until recently, ISCs were a rather elusive entity at the bottom of the intestinal crypt; only in the last few years, important efforts have been made in the stem cell research field to characterize their existence, position and function. In 2007 Van der Flier et al[18], through both a Wnt transcriptome analysis and in situ studies, identified a panel of 17 putative stem cell markers expressed at the crypt base[17]. Two genes displayed the strongest relationship with the Wnt signaling: Leucine rich repeat containing G protein coupled receptor 5 (Lrg5) and achaete-scute like2 (Ascl2). Lgr5 was subsequently localized at the crypt base of the mouse small intestine and positive cells were shown to be able to differentiate into all epithelial cell lineages. These studies suggest that Lgr5 and Ascl2 are intestinal-specific stem cell markers and emphasize a “crucial role” of the Wnt cascade not only during embryonic development but also in adult organs, particularly in tissue homeostasis, cell renewal and ISC maintenance[18-20]. Wnt is the “fusion of two terms” the segment polarity gene wingless (wg) discovered in Drosophila and the proto-oncogene integration-1 (int-1). The first direct connection of the Wnt pathway with CRC came out in the early 1990s. The APC gene was found to be involved in a hereditary cancer syndrome, termed familial adenomatous polyposis (FAP) but also in sporadic CRC[4,5]. Soon thereafter, the large “scaffold” cytoplasmic APC protein was found to interact with β-catenin providing the molecular basis of the seminal work by Fearon and Vogelstein: CRC develops as a stepwise accumulation of genetic hits in specific genes and pathways[4,5]. From that time, many additional components of the pathway and disease connections have been identified so that the list of new target genes, as well as new interacting pathways, constantly grows. Recent studies have disclosed that the interplay between Wnt and Hippo signaling pathways is indispensable to coordinate proliferation and differentiation during organ growth. Interaction of Wnt ligands with their receptor complexes triggers two major intracellular signaling cascades that are traditionally indicated according to the role played by β-catenin[21,22]. The Wnt “canonical” signaling is “activated” upon the binding of one of multiple Wnt factors, a family of soluble secreted proteins, to one of ten possible Frizzled receptors (Fzd) in the presence of a low density lipoprotein receptor related co-receptor (LRP5 or 6). This interaction generates a cascade of events involving the cytosolic adapter protein, Disheveled (Dvl), that promotes the dissociation of a multiprotein “destruction complex” resulting in the stabilization of β-catenin and its translocation to the nucleus. Upon displacement of the transcriptional repressor Groucho, β-catenin interacts with and activates members of the T cells factors and lymphoid enhancing factors (TCF/LEF) to promote the expression of target genes involved in cell differentiation and proliferation such as c-myc (MYC), cyclin D1 (CCND1), axin 2 (AXIN2), CD44 and Survivin (Figure 1A)[23]. When the Wnt pathway is “inactive”, β-catenin binds to its destruction complex formed by the scaffold proteins, APC and axin, and is phosphorylated by the specific kinases glycogen synthase kinase 3β (GSK3β) and casein kinase 1 (CK1)[23,24]. This leads to its ubiquitination and subsequent targeting to the proteasome for degradation. β-catenin consists of three main domains: the N-terminal region of 141 aminoacids, a central core domain of 513 aminoacids, and a C-terminus of 107 aminoacids, which contains the transactivation domain. Phosphorylation of β-catenin by GSK3β and CK1 occurs at aminoacids S33, S37, T41 and S45, generating a recognition tag for ubiquitination and subsequent proteasomal degradation[25,26]. The central “core region” contains 12 imperfect armadillo repeats each formed by 42 aminoacids; the repeats consist of three α-helices and, together, the twelve repeats form a superhelix containing a long positively charged groove[27]. This structure appears to facilitate the binding to the negatively charged β-catenin binding domains (CBD) within TCF/LEF[28,29] or other interacting proteins such as APC, axin, and cadherins. Recent studies have identified two lysines, K312 and K435, defined “charged buttons”, within the armadillo repeats 5 to 9 of β-catenin, that form salt bridges with negatively charged glutamate or aspartate in the CBD of the interacting proteins[28]. Mutations of one of the components of the pathway (APC, axin, β-catenin) or autocrine signaling due to constitutive Wnt production by tumor cells cause an “active” Wnt signaling. In most cases, APC loss-of-function mutations result in a truncated and inactive protein; in other cases, mutations in β-catenin phosphoacceptor sites turn into an active oncogene; for instance, S37A β-catenin is expressed at high levels in several human carcinomas[30]. β-catenin phosphorylation is also hampered through GSK3β sequestration into multivesicular compartments and/or other still unknown mechanisms.
Figure 1.
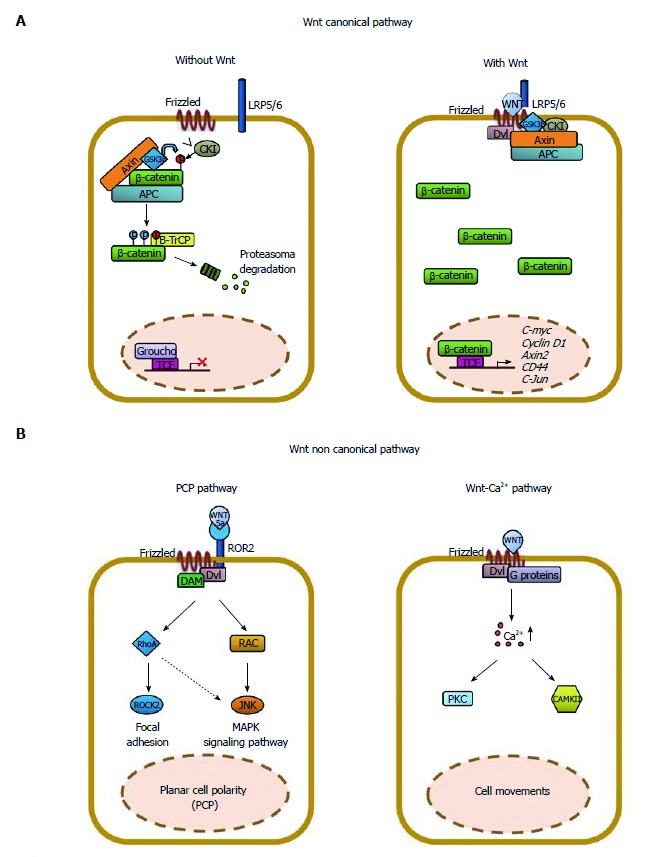
Schematic representation of the Wnt/β-catenin signaling in epithelial cells. The Wnt signaling pathway can be subdivided into a “canonical” or β-catenin-dependent and “non-canonical” or β-catenin-independent. A: In the absence of Wnt ligands, a multi-subunit destruction complex, composed by adenomatous polyposis coli (APC), Axin, GSK3β, CKI, binds and phosphorylates β-catenin tagging for ubiquitination and subsequent proteasomal degradation (βTrCP). The “canonical” Wnt signaling is initiated by the binding of one of 19 Wnt ligands to one of 10 Frizzled receptors (Fzd), in the presence of the co-receptor LRP5 or 6. This leads to recruitment of Disheveled and inhibition of the APC destruction complex. Accumulation of β-catenin in the cytoplasm leads to its translocation to the nucleus where it interacts with TCF/LEF to drive transcription of Wnt target genes including c-myc, cyclin D1, axin2 and others; B: The “non-canonical” Wnt signaling is initiated by the binding of Wnt5a to ROR2, alone or in combination, with a Frizzled receptor leading to the activation of the planar-cell polarity (PCP) pathway through Rock2, RhoA, Rac or JNK. Alternatively, Wnt11 can bind a Frizzled receptor alone and activate the Wnt/calcium pathway that involves the calcium/calmodulin dependent Kinase II (CamKII), protein-kinase-C (PKC) and nuclear factor of activated T cells (NFAT). Importantly, the “non-canonical” Wnt pathway inhibits the “canonical“ one either impairing β-catenin accumulation in the cytoplasm or the β-catenin/TCF/LEF complex formation.
The critical step in the Wnt canonical pathway appears then to be the ratio of cytosolic and/or membrane-associated β-catenin levels vs its nuclear counterpart[30]. Consistently, nuclear β-catenin is an indicator of an active Wnt signaling, likely operating in cancer initiating cells, and is a useful biomarker associated with CRC disease progression and poor prognosis; more recently, it has predominantly been observed at the invasive front of CRC tissues. According to these data, a recent meta-analysis suggests that increased cytoplasmic expression of β-catenin, not accompanied by nuclear accumulation, has no relationship with the prognosis[31]. Finally, growing evidence indicates that aberrant activation of the Wnt cascade leads to stem cell expansion, proliferation and disturbed tissue architecture (Figure 1A).
The so-called Wnt “non-canonical” signaling is independent of β-catenin function and is less characterized than the “canonical” one. It is initiated by the binding of Wnt5a to receptor tyrosine kinase-like orphan receptor 2 (ROR2), alone or in combination with a Frizzled receptor, leading to the activation of the planar-cell polarity (PCP) pathway through Rock2, RhoA, Rac or JNK. Alternatively, Wnt11 can bind a Frizzled receptor alone and activate the Wnt/calcium pathway that involves the calcium/calmodulin-dependent kinase II (CamKII), protein-kinase-C (PKC) and nuclear factor of activated T cells (NFAT) (Figure 1B)[32]. Importantly, the Wnt “non-canonical” pathway inhibits the canonical one either impairing β-catenin accumulation in the cytoplasm or the β-catenin/TCF/LEF complex formation. In this review, for space reasons, we will focus only on the Wnt “canonical”, β-catenin-dependent signaling. In epithelial cells, membrane-bound β-catenin interacts with E-cadherin forming cell adhesion complexes that anchor the extracellular matrix to the cytoskeleton[33]. Upon β-catenin nuclear translocation, the interactions with E-cadherin are reduced, impairing cell-cell interactions and providing cells a migration and invasion potential into the neighbouring tissues and, eventually, into the circulation. These events are the basis of the epithelial mesenchymal transition (EMT), a process implicated in tumor progression and metastasis[34]. Both activation of the Wnt/β-catenin signaling and E-cadherin loss are important effectors of EMT in CRC. β-catenin nuclear accumulation at the invasive front of CRCs has been associated with migrating cancer stem cells (MCSCs), metastatic spreading and EMT. Conversely, the serrated pathway has been associated with a lower frequency of nuclear β-catenin localization or reduced membrane-bound β-catenin expression than the traditional one (adenoma-carcinoma sequence), suggesting that the Wnt “non-canonical” pathway may influence metastasis formation especially in right-sided tumors[6,14].
PPARγ SIGNALING PATHWAY IN CRC INITIATION AND PROGRESSION
PPARs are ligand-activated transcription factors belonging to the nuclear receptor superfamily. PPARs activate transcription by recognizing specific sequence motifs, defined PPRE (peroxisome proliferator response elements), located in the regulatory regions of target genes as heterodimers with the retinoid X receptors (RXR)[12,35-37]. In the absence of ligand, PPARs are complexed with corepressor proteins such as the nuclear receptor corepressor (NCoR) or silencing mediator of retinoid and thyroid receptors (SMRT) and act as transcriptional repressors. Ligand binding induces conformational changes that allow displacement of the corepressor complexes and recruitment of transcriptional coactivators, including members of the steroid receptor coactivator (SRC) family and histone acetyltransferases, such as p300/CBP (Figure 2)[12,35-37]. A variety of endogenous and exogenous lipophilic molecules, such as polyunsaturated fatty acids, prostaglandines, leukotrienes and hypolipidemic drugs, have been identified as PPAR ligands. The structural heterogeneity of these compounds seems to reflect the conformation of the ligand binding domain (LBD), that forms a large Y-shaped hydrophobic pocket with relatively low ligand specificity (Figure 3A). Yet these compounds display selective binding for each of the three PPAR isotypes identified so far PPARα (NR1C1), PPARβ/δ (NR1C2) and PPARγ (NR1C3) that, in addition to their ligand specificity, display distinct tissue expression patterns. PPARγ, in particular, is expressed in adipose tissue, muscle, gastrointestinal tract, blood cells, macrophages and liver. PPARγ modulates cellular and whole-body glucose and lipid homeostasis, increases insulin sensitivity in adipose and muscle tissues following activation by the antidiabetic agents thiazolidinediones (TZDs)[12,35-37]. PPARγ has also been implicated in the modulation of immune and inflammatory processes, vascular homeostasis and cell differentiation both in normal and neoplastic tissues[13,37,38]. In line with this, PPARγ is expressed in a variety of tumors and its role in cancer initiation/progression has been debated for long time[13,37]. In vitro studies have shown that PPARγ activation induces growth arrest of epithelial-derived cancer cell lines, including those from thyroid, lung, prostate, breast, pituitary and colon[36]. Consistently, some PPARγ downstream targets, such as p18, p21, and p27 are induced, determining a cell cycle block[39,40]. PPARγ-dependent upregulation of the tumor suppressor gene phosphatase and tensin homolog (PTEN) inhibits PI3-kinase and AKT phosphorylation reducing cell migration and proliferation[41]. The anti-proliferative effects are reinforced by downregulation of the anti-apoptotic protein B-cell CLL/lymphoma 2 (Bcl-2)[42]. More recently, PPARγ has been endowed with anti-angiogenic activity through inhibition of VEGF and its receptors in various cells[43] and with anti-inflammatory activity through inhibition of NFκB-mediated gene transcription[44]. Finally, PPARγ hampers the EMT and thus metastasis formation[45]. All these data strongly support for PPARγ a role as a tumor suppressor; other studies, in contrast, support a role as tumor promoter[46,47]. A more recent work suggests for PPARγ a dual function as a tumor promoting factor in neuroblastoma cells and tumor suppressor in breast cancer cells[48]. Also in vivo, contentious results have been reported: administration of PPARγ ligands increases the incidence of colon tumors in Apc+/Min mice[49,50]. In contrast, PPARγ has no effects on tumor incidence in Apc/1638N and 1309 mice, using both genetic and pharmacological models[51,52]. Recently, exposure to pioglitazone, a TZD family member, suppresses colon tumor growth in Apc+/Min mice[52]. Data obtained by a Pparg tissue-specific biallelic knockout in ApcMin/+ mice have apparently solved these contraddictory observations. In this mouse strain, an increased tumor incidence and tumor size is observed, consistent with the in vitro data: PPARγ ligands inhibit cell growth even in the presence of APC mutations[53]. In azoxymethane (AOM)-treated mice, the most widely used preclinical model of sporadic CRC in rodents, PPARγ inhibits colon carcinogenesis and TZDs act as potent suppressors of tumor formation[54]. Of note, some of the effects attributed to TZDs can be due to PPARγ-independent effects[55]. A direct role of PPARγ as tumor suppressor is confirmed by the observation that hemizygous Pparg colon-specific knockout mice display a significantly higher incidence of colon tumors following AOM treatment[56]. Epidemiological studies in humans have clearly established a link between chronic inflammatory conditions, such as inflammatory bowel diseases (IBD), and a higher risk of CRC[57]. In colitis-associated cancers (CAC), tumor promotion is mainly due to the presence of a leucocyte infiltration and to inflammatory mediators; moreover, administration of nonsteroidal anti-inflammatory drugs to IBD patients reduces the risk of CRC development[57,58]. In spite of the results obtained in murine models, evidence of a PPARγ involvement in human colon carcinogenesis is still circumstantial. PPARγ is expressed at high levels in about 60% of sporadic human CRCs and specific loss-of-function gene mutations have been reported in 8% of primary CRCs[59]. Increasing evidence suggests that PPARγ activity is attenuated during the transition from adenoma to carcinoma, likely explaining why PPARγ agonists can block the early stages of tumorigenesis, inhibiting the aberrant crypt focus (ACF) formation but with little or no effect on advanced tumor stages[36]. PPARγ phosphorylation operated by the mitogen activated kinases ERK1 and 2 and its ligand-independent SUMOylation negatively regulate its function[44,60]. Both loss-of-function mutations and the reduced activity due to posttranslational modifications, however, do not fully explain the low PPARγ levels found in 35%-40% of sporadic CRCs[61]. Interestingly, they are associated with a more aggressive course, EMT activation, and patients’ worse prognosis, suggesting that PPARγ can be considered an independent prognostic factor[16,62]. PPARG has recently been shown to be post-transcriptionally modulated by miRNAs[48,63].
Figure 2.
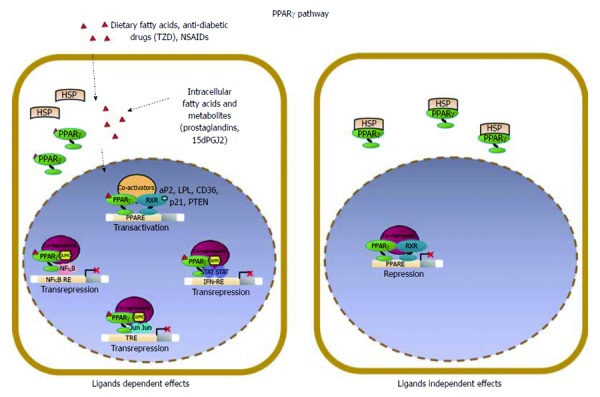
Schematic model of PPARγ signaling in epithelial cells. PPARγ acts as a pro-differentiating transcription factor in colonic epithelial cells where it is abundantly expressed. A variety of endogenous and exogenous compounds, including lipophilic molecules such as polyunsaturated fatty acids and prostaglandines, have been identified as PPARγ ligands; in particular, 15-deoxy-Δ12,14-PGJ2 (15dPGJ2), is considered a natural ligand for PPARγ. Two molecular mechanisms have been proposed to explain PPARγ effects in maintaining cellular differentiation and homeostasis referred to as PPARγ ligand-dependent or PPARγ ligand-independent effects. (1) in the ligand-dependent transactivation, PPARγ binds the cognate PPRE as heterodimer with RXR and activates target gene expression (PTEN, p21, CDH1) through the recruitment of coactivators; (2) an alternative mode of action is known as ligand-dependent trans-repression, in which the SUMOylated form of the receptor interacts with transcription factors such as NFκB, STAT or JUN and represses their target genes transcription. This is attained through the recruitment and stabilization of corepressor complexes at the promoter regions of proinflammatory or protumorigenic genes by a functionally distinct pool of PPARγ that is specifically SUMOylated at susceptible aminoacid residues in the presence of selected agonists.
Figure 3.
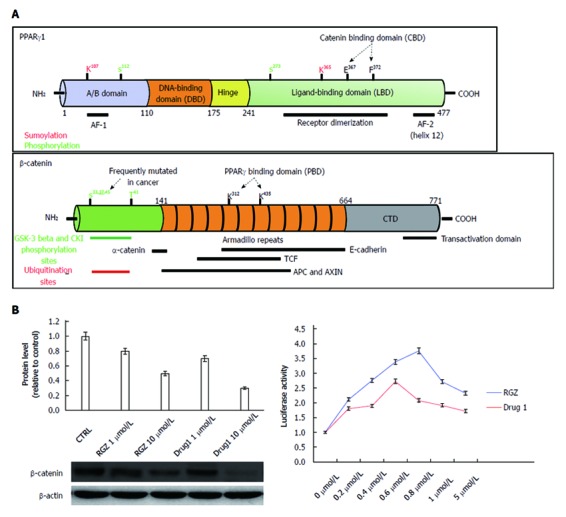
Structural and functional domains of PPARγ and β-catenin. A: The mature PPARγ protein consists of four structural/functional domains: (1) the variable A/B region at the N terminus contains the ligand-independent transactivation domain AF1 (residues 1–71 of PPARγ1); lysine 79 and serine 84 residues are targets of SUMOylation and phosphorylation events, respectively; (2) the C region is the DNA binding domain, characterized by two C4 Zinc-finger motifs, that interact with the major groove of the DNA; (3) the D or hinge region allows receptor dimerization and DNA binding; and (4) the E/F region is the ligand binding domain (LBD) constituted by 12 α-helices and 4 β-strands where the agonist accommodates. This region (helices 7 and 8) includes a β-catenin binding domain (CBD) essential for the interaction with β-catenin. The most important aminoacid residues implicated in PPARγ activity regulation are shown. The full length β-catenin is essentially composed by three domains: (1) the N-terminal domain involved in the ubiquitin-mediated degradation; (2) the arm repeat domain, containing 12 armadillo repeats that mediate the binding with cadherins, APC, TCF/LEF, CREB binding protein (CBP) and PPARγ; and (3) the carboxy terminal (CTD) or transactivating domain interacts with coactivators such as CBP or corepressors such as β-catenin inhibitor and TCF-4 (ICAT). The most important aminoacid residues implicated in β-catenin activity regulation are shown; B: Luciferase activity from HEK293T cells transfected with a PPRE-driven luciferase reporter gene and exposed to the compound indicated as Drug1 is lower than that obtained from cells exposed to rosiglitazone, indicating a reduced transactivation potential in line with the notion of a partial agonist. HT29 colon cancer cells treated with Drug1 exhibit inhibition of cell growth and a 40% higher ability to downregulate β-catenin than rosiglitazone, likely through a mechanism involving β-catenin nuclear export and proteasome-mediated degradation.
WNT/β-CATENIN AND PPARγ SIGNALING CROSSTALK IN CRC
Recent reports suggest that, in addition to members of the canonical Wnt signaling pathway, β-catenin can interact with a variety of coactivators and transcription factors, implying an even wider involvement in physiologic and pathologic processes[17,22]. Specific interactions of β-catenin with nuclear receptors signaling pathways seem to be fundamental in gut physiology. Such cross-regulation, in addition, provides a molecular platform to evaluate alterations in cell adhesion and transcription occurring during tumor progression[64]. All components of the Wnt pathway can affect nuclear receptors functions by events including transcriptional activation or repression and protein phosphorylation. Conversely, nuclear receptors and their ligands confer a dynamic impact upon Wnt functions as shown by the effects on their target genes. In this context, liver receptor homologue 1 (LRH-1) is activated upon association with β-catenin, promotes cyclin D1 and cyclin E transcriptional activation and governs the self-renewal of intestinal crypt cells. Proliferation of epithelial cells is thus enhanced, contributing to CRC development[65]. In contrast, β-catenin activity is repressed by association with retinoic acid receptor, vitamin D receptor and androgen receptor. Retinoic acid and its synthetic derivatives, 1α, 25-dihydroxyvitamin D3, the active form of vitamin D and its synthetic derivatives, have shown chemopreventive effects in animal models of CRC and are employed in cancer therapy[64]. Although the mechanism by which these receptors inhibit the Wnt/β-catenin pathway is not fully understood, several hypotheses have been put forward. In the case of the androgen receptor, the complex with β-catenin represses β-catenin activity and tumor formation in some target tissues[66]. Molecular interactions between components of the Wnt/β-catenin and PPARγ signalings have been reported in several studies, suggesting the potential for cross-regulation at different levels (Figure 3A). PPARγ protein is generally elevated in human CRC specimens and altered in colon tissues from the APC/Min mice; these results are correlated with and attributed to high β-catenin levels and activation[25,56,67,68]. Girnun and collaborators provided the first evidence that PPARγ is capable of inhibiting colon carcinogenesis by suppressing β-catenin in cells that express a functional Wnt/β-catenin pathway[56]. Loss of one Pparg allele is sufficient to increase sensitivity to chemical carcinogenesis likely due to the higher β-catenin levels that may prime the colonic epithelium to respond more rapidly to a carcinogenic insult. Addition of PPARγ selective ligands can alter the balance between this nuclear receptor and β-catenin in preadipocytes by activating PPARγ and inducing β-catenin proteasomal degradation in a GSK3β-dependent manner[68]. Oncogenic β-catenin mutants in the phosphoacceptor sites at residues S33, S37, T41 and S45 of the N terminal region of the protein, escape phosphorylation by GSK3β and proteasomal degradation by several hypothesized molecular routes[25] (Figure 3A). The accumulated protein can translocate to the nucleus and suppress PPARγ activity as assessed by lack of transcription of selected target genes[25]. Interestingly, despite the reduced activity, the total amount of PPARγ in these cells is higher likely due to posttranscriptional and not transcriptional events[67]. The selective inhibition of target genes expression may alternatively be ascribed to the interaction of β-catenin with PPARγ-associated transcriptional complexes recruited on the DNA that results in transcription inhibition or squelching of critical PPARγ coactivators[25] (Figure 3A). Notably, β-catenin interaction with the PPARγ transcriptional complexes involves the same sequence motifs of the central region that are required for binding to TCF/LEF without affecting the transcriptional activation of the target genes of this latter complex[25]. TCF factors may form binary complexes with β-catenin or ternary complexes including PPARγ. Indeed, ternary complexes containing β-catenin/PPARγ/TCF4 have already been found in tumor tissues although their biological significance remains elusive[67]. In cells with an intact form of APC or β-catenin, a dominant model can be hypothesized whereby PPARγ suppresses tumorigenesis by activating transcription of its own target genes but also facilitating the GSK3β-dependent degradation of β-catenin. In CRC-derived HT29 cells, that harbor a mutated APC and a wild type β-catenin, selective ligands activate PPARγ stimulating not only target gene expression but also interaction with β-catenin leading to its proteasomal degradation (our unpublished data). Given the fact that the canonical Wnt signaling pathway may be altered at multiple levels, PPARγ could exert its tumor suppressive activity in a context-dependent manner. Data in the literature, in fact, suggest that the cellular response to extracellular effectors and intracellular signalings depends on the relative amounts of β-catenin and PPARγ present in a given cell and on changes of this ratio that can influence their crosstalk (Figure 4). Furthermore, the molecular mechanisms by which PPARγ ligands can elicit transcription of different target genes owing to the differential recruitment of co-activators have not been completely elucidated and the reciprocal effects of PPARγ activation on Wnt signaling pathway are only at the beginning to emerge. We and others have shown that the overall survival of CRC patients is markedly better when PPARγ expression in primary tumours is detectable[14,16,61,62]. Notably, reduced PPARγ expression is not correlated with “activated” β-catenin (i.e. nuclear β-catenin) suggesting that these pathways signal differently in cancer and that there may be subtle tissue specific differences in their regulation. Defining how PPARγ influences the Wnt/β-catenin activities will be important to modulate downstream effectors as possible treatment of intestinal diseases.
Figure 4.
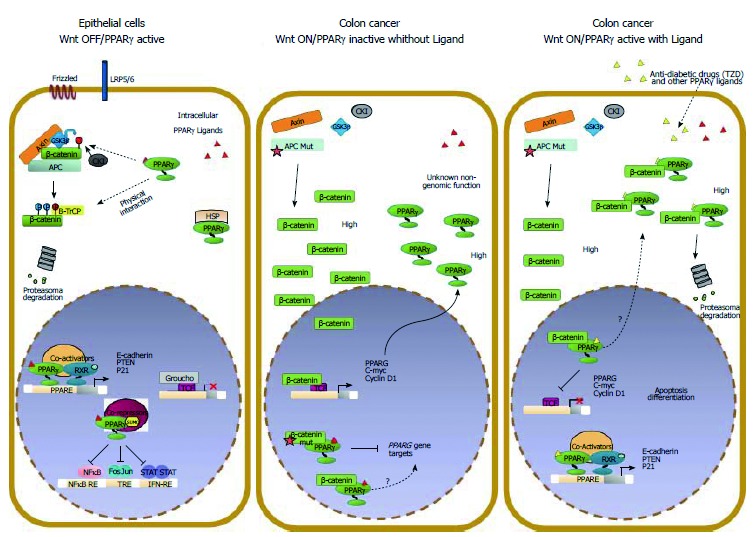
Molecular interactions between Wnt/β-catenin and PPARγ signaling in colorectal cancer cells. The Wnt/β-catenin and PPARγ signal transduction pathways likely act in a coordinated manner to ensure epithelial cells a balance between growth and differentiation. In this condition, β-catenin is targeted by PPARγ for phosphorylation and subsequent degradation. In CRC, the Wnt pathway is generally overactive and β-catenin is stabilized and translocates to the nucleus to activate Wnt target genes. In a “Wnt on” state, PPARγ protein is generally elevated likely due to high β-catenin levels. The selective inhibition of PPARγ target genes expression may be ascribed to different mechanisms: interaction of β-catenin with PPARγ-associated transcriptional complexes recruited on the DNA that results in transcription inhibition or in squelching of critical PPARγ coactivators through the alternative binding with β-catenin. Our data and those already published suggest a hypothetical model whereby a ligand-bound PPARγ suppresses Wnt/β-catenin signaling in a cancer-cell context dependent manner by: (1) activating transcription of its own target genes; (2) facilitating the GSK3β-dependent degradation of β-catenin; and (3) competing in the nucleus with transcription factors such as LEF/TCFs in blocking prosurvival β-catenin target genes also in cells harboring a mutated APC.
OLD AND NEW COMPOUNDS TARGETING BOTH β-CATENIN AND PPARγ PATHWAYS
In this section we discuss how natural and synthetic compounds can affect β-catenin and PPARγ activity. The rationale for targeting β-catenin stems from the important functions that this protein serves in cell-adhesion and in the Wnt signaling. Thus, drugs targeting aberrantly activated members of the pathway have the potential as cancer therapeutics. In addition to the known mutations, the Cancer Genome Atlas Network has identified mutations in other genes of the Wnt pathway such as transcription factor 7-like 2 (TCF7L2; previously known as TCF4), SOX9, CTNNB1 and Wilms tumour gene on the X chromosome (WTX; also known as FAM123B) known to activate the Wnt/β-catenin signaling[11]. Furthermore, many of the cell surface markers (including LGR5/GPR49, CD44, CD24 and Epcam) used to identify tumor stem cell populations are Wnt direct targets[17]. Similar considerations underlie PPARγ targeting. Given that PPARγ acts as a tumor suppressor in CRC, attempts have been made to identify and/or synthesize new molecules that can enhance this activity. The list of natural or synthetic compounds able to interfere with either Wnt/β-catenin and/or PPARγ signaling is long and constantly growing (Table 1). Herein we will describe the effects of only some of them.
Table 1.
Natural and synthetic compounds targeting Wnt/β-catenin and PPARγ pathways
| Subcategory | Drug | Pathways | Effects | Target tissues | Ref. | |
| Natural ligands | Polyphenols | Quercetin EGCG Curcumin Resveratrol DIF | β-catenin/TCF WNT GSK-3β | β-catenin, TCF, cMyc, cyclin D1, survivin, conductin reduction | CRC, Adipose Tissue, Kidney | [81,91-101] |
| Phytochemicals | Capsaicin Cladosporol Thymoquinone | β-catenin GSK-3β | β-catenin, cMyc reduction; GSK-3β, PPARγ activation | CRC | [89,102-105] | |
| Vitamins | Retinoids 1α25,-dihydroxy Vitamin D3 | β-catenin Dab2 | β-catenin reduction | CRC | [79,106-108] | |
| Synthetic ligands | NSAIDs | Aspirin Sulindac Celecoxib Indometacin Diclofenac NS398 | β-catenin, TCF, PPARγ | β-catenin, TCF cMyc reduction; PPARγ activation | CRC | [72-74,76,77,109-116] |
| Small Molecules | PNU 74654 2,4-diamino-quinazoline ICG-001 FH535 Others | β-catenin/TCF CBP Dv1 Axin Tankyrase 1,2 | Block Wnt Wnt/β-catenin suppression | CRC | [71-80] | |
| PPARγ ligands | TZDs Lutein | β-catenin/TCF | β-catenin degradation; PPARγ activation | CRC | [82-86,89] | |
| Partial PPARγ Agonists | Drug1 | β-catenin/TCF | β-catenin degradation; PPARγ activation | CRC | [86-90] |
NSAIDs: Non-steroidal anti-inflammatory drugs; CRC: Colorectal cancer.
A large number of natural compounds derived from dietary intake, plants and marine organisms, fungi and microrganisms, display chemopreventive and/or chemotherapeutic activity through modulation of the Wnt/β-catenin signaling[69-71]. Direct targeting of this pathway has been difficult, largely owing to the lack of pathway-specific targets and the potential redundancy of many components; moreover, inhibition of β-catenin signaling could cause side effects in normal, adult cells. To overcome these difficulties, it has been suggested to specifically target Wnt factors and Wnt receptors as an attractive cancer therapeutic strategy. Promising results have been obtained with Wnt3A or FZD7-neutralizing antibodies in vivo.
Among the small-molecule inhibitors, the non-steroidal anti-inflammatory drugs (NSAIDs) (indomethacin, sulindac, aspirin) and the selective COX-2 inhibitor (celecoxib) prevent β-catenin-dependent transcription in colorectal cells. Other existing drugs include molecular targeted agents such as the CBP/β-catenin antagonist ICG-001[71]. The common mechanism by which NSAIDs and their derivatives act is through inhibition of β-catenin/TCF pathway transcriptional activity and, consequently, down-regulation of target genes such as cyclin D1.
Indomethacin is a COX-1 and COX-2 inhibitor and exhibits anti-inflammatory and analgesic properties. In addition to the more general inhibition of the β-catenin/TCF pathway mentioned above, indomethacin impairs β-catenin gene expression itself at early times, as shown by the significant reduction of the corresponding mRNA. Furthermore, indomethacin stimulates β-catenin degradation in an APC/GSK3β and proteasome-independent manner (Wnt “non-canonical” pathway) even in cells bearing a mutated APC or β-catenin. These results support the potential chemotherapeutic activity of the molecule[71-73].
Sulindac inhibits β-catenin/TCF pathway and reduces β-catenin levels in human colon cancer cells. The antiproliferative effects of sulindac and its derivatives are confirmed in different mouse models of multiple intestinal adenomas and also in human colorectal adenomas. Like indomethacin, also sulindac causes β-catenin degradation mainly through an APC/GSK3β-independent mechanism, while the canonical pathway and, in turn, the proteasomal degradation are activated at late times, especially after induction of apoptosis[71,74]. Finally, a third alternative degradation mechanism in CRC cells is mediated by an increase of cGMP levels due to the cGMP phosphodiesterase (PDE) inhibition. High cGMP levels activate the cGMP-dependent kinase (PKG) that, in turn, stimulates β-catenin phosphorylation reducing its protein levels. It has been proposed that phosphorylation by PKG is an alternative way to induce proteasomal-mediated β-catenin degradation in cells with an inactive APC/GSK3β-destruction complex[75].
Aspirin also down-regulates the Wnt/β-catenin pathway in CRC cells leading to reduced transcription of target genes. Unlike other NSAIDs, this effect seems to be mediated by stabilization of β-catenin in its transcriptionally inactive form (i.e., phosphorylated form), hampering its activity as transcription factor[76].
All NSAIDs, in addition to their effects on β-catenin and related pathway, act as PPARγ ligands[77]. As such, they stimulate PPARγ-dependent effects as cell cycle block, differentiation and apoptosis, adding to those reported on β-catenin and providing the basis of a double benefit in cancer therapy.
The active form of vitamin D, 1α, 25-dihydroxyvitamin D3, plays a relevant role in chemoprevention in animal models of colon cancer by targeting the Wnt/β-catenin pathway. Although the molecular mechanisms have not been elucidated yet, the vitamin D receptor has been proposed to specifically interact with β-catenin competing for TCFs binding. The resulting complex is no longer able to activate transcription of β-catenin target genes. Indeed ternary complexes have been reported and their putative functions suggested; in the case of the vitamin D receptor, the active complex is no longer formed, the novel is not functional thus explaining the negative results[78,79]. Also small molecules, such as PKF115-584, CGP049090, PKF222-815, derived from fungi, and PKF118-744, PKF222-310 from actinomycete strains, inhibit colon cancer cell proliferation by blocking β-catenin/TCF4 interaction and, subsequently, repressing their target genes[80].
Resveratrol, a polyphenol belonging to the stilbene phytochemical family, is found in dark grapes, red wine, peanuts and shown to block colon cancer cell proliferation through inhibition of the Wnt/β-catenin pathway. Specifically, low (subapoptotic) concentrations of resveratrol reduce the expression of Bcl9, Pygo I and II and interfere with β-catenin nuclear localization[81]. In addition, resveratrol displays a PPARγ agonist effect inducing cell growth arrest and apoptosis doubling its beneficial antitumor activity. The molecular mechanisms underlying these activities are currently under investigation.
Different classes of new molecules have been isolated and/or synthesized as putative PPARγ ligands. When administered to cells in culture they are assessed for PPARγ-dependent effects and compared with full agonists. Among all molecules tested, only TZDs can interfere with the Wnt/β-catenin pathway, indicating that they function not only as PPARγ transcriptional activators[82]. Therefore, it would be useful to examine whether other partial agonists could display such a repressive effects on β-catenin signaling in order to establish a structure/function relationship. As mentioned, PPARγ displays a Y shaped ligand binding domain in which molecules with heterogeneous structures can accommodate. The aminoacid residues of the receptor involved in the interactions with full agonists have been identified as well as the conformational changes implicated[83-85]. The new partial agonists could accommodate in a distinct binding pocket, induce different conformational changes of the LBD that, in turn, result in the activation of only a subset of PPARγ target genes. Such an alternative mode of action has been reported for luteolin and appears to be carried out by an additional ligand whereby only genes involved in lipid and glucose metabolism are transcribed, through a differential recruitment of coactivators[86].
On the basis of these considerations, attempts are currently made to synthesize or isolate novel molecules able to act as PPARγ partial agonists[87,88]. These should only partially transactivate the receptor, promote transcription of genes involved in cell growth arrest, differentiation and/or apoptosis and, additionally, exhibit inhibition of β-catenin at different levels. In line with this, we have recently shown that cladosporol A, a secondary metabolite from the fungus Cladosporium tenuissimum, displays antiproliferative properties in human colorectal cancer cells through up-regulation of p21waf1/cip1 and down-regulation of cyclin D1, cyclin E, CDK2 and CDK4[89]. The effects observed are mediated through activation of PPARγ as a partial agonist. Interestingly, cladosporol A causes β-catenin nuclear export and its proteasomal-mediated degradation. Consistently, also cyclin D1 and c-Myc are reduced, indicating that the β-catenin/TCF pathway is inhibited, further strenghtening the antiproliferative properties of this drug (Zurlo et al[89] submitted for publication). We have also tested a new compound belonging to the class of chiral phenoxyacetic acids for its ability to act as a PPARγ ligand and activate downstream genes[90]. As shown in Figure 3B, Drug 1 acts as a partial agonist displaying a transactivation potential that is only 60% of that obtained with rosiglitazone. Interestingly, when tested for the ability to downregulate β-catenin, Drug 1 shows a 40% higher ability than rosiglitazone, likely through the proteasomal destruction machinery. These results suggest that it is possible to design novel PPARγ partial agonists with the ability to recruit selected coactivators and stimulate transcription of a subset of genes. The possibility to enhance β-catenin degradation and counteract an active and oncogenic Wnt signaling is an added value that should further stimulate the search for such compounds as novel drugs in cancer therapy.
CONCLUSION
CRC remains the fourth most common cause of cancer related death in western countries despite the discovery of a number of key genetic and epigenetic alterations involved in its initiation/development and the progress in the treatment. Therefore, an interdisciplinary approach that combines new techniques aimed at the identification of novel biomarkers with the treatment of advanced CRCs will aid in guiding future therapeutic interventions. Recent genome-wide studies and the integrative analysis of genomic data support the notion that clinically distinct CRC subtypes exist and provide insights into the pathways that are dysregulated in this malignancy. The Wnt/β-catenin signaling is one of the most frequently modified pathways in CRC, suggestig that drugs targeting aberrantly activated members have the potential as cancer therapeutics. PPARγ is emerging as a growth-limiting, differentiation-promoting signal with known potential link with Wnt/β-catenin signaling. Accumulating evidence suggests that PPARγ overexpression has prominent suppressive activities in CRC growth, acting as an independent biomarker of good prognosis. Naturally occurring and novel synthetic agonists capable of differently modulating PPARγ signaling and interfering with related pathways show great promise in animal models and in preclinical studies. Specifically, novel PPARγ agonists, endowed with Wnt/β-catenin pathway inhibitory activities, are currently designed and investigated to provide new molecular target therapies in CRC.
Footnotes
Supported by Ministero dell’Istruzione, Università e Ricerca, MIUR-PRIN 2010-2011, No. prot. 2010W7YRLZ_003
P- Reviewers: Barreto S, Hung LY S- Editor: Ma YJ L- Editor: A E- Editor: Liu XM
References
- 1.Schoen RE, Pinsky PF, Weissfeld JL, Yokochi LA, Church T, Laiyemo AO, Bresalier R, Andriole GL, Buys SS, Crawford ED, et al. Colorectal-cancer incidence and mortality with screening flexible sigmoidoscopy. N Engl J Med. 2012;366:2345–2357. doi: 10.1056/NEJMoa1114635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centelles JJ. General aspects of colorectal cancer. ISRN Oncol. 2012;2012:139268. doi: 10.5402/2012/139268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 4.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 5.Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 6.Pancione M, Remo A, Colantuoni V. Genetic and epigenetic events generate multiple pathways in colorectal cancer progression. Patholog Res Int. 2012;2012:509348. doi: 10.1155/2012/509348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Issa JP. Colon cancer: it’s CIN or CIMP. Clin Cancer Res. 2008;14:5939–5940. doi: 10.1158/1078-0432.CCR-08-1596. [DOI] [PubMed] [Google Scholar]
- 8.De Sousa E Melo F, Wang X, Jansen M, Fessler E, Trinh A, de Rooij LP, de Jong JH, de Boer OJ, van Leersum R, Bijlsma MF, et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat Med. 2013;19:614–618. doi: 10.1038/nm.3174. [DOI] [PubMed] [Google Scholar]
- 9.Sadanandam A, Lyssiotis CA, Homicsko K, Collisson EA, Gibb WJ, Wullschleger S, Ostos LC, Lannon WA, Grotzinger C, Del Rio M, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19:619–625. doi: 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 13.Michalik L, Desvergne B, Wahli W. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nat Rev Cancer. 2004;4:61–70. doi: 10.1038/nrc1254. [DOI] [PubMed] [Google Scholar]
- 14.Pancione M, Sabatino L, Fucci A, Carafa V, Nebbioso A, Forte N, Febbraro A, Parente D, Ambrosino C, Normanno N, et al. Epigenetic silencing of peroxisome proliferator-activated receptor γ is a biomarker for colorectal cancer progression and adverse patients’ outcome. PLoS One. 2010;5:e14229. doi: 10.1371/journal.pone.0014229. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Sabatino L, Fucci A, Pancione M, Carafa V, Nebbioso A, Pistore C, Babbio F, Votino C, Laudanna C, Ceccarelli M, et al. UHRF1 coordinates peroxisome proliferator activated receptor gamma (PPARG) epigenetic silencing and mediates colorectal cancer progression. Oncogene. 2012;31:5061–5072. doi: 10.1038/onc.2012.3. [DOI] [PubMed] [Google Scholar]
- 16.Ogino S, Shima K, Baba Y, Nosho K, Irahara N, Kure S, Chen L, Toyoda S, Kirkner GJ, Wang YL, et al. Colorectal cancer expression of peroxisome proliferator-activated receptor gamma (PPARG, PPARgamma) is associated with good prognosis. Gastroenterology. 2009;136:1242–1250. doi: 10.1053/j.gastro.2008.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clevers H. The intestinal crypt, a prototype stem cell compartment. Cell. 2013;154:274–284. doi: 10.1016/j.cell.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 18.Van der Flier LG, Sabates-Bellver J, Oving I, Haegebarth A, De Palo M, Anti M, Van Gijn ME, Suijkerbuijk S, Van de Wetering M, Marra G, et al. The Intestinal Wnt/TCF Signature. Gastroenterology. 2007;132:628–632. doi: 10.1053/j.gastro.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 19.Fearon ER. PARsing the phrase “all in for Axin”- Wnt pathway targets in cancer. Cancer Cell. 2009;16:366–368. doi: 10.1016/j.ccr.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 20.Ziskin JL, Dunlap D, Yaylaoglu M, Fodor IK, Forrest WF, Patel R, Ge N, Hutchins GG, Pine JK, Quirke P, et al. In situ validation of an intestinal stem cell signature in colorectal cancer. Gut. 2013;62:1012–1023. doi: 10.1136/gutjnl-2011-301195. [DOI] [PubMed] [Google Scholar]
- 21.Varelas X, Miller BW, Sopko R, Song S, Gregorieff A, Fellouse FA, Sakuma R, Pawson T, Hunziker W, McNeill H, et al. The Hippo pathway regulates Wnt/beta-catenin signaling. Dev Cell. 2010;18:579–591. doi: 10.1016/j.devcel.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 22.Imajo M, Miyatake K, Iimura A, Miyamoto A, Nishida E. A molecular mechanism that links Hippo signalling to the inhibition of Wnt/β-catenin signalling. EMBO J. 2012;31:1109–1122. doi: 10.1038/emboj.2011.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 24.Verheyen EM, Gottardi CJ. Regulation of Wnt/beta-catenin signaling by protein kinases. Dev Dyn. 2010;239:34–44. doi: 10.1002/dvdy.22019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J, Wang H, Zuo Y, Farmer SR. Functional interaction between peroxisome proliferator-activated receptor gamma and beta-catenin. Mol Cell Biol. 2006;26:5827–5837. doi: 10.1128/MCB.00441-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Willert K, Nusse R. Beta-catenin: a key mediator of Wnt signaling. Curr Opin Genet Dev. 1998;8:95–102. doi: 10.1016/s0959-437x(98)80068-3. [DOI] [PubMed] [Google Scholar]
- 27.Huber AH, Nelson WJ, Weis WI. Three-dimensional structure of the armadillo repeat region of beta-catenin. Cell. 1997;90:871–882. doi: 10.1016/s0092-8674(00)80352-9. [DOI] [PubMed] [Google Scholar]
- 28.Graham TA, Weaver C, Mao F, Kimelman D, Xu W. Crystal structure of a beta-catenin/Tcf complex. Cell. 2000;103:885–896. doi: 10.1016/s0092-8674(00)00192-6. [DOI] [PubMed] [Google Scholar]
- 29.Huber AH, Weis WI. The structure of the beta-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by beta-catenin. Cell. 2001;105:391–402. doi: 10.1016/s0092-8674(01)00330-0. [DOI] [PubMed] [Google Scholar]
- 30.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- 31.Chen Z, He X, Jia M, Liu Y, Qu D, Wu D, Wu P, Ni C, Zhang Z, Ye J, et al. β-catenin overexpression in the nucleus predicts progress disease and unfavourable survival in colorectal cancer: a meta-analysis. PLoS One. 2013;8:e63854. doi: 10.1371/journal.pone.0063854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rao TP, Kühl M. An updated overview on Wnt signaling pathways: a prelude for more. Circ Res. 2010;106:1798–1806. doi: 10.1161/CIRCRESAHA.110.219840. [DOI] [PubMed] [Google Scholar]
- 33.Conacci-Sorrell M, Zhurinsky J, Ben-Ze’ev A. The cadherin-catenin adhesion system in signaling and cancer. J Clin Invest. 2002;109:987–991. doi: 10.1172/JCI15429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sánchez-Tilló E, de Barrios O, Siles L, Cuatrecasas M, Castells A, Postigo A. β-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc Natl Acad Sci USA. 2011;108:19204–19209. doi: 10.1073/pnas.1108977108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lehrke M, Lazar MA. The many faces of PPARgamma. Cell. 2005;123:993–999. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- 36.Sabatino L, Fucci A, Pancione M, Colantuoni V. PPARG Epigenetic Deregulation and Its Role in Colorectal Tumorigenesis. PPAR Res. 2012;2012:687492. doi: 10.1155/2012/687492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feige JN, Gelman L, Michalik L, Desvergne B, Wahli W. From molecular action to physiological outputs: peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog Lipid Res. 2006;45:120–159. doi: 10.1016/j.plipres.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 38.Wang D, Dubois RN. Peroxisome proliferator-activated receptors and progression of colorectal cancer. PPAR Res. 2008;2008:931074. doi: 10.1155/2008/931074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koga H, Sakisaka S, Harada M, Takagi T, Hanada S, Taniguchi E, Kawaguchi T, Sasatomi K, Kimura R, Hashimoto O, et al. Involvement of p21(WAF1/Cip1), p27(Kip1), and p18(INK4c) in troglitazone-induced cell-cycle arrest in human hepatoma cell lines. Hepatology. 2001;33:1087–1097. doi: 10.1053/jhep.2001.24024. [DOI] [PubMed] [Google Scholar]
- 40.Motomura W, Okumura T, Takahashi N, Obara T, Kohgo Y. Activation of peroxisome proliferator-activated receptor gamma by troglitazone inhibits cell growth through the increase of p27KiP1 in human. Pancreatic carcinoma cells. Cancer Res. 2000;60:5558–5564. [PubMed] [Google Scholar]
- 41.Zhang W, Wu N, Li Z, Wang L, Jin J, Zha XL. PPARgamma activator rosiglitazone inhibits cell migration via upregulation of PTEN in human hepatocarcinoma cell line BEL-7404. Cancer Biol Ther. 2006;5:1008–1014. doi: 10.4161/cbt.5.8.2887. [DOI] [PubMed] [Google Scholar]
- 42.Chen GG, Lee JF, Wang SH, Chan UP, Ip PC, Lau WY. Apoptosis induced by activation of peroxisome-proliferator activated receptor-gamma is associated with Bcl-2 and NF-kappaB in human colon cancer. Life Sci. 2002;70:2631–2646. doi: 10.1016/s0024-3205(02)01510-2. [DOI] [PubMed] [Google Scholar]
- 43.Panigrahy D, Singer S, Shen LQ, Butterfield CE, Freedman DA, Chen EJ, Moses MA, Kilroy S, Duensing S, Fletcher C, et al. PPARgamma ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. J Clin Invest. 2002;110:923–932. doi: 10.1172/JCI15634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reka AK, Kurapati H, Narala VR, Bommer G, Chen J, Standiford TJ, Keshamouni VG. Peroxisome proliferator-activated receptor-gamma activation inhibits tumor metastasis by antagonizing Smad3-mediated epithelial-mesenchymal transition. Mol Cancer Ther. 2010;9:3221–3232. doi: 10.1158/1535-7163.MCT-10-0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tachibana K, Yamasaki D, Ishimoto K, Doi T. The Role of PPARs in Cancer. PPAR Res. 2008;2008:102737. doi: 10.1155/2008/102737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kristiansen G, Jacob J, Buckendahl AC, Grützmann R, Alldinger I, Sipos B, Klöppel G, Bahra M, Langrehr JM, Neuhaus P, et al. Peroxisome proliferator-activated receptor gamma is highly expressed in pancreatic cancer and is associated with shorter overall survival times. Clin Cancer Res. 2006;12:6444–6451. doi: 10.1158/1078-0432.CCR-06-0834. [DOI] [PubMed] [Google Scholar]
- 48.Lee JJ, Drakaki A, Iliopoulos D, Struhl K. MiR-27b targets PPARγ to inhibit growth, tumor progression and the inflammatory response in neuroblastoma cells. Oncogene. 2012;31:3818–3825. doi: 10.1038/onc.2011.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saez E, Tontonoz P, Nelson MC, Alvarez JG, Ming UT, Baird SM, Thomazy VA, Evans RM. Activators of the nuclear receptor PPARgamma enhance colon polyp formation. Nat Med. 1998;4:1058–1061. doi: 10.1038/2042. [DOI] [PubMed] [Google Scholar]
- 50.Lefebvre AM, Chen I, Desreumaux P, Najib J, Fruchart JC, Geboes K, Briggs M, Heyman R, Auwerx J. Activation of the peroxisome proliferator-activated receptor gamma promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat Med. 1998;4:1053–1057. doi: 10.1038/2036. [DOI] [PubMed] [Google Scholar]
- 51.Niho N, Takahashi M, Kitamura T, Shoji Y, Itoh M, Noda T, Sugimura T, Wakabayashi K. Concomitant suppression of hyperlipidemia and intestinal polyp formation in Apc-deficient mice by peroxisome proliferator-activated receptor ligands. Cancer Res. 2003;63:6090–6095. [PubMed] [Google Scholar]
- 52.Niho N, Takahashi M, Shoji Y, Takeuchi Y, Matsubara S, Sugimura T, Wakabayashi K. Dose-dependent suppression of hyperlipidemia and intestinal polyp formation in Min mice by pioglitazone, a PPAR gamma ligand. Cancer Sci. 2003;94:960–964. doi: 10.1111/j.1349-7006.2003.tb01385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McAlpine CA, Barak Y, Matise I, Cormier RT. Intestinal-specific PPARgamma deficiency enhances tumorigenesis in ApcMin/+ mice. Int J Cancer. 2006;119:2339–2346. doi: 10.1002/ijc.22115. [DOI] [PubMed] [Google Scholar]
- 54.Osawa E, Nakajima A, Wada K, Ishimine S, Fujisawa N, Kawamori T, Matsuhashi N, Kadowaki T, Ochiai M, Sekihara H, et al. Peroxisome proliferator-activated receptor gamma ligands suppress colon carcinogenesis induced by azoxymethane in mice. Gastroenterology. 2003;124:361–367. doi: 10.1053/gast.2003.50067. [DOI] [PubMed] [Google Scholar]
- 55.Palakurthi SS, Aktas H, Grubissich LM, Mortensen RM, Halperin JA. Anticancer effects of thiazolidinediones are independent of peroxisome proliferator-activated receptor gamma and mediated by inhibition of translation initiation. Cancer Res. 2001;61:6213–6218. [PubMed] [Google Scholar]
- 56.Girnun GD, Smith WM, Drori S, Sarraf P, Mueller E, Eng C, Nambiar P, Rosenberg DW, Bronson RT, Edelmann W, et al. APC-dependent suppression of colon carcinogenesis by PPARgamma. Proc Natl Acad Sci USA. 2002;99:13771–13776. doi: 10.1073/pnas.162480299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 58.Danese S, Malesci A, Vetrano S. Colitis-associated cancer: the dark side of inflammatory bowel disease. Gut. 2011;60:1609–1610. doi: 10.1136/gutjnl-2011-300953. [DOI] [PubMed] [Google Scholar]
- 59.Sarraf P, Mueller E, Smith WM, Wright HM, Kum JB, Aaltonen LA, de la Chapelle A, Spiegelman BM, Eng C. Loss-of-function mutations in PPAR gamma associated with human colon cancer. Mol Cell. 1999;3:799–804. doi: 10.1016/s1097-2765(01)80012-5. [DOI] [PubMed] [Google Scholar]
- 60.Burgermeister E, Seger R. MAPK kinases as nucleo-cytoplasmic shuttles for PPARgamma. Cell Cycle. 2007;6:1539–1548. doi: 10.4161/cc.6.13.4453. [DOI] [PubMed] [Google Scholar]
- 61.Pancione M, Forte N, Sabatino L, Tomaselli E, Parente D, Febbraro A, Colantuoni V. Reduced beta-catenin and peroxisome proliferator-activated receptor-gamma expression levels are associated with colorectal cancer metastatic progression: correlation with tumor-associated macrophages, cyclooxygenase 2, and patient outcome. Hum Pathol. 2009;40:714–725. doi: 10.1016/j.humpath.2008.08.019. [DOI] [PubMed] [Google Scholar]
- 62.Dai Y, Wang WH. Peroxisome proliferator-activated receptor γ and colorectal cancer. World J Gastrointest Oncol. 2010;2:159–164. doi: 10.4251/wjgo.v2.i3.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Colangelo T, Fucci A, Votino C, Sabatino L, Pancione M, Laudanna C, Binaschi M, Bigioni M, Maggi CA, Parente D, et al. MicroRNA-130b promotes tumor development and is associated with poor prognosis in colorectal cancer. Neoplasia. 2013;15:1086–1099. doi: 10.1593/neo.13998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mulholland DJ, Dedhar S, Coetzee GA, Nelson CC. Interaction of nuclear receptors with the Wnt/beta-catenin/Tcf signaling axis: Wnt you like to know? Endocr Rev. 2005;26:898–915. doi: 10.1210/er.2003-0034. [DOI] [PubMed] [Google Scholar]
- 65.Botrugno OA, Fayard E, Annicotte JS, Haby C, Brennan T, Wendling O, Tanaka T, Kodama T, Thomas W, Auwerx J, et al. Synergy between LRH-1 and beta-catenin induces G1 cyclin-mediated cell proliferation. Mol Cell. 2004;15:499–509. doi: 10.1016/j.molcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 66.Yang F, Li X, Sharma M, Sasaki CY, Longo DL, Lim B, Sun Z. Linking beta-catenin to androgen-signaling pathway. J Biol Chem. 2002;277:11336–11344. doi: 10.1074/jbc.M111962200. [DOI] [PubMed] [Google Scholar]
- 67.Jansson EA, Are A, Greicius G, Kuo IC, Kelly D, Arulampalam V, Pettersson S. The Wnt/beta-catenin signaling pathway targets PPARgamma activity in colon cancer cells. Proc Natl Acad Sci USA. 2005;102:1460–1465. doi: 10.1073/pnas.0405928102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu J, Farmer SR. Regulating the balance between peroxisome proliferator-activated receptor gamma and beta-catenin signaling during adipogenesis. A glycogen synthase kinase 3beta phosphorylation-defective mutant of beta-catenin inhibits expression of a subset of adipogenic genes. J Biol Chem. 2004;279:45020–45027. doi: 10.1074/jbc.M407050200. [DOI] [PubMed] [Google Scholar]
- 69.Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3:768–780. doi: 10.1038/nrc1189. [DOI] [PubMed] [Google Scholar]
- 70.Teiten MH, Gaascht F, Dicato M, Diederich M. Targeting the wingless signaling pathway with natural compounds as chemopreventive or chemotherapeutic agents. Curr Pharm Biotechnol. 2012;13:245–254. doi: 10.2174/138920112798868593. [DOI] [PubMed] [Google Scholar]
- 71.Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13:11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- 72.Hawcroft G, D’Amico M, Albanese C, Markham AF, Pestell RG, Hull MA. Indomethacin induces differential expression of beta-catenin, gamma-catenin and T-cell factor target genes in human colorectal cancer cells. Carcinogenesis. 2002;23:107–114. doi: 10.1093/carcin/23.1.107. [DOI] [PubMed] [Google Scholar]
- 73.Dihlmann S, Siermann A, von Knebel Doeberitz M. The nonsteroidal anti-inflammatory drugs aspirin and indomethacin attenuate beta-catenin/TCF-4 signaling. Oncogene. 2001;20:645–653. doi: 10.1038/sj.onc.1204123. [DOI] [PubMed] [Google Scholar]
- 74.Rice PL, Kelloff J, Sullivan H, Driggers LJ, Beard KS, Kuwada S, Piazza G, Ahnen DJ. Sulindac metabolites induce caspase- and proteasome-dependent degradation of beta-catenin protein in human colon cancer cells. Mol Cancer Ther. 2003;2:885–892. [PubMed] [Google Scholar]
- 75.Thompson WJ, Piazza GA, Li H, Liu L, Fetter J, Zhu B, Sperl G, Ahnen D, Pamukcu R. Exisulind induction of apoptosis involves guanosine 3’,5’-cyclic monophosphate phosphodiesterase inhibition, protein kinase G activation, and attenuated beta-catenin. Cancer Res. 2000;60:3338–3342. [PubMed] [Google Scholar]
- 76.Dihlmann S, Klein S, Doeberitz Mv Mv. Reduction of beta-catenin/T-cell transcription factor signaling by aspirin and indomethacin is caused by an increased stabilization of phosphorylated beta-catenin. Mol Cancer Ther. 2003;2:509–516. [PubMed] [Google Scholar]
- 77.Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J Biol Chem. 1997;272:3406–3410. doi: 10.1074/jbc.272.6.3406. [DOI] [PubMed] [Google Scholar]
- 78.Shah S, Hecht A, Pestell R, Byers SW. Trans-repression of beta-catenin activity by nuclear receptors. J Biol Chem. 2003;278:48137–48145. doi: 10.1074/jbc.M307154200. [DOI] [PubMed] [Google Scholar]
- 79.Pálmer HG, González-Sancho JM, Espada J, Berciano MT, Puig I, Baulida J, Quintanilla M, Cano A, de Herreros AG, Lafarga M, et al. Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J Cell Biol. 2001;154:369–387. doi: 10.1083/jcb.200102028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lepourcelet M, Chen YN, France DS, Wang H, Crews P, Petersen F, Bruseo C, Wood AW, Shivdasani RA. Small-molecule antagonists of the oncogenic Tcf/beta-catenin protein complex. Cancer Cell. 2004;5:91–102. doi: 10.1016/s1535-6108(03)00334-9. [DOI] [PubMed] [Google Scholar]
- 81.Hope C, Planutis K, Planutiene M, Moyer MP, Johal KS, Woo J, Santoso C, Hanson JA, Holcombe RF. Low concentrations of resveratrol inhibit Wnt signal throughput in colon-derived cells: implications for colon cancer prevention. Mol Nutr Food Res. 2008;52 Suppl 1:S52–S61. doi: 10.1002/mnfr.200700448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lu D, Carson DA. Repression of beta-catenin signaling by PPAR gamma ligands. Eur J Pharmacol. 2010;636:198–202. doi: 10.1016/j.ejphar.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Burris TP, Busby SA, Griffin PR. Targeting orphan nuclear receptors for treatment of metabolic diseases and autoimmunity. Chem Biol. 2012;19:51–59. doi: 10.1016/j.chembiol.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ondrey F. Peroxisome proliferator-activated receptor gamma pathway targeting in carcinogenesis: implications for chemoprevention. Clin Cancer Res. 2009;15:2–8. doi: 10.1158/1078-0432.CCR-08-0326. [DOI] [PubMed] [Google Scholar]
- 85.Menendez-Gutierrez MP, Roszer T, Ricote M. Biology and therapeutic applications of peroxisome proliferator- activated receptors. Curr Top Med Chem. 2012;12:548–584. doi: 10.2174/156802612799436669. [DOI] [PubMed] [Google Scholar]
- 86.Puhl AC, Bernardes A, Silveira RL, Yuan J, Campos JL, Saidemberg DM, Palma MS, Cvoro A, Ayers SD, Webb P, et al. Mode of peroxisome proliferator-activated receptor γ activation by luteolin. Mol Pharmacol. 2012;81:788–799. doi: 10.1124/mol.111.076216. [DOI] [PubMed] [Google Scholar]
- 87.Pochetti G, Mitro N, Lavecchia A, Gilardi F, Besker N, Scotti E, Aschi M, Re N, Fracchiolla G, Laghezza A, et al. Structural insight into peroxisome proliferator-activated receptor gamma binding of two ureidofibrate-like enantiomers by molecular dynamics, cofactor interaction analysis, and site-directed mutagenesis. J Med Chem. 2010;53:4354–4366. doi: 10.1021/jm9013899. [DOI] [PubMed] [Google Scholar]
- 88.Laghezza A, Pochetti G, Lavecchia A, Fracchiolla G, Faliti S, Piemontese L, Di Giovanni C, Iacobazzi V, Infantino V, Montanari R, et al. New 2-(aryloxy)-3-phenylpropanoic acids as peroxisome proliferator-activated receptor α/γ dual agonists able to upregulate mitochondrial carnitine shuttle system gene expression. J Med Chem. 2013;56:60–72. doi: 10.1021/jm301018z. [DOI] [PubMed] [Google Scholar]
- 89.Zurlo D, Leone C, Assante G, Salzano S, Renzone G, Scaloni A, Foresta C, Colantuoni V, Lupo A. Cladosporol a stimulates G1-phase arrest of the cell cycle by up-regulation of p21(waf1/cip1) expression in human colon carcinoma HT-29 cells. Mol Carcinog. 2013;52:1–17. doi: 10.1002/mc.20872. [DOI] [PubMed] [Google Scholar]
- 90.Fracchiolla G, Laghezza A, Piemontese L, Carbonara G, Lavecchia A, Tortorella P, Crestani M, Novellino E, Loiodice F. Synthesis, biological evaluation, and molecular modeling investigation of chiral phenoxyacetic acid analogues with PPARalpha and PPARgamma agonist activity. ChemMedChem. 2007;2:641–654. doi: 10.1002/cmdc.200600307. [DOI] [PubMed] [Google Scholar]
- 91.Park CH, Chang JY, Hahm ER, Park S, Kim HK, Yang CH. Quercetin, a potent inhibitor against beta-catenin/Tcf signaling in SW480 colon cancer cells. Biochem Biophys Res Commun. 2005;328:227–234. doi: 10.1016/j.bbrc.2004.12.151. [DOI] [PubMed] [Google Scholar]
- 92.Shan BE, Wang MX, Li RQ. Quercetin inhibit human SW480 colon cancer growth in association with inhibition of cyclin D1 and survivin expression through Wnt/beta-catenin signaling pathway. Cancer Invest. 2009;27:604–612. doi: 10.1080/07357900802337191. [DOI] [PubMed] [Google Scholar]
- 93.Lee H, Bae S, Yoon Y. The anti-adipogenic effects of (-)epigallocatechin gallate are dependent on the WNT/β-catenin pathway. J Nutr Biochem. 2013;24:1232–1240. doi: 10.1016/j.jnutbio.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 94.Dashwood WM, Orner GA, Dashwood RH. Inhibition of beta-catenin/Tcf activity by white tea, green tea, and epigallocatechin-3-gallate (EGCG): minor contribution of H(2)O(2) at physiologically relevant EGCG concentrations. Biochem Biophys Res Commun. 2002;296:584–588. doi: 10.1016/s0006-291x(02)00914-2. [DOI] [PubMed] [Google Scholar]
- 95.Kim J, Zhang X, Rieger-Christ KM, Summerhayes IC, Wazer DE, Paulson KE, Yee AS. Suppression of Wnt signaling by the green tea compound (-)-epigallocatechin 3-gallate (EGCG) in invasive breast cancer cells. Requirement of the transcriptional repressor HBP1. J Biol Chem. 2006;281:10865–10875. doi: 10.1074/jbc.M513378200. [DOI] [PubMed] [Google Scholar]
- 96.Park CH, Hahm ER, Park S, Kim HK, Yang CH. The inhibitory mechanism of curcumin and its derivative against beta-catenin/Tcf signaling. FEBS Lett. 2005;579:2965–2971. doi: 10.1016/j.febslet.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 97.Jaiswal AS, Marlow BP, Gupta N, Narayan S. Beta-catenin-mediated transactivation and cell-cell adhesion pathways are important in curcumin (diferuylmethane)-induced growth arrest and apoptosis in colon cancer cells. Oncogene. 2002;21:8414–8427. doi: 10.1038/sj.onc.1205947. [DOI] [PubMed] [Google Scholar]
- 98.Chen HJ, Hsu LS, Shia YT, Lin MW, Lin CM. The β-catenin/TCF complex as a novel target of resveratrol in the Wnt/β-catenin signaling pathway. Biochem Pharmacol. 2012;84:1143–1153. doi: 10.1016/j.bcp.2012.08.011. [DOI] [PubMed] [Google Scholar]
- 99.Li H, Wu WK, Li ZJ, Chan KM, Wong CC, Ye CG, Yu L, Sung JJ, Cho CH, Wang M. 2,3’,4,4’,5’-Pentamethoxy-trans-stilbene, a resveratrol derivative, inhibits colitis-associated colorectal carcinogenesis in mice. Br J Pharmacol. 2010;160:1352–1361. doi: 10.1111/j.1476-5381.2010.00785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jingushi K, Takahashi-Yanaga F, Yoshihara T, Shiraishi F, Watanabe Y, Hirata M, Morimoto S, Sasaguri T. DIF-1 inhibits the Wnt/β-catenin signaling pathway by inhibiting TCF7L2 expression in colon cancer cell lines. Biochem Pharmacol. 2012;83:47–56. doi: 10.1016/j.bcp.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 101.Takahashi-Yanaga F, Mori J, Matsuzaki E, Watanabe Y, Hirata M, Miwa Y, Morimoto S, Sasaguri T. Involvement of GSK-3beta and DYRK1B in differentiation-inducing factor-3-induced phosphorylation of cyclin D1 in HeLa cells. J Biol Chem. 2006;281:38489–38497. doi: 10.1074/jbc.M605205200. [DOI] [PubMed] [Google Scholar]
- 102.Lee SH, Richardson RL, Dashwood RH, Baek SJ. Capsaicin represses transcriptional activity of β-catenin in human colorectal cancer cells. J Nutr Biochem. 2012;23:646–655. doi: 10.1016/j.jnutbio.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lee SH, Krisanapun C, Baek SJ. NSAID-activated gene-1 as a molecular target for capsaicin-induced apoptosis through a novel molecular mechanism involving GSK3beta, C/EBPbeta and ATF3. Carcinogenesis. 2010;31:719–728. doi: 10.1093/carcin/bgq016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lang M, Borgmann M, Oberhuber G, Evstatiev R, Jimenez K, Dammann KW, Jambrich M, Khare V, Campregher C, Ristl R, et al. Thymoquinone attenuates tumor growth in ApcMin mice by interference with Wnt-signaling. Mol Cancer. 2013;12:41. doi: 10.1186/1476-4598-12-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Woo CC, Kumar AP, Sethi G, Tan KH. Thymoquinone: potential cure for inflammatory disorders and cancer. Biochem Pharmacol. 2012;83:443–451. doi: 10.1016/j.bcp.2011.09.029. [DOI] [PubMed] [Google Scholar]
- 106.Dillard AC, Lane MA. Retinol decreases beta-catenin protein levels in retinoic acid-resistant colon cancer cell lines. Mol Carcinog. 2007;46:315–329. doi: 10.1002/mc.20280. [DOI] [PubMed] [Google Scholar]
- 107.Jiang Y, Prunier C, Howe PH. The inhibitory effects of Disabled-2 (Dab2) on Wnt signaling are mediated through Axin. Oncogene. 2008;27:1865–1875. doi: 10.1038/sj.onc.1210829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pendás-Franco N, Aguilera O, Pereira F, González-Sancho JM, Muñoz A. Vitamin D and Wnt/beta-catenin pathway in colon cancer: role and regulation of DICKKOPF genes. Anticancer Res. 2008;28:2613–2623. [PubMed] [Google Scholar]
- 109.Thun MJ, Namboodiri MM, Heath CW. Aspirin use and reduced risk of fatal colon cancer. N Engl J Med. 1991;325:1593–1596. doi: 10.1056/NEJM199112053252301. [DOI] [PubMed] [Google Scholar]
- 110.Chan TA. Nonsteroidal anti-inflammatory drugs, apoptosis, and colon-cancer chemoprevention. Lancet Oncol. 2002;3:166–174. doi: 10.1016/s1470-2045(02)00680-0. [DOI] [PubMed] [Google Scholar]
- 111.Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst. 2002;94:252–266. doi: 10.1093/jnci/94.4.252. [DOI] [PubMed] [Google Scholar]
- 112.Sandler RS, Halabi S, Baron JA, Budinger S, Paskett E, Keresztes R, Petrelli N, Pipas JM, Karp DD, Loprinzi CL, et al. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N Engl J Med. 2003;348:883–890. doi: 10.1056/NEJMoa021633. [DOI] [PubMed] [Google Scholar]
- 113.Baron JA, Cole BF, Sandler RS, Haile RW, Ahnen D, Bresalier R, McKeown-Eyssen G, Summers RW, Rothstein R, Burke CA, et al. A randomized trial of aspirin to prevent colorectal adenomas. N Engl J Med. 2003;348:891–899. doi: 10.1056/NEJMoa021735. [DOI] [PubMed] [Google Scholar]
- 114.Li N, Xi Y, Tinsley HN, Gurpinar E, Gary BD, Zhu B, Li Y, Chen X, Keeton AB, Abadi AH, et al. Sulindac selectively inhibits colon tumor cell growth by activating the cGMP/PKG pathway to suppress Wnt/β-catenin signaling. Mol Cancer Ther. 2013;12:1848–1859. doi: 10.1158/1535-7163.MCT-13-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Boon EM, Keller JJ, Wormhoudt TA, Giardiello FM, Offerhaus GJ, van der Neut R, Pals ST. Sulindac targets nuclear beta-catenin accumulation and Wnt signalling in adenomas of patients with familial adenomatous polyposis and in human colorectal cancer cell lines. Br J Cancer. 2004;90:224–229. doi: 10.1038/sj.bjc.6601505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sakoguchi-Okada N, Takahashi-Yanaga F, Fukada K, Shiraishi F, Taba Y, Miwa Y, Morimoto S, Iida M, Sasaguri T. Celecoxib inhibits the expression of survivin via the suppression of promoter activity in human colon cancer cells. Biochem Pharmacol. 2007;73:1318–1329. doi: 10.1016/j.bcp.2006.12.033. [DOI] [PubMed] [Google Scholar]