

Abstract
There have been considerable recent advances towards a better understanding of the complex cellular and molecular network underlying liver fibrogenesis. Recent data indicate that the termination of fibrogenic processes and the restoration of deficient fibrolytic pathways may allow the reversal of advanced fibrosis and even cirrhosis. Therefore, efforts have been made to better clarify the cellular and molecular mechanisms that are involved in liver fibrosis. Activation of hepatic stellate cells (HSCs) remains a central event in fibrosis, complemented by other sources of matrix-producing cells, including portal fibroblasts, fibrocytes and bone marrow-derived myofibroblasts. These cells converge in a complex interaction with neighboring cells to provoke scarring in response to persistent injury. Defining the interaction of different cell types, revealing the effects of cytokines on these cells and characterizing the regulatory mechanisms that control gene expression in activated HSCs will enable the discovery of new therapeutic targets. Moreover, the characterization of different pathways associated with different etiologies aid in the development of disease-specific therapies. This article outlines recent advances regarding the cellular and molecular mechanisms involved in liver fibrosis that may be translated into future therapies. The pathogenesis of liver fibrosis associated with alcoholic liver disease, non-alcoholic fatty liver disease and viral hepatitis are also discussed to emphasize the various mechanisms involved in liver fibrosis.
Keywords: Liver, Liver fibrosis, Cirrhosis, Fibrogenesis, Hepatic stellate cells, Myofibroblast, Extracellular matrix
Core tip: Liver fibrosis is a dynamic process that results from an imbalance between the production and dissolution of the extracellular matrix. Development of liver fibrosis is orchestrated by many cell types, including hepatic stellate cells (HSCs). The activation of HCSs is a complex process, leading to multiple potential sites for therapeutic interventions. Additionally, the differences between the pathogenesis of liver fibrosis associated with different etiologies may provide the determination of new therapeutic approaches. This review summarizes the most significant data that has contributed to the understanding of the cellular and molecular pathogenesis of liver fibrosis, which may be translated into future therapeutic strategies.
INTRODUCTION
Liver fibrosis is a common pathological consequence of a variety of chronic stimuli, including viral, autoimmune, drug induced, cholestatic and metabolic diseases[1-4]. Liver fibrosis can be defined as a result of the progressive accumulation and decreased remodeling of the extracellular matrix (ECM), which disrupts the normal architecture of the liver[2]. If left untreated, fibrosis can progress to liver cirrhosis, ultimately leading to organ failure and death. The characterization of the underlying mechanisms of liver fibrogenesis has indicated that fibrosis is driven by a dynamic process involving the increased synthesis of matrix components and a failure of physiological mechanisms of matrix turnover. Moreover, the capacity of the liver to undergo fibrosis regression following cessation of the liver insult has been highlighted[4-6]. These findings have provided progressed the understanding of the pathogenesis of chronic liver diseases and have presented opportunities for novel therapeutic approaches for the management of liver fibrosis.
This review presents key advances in the new insights into the cellular and molecular mechanisms that regulate liver fibrosis, which may represent future therapeutic targets.
ECM IN LIVER FIBROSIS
During chronic liver injury, an increase of fibril-forming collagen and the replacement of the low density, basement membrane-like interstitial matrix occurs[4,6,7]. There is also an accumulation of other matrix proteins, including elastin, hyaluronan, proteoglycans and fibronectin. This type of matrix has the capacity to activate quiescent HSCs, leading to the loss of hepatocyte microvilli and the disappearance of endothelial fenestrations (Figure 1)[4,7,8]. This architectural change of endothelial cells also impairs the transport of solutes from the sinusoid to the hepatocytes, further contributing to hepatocyte dysfunction[7]. Moreover, the accumulation of ECM itself provokes positive feedback pathways that further amplify fibrosis[8]. The alteration of ECM proteins influences cellular behavior via cell membrane receptors. The most potent proteins are integrins that permit communication between the ECM and the cytoskeleton[9-11]. Patsenker et al[11] demonstrated that the inhibition of integrin alpha-V-beta slows the progression of biliary fibrosis and suggested that this inhibition could have potential therapeutic utility.
Figure 1.
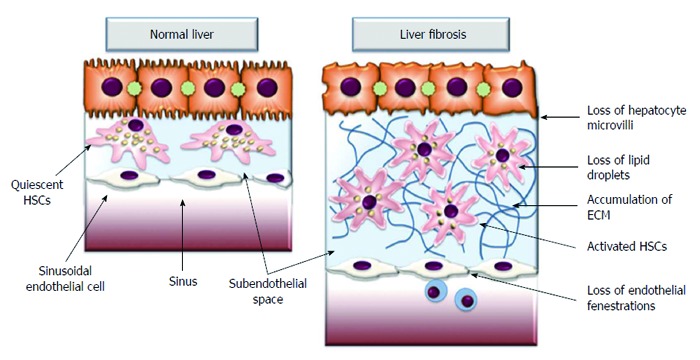
Extracellular matrix accumulation in subendothelial space activates quiescent hepatic stellate cells leading to the loss of hepatocyte microvilli and disappearance of endothelial fenestrations. These architectural changes impair transport of solutes from the sinusoid to the hepatocytes, further contributing to the hepatocyte damage. ECM: Extracellular matrix; HSCs: Hepatic stellate cells.
ECM remodeling is critical in the preservation of homeostasis during liver injury. This homeostasis depends on the fine balance between matrix metalloproteinases (MMPs) and their inhibitors, tissue inhibitors of matrix metalloproteinases (TIMPs). While the excessive increase in the ECM is controlled by MMPs (especially MMP-1, 2, 8 and 13), progressive fibrosis is correlated with the marked increase of TIMPs (TIMP-1 and TIMP-2)[12,13]. Moreover, because TIMP-1 has also anti-apoptotic effects on HSCs, it induces fibrogenesis by promoting fibrogenic cell survival. Several studies have reported that the regulation of TIMPs in HSCs may accelerate the elimination of fibrotic liver tissue and the reversal of fibrosis[14,15]. Enhancing the degradation of excess ECM by increasing the activity of MMPs or decreasing that of TIMPs is an additional approach in the development of antifibrotic drugs.
Angiogenesis is another response to chronic liver injury that leads to sinusoidal remodeling and pericyte amplification[16-18]. Consequently, many potent angiogenic mediators are involved in the exaggerated wound healing response to chronic liver injury, leading to an excessive accumulation of ECM[17,18]. The ECM can also affect cell function indirectly by releasing cytokines. These include transforming growth factor β (TGF-β), platelet derived growth factor (PDGF), hepatocyte growth factor (HGF), connective tissue growth factor (CTGF), tumor necrosis factor-α (TNF-α), basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF)[19].
CELL TYPES INVOLVED IN THE PATHOGENESIS OF LIVER FIBROSIS
Although the cellular source of ECM components in fibrotic liver has been a matter of controversy for many years, recent investigations have revealed that ECM accumulation during chronic liver injury is driven by a heterogeneous population of cells. Currently, it is accepted that liver fibrogenic cells (myofibroblasts) play a central role during liver fibrosis. Their origin has been extensively studied, and several sources of myofibroblasts (MFs) have been identified[3,20-27]. Because HSCs are the main ECM-producing cells in the injured liver[20] they are currently considered to be the major source of MFs[3,20-22]. Hepatic MFs may also originate from portal fibroblasts and bone marrow derived mesenchymal cells[24,28]. Two other minor contributors of fibrogenic cells are the epithelial-mesenchymal transition (EMT)[29,30] and endothelial to mesenchymal transition (Figure 2)[31,32].
Figure 2.
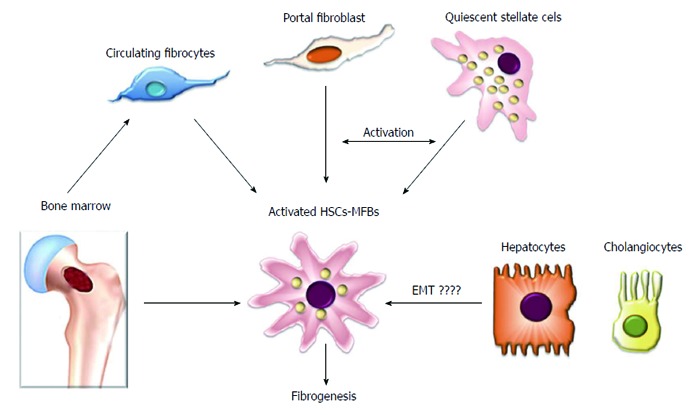
Hepatic myofibroblasts are a heterogenous population of fibrogenic cells. Hepatic stellate cells are considered to be a major source of liver fibrogenic cells followed by portal fibroblasts that play an important role in the fibrogenic process during cholestatic liver diseases. Other sources of hepatic myofibroblasts include circulating fibrocytes and bone marrow-derived cells that constitute a minor proportion of liver fibrogenic cells. The epithelial origin of liver fibrogenic cells is unlikely. EMT: Epithelial mesenchymal transition; MFBs: Myofibroblasts; HSCs: Hepatic stellate cells.
HSCs
Activation of HSCs is recognized as a central event during liver fibrosis, and the molecular mechanisms of this cellular alteration continue to attract increasing attention, creating many new findings[33,34]. However, there is limited knowledge about HSC activation from the viewpoint of cell fate or lineage regulation[35-37]. Recently, many studies have shown that HSCs are derived from mesodermal-derived multipotent mesenchymal progenitor cells (MMPC), which also give rise to neural cells and other mesenchymal cells[38,39]. Supporting these findings, HSCs also express neural and mesenchymal lineage markers. Because cell types derived from MMPC may undergo transdifferentiation within their lineages, the notion that HSC transdifferentiation may reside in these mesenchymal lineages is reasonable[39]. In normal liver tissue, HSCs exist in a quiescent state, storing retinoids and synthesizing glial fibrillary acidic protein (GFAP)[40-43]. Following liver injury, HSCs are activated with a gradual loss of retinoids and GFAP, leading to a reduction in the expression of adipogenic/lipogenic factors. Meanwhile, a complex network of autocrine/paracrine fibrogenic signals promotes the transdifferentiation of HSCs to a myofibroblastic phenotype.
Portal fibroblasts
Portal fibroblasts are spindle shaped cells of mesenchymal origin that undergo myofibroblastic differentiation, primarily in biliary and cholestatic liver injuries[44-46]. Although they possess biological similarities with activated HSCs, portal fibroblasts have different genetic profiles and signaling responses[45,46]. The latter could enable the development of disease specific antifibrotic therapies targeting these cells.
Fibrocytes
Fibrocytes originate from hematopoietic stem cells and have the ability to differentiate into MFs. In cases of tissue damage, fibrocytes proliferate and migrate to the injured organ and secrete growth factors that promote deposition of the ECM[47-49]. Several studies have suggested that the extent of fibrocyte differentiation into MFs depends on the organ and the type of injury[48,49]. Other studies have demonstrated that liver injury induces migration of fibrocytes to lymphoid organs[49], suggesting that the function of these cells may not be limited to ECM deposition.
Bone marrow-derived MFs
A fraction of hepatic MFs can also arise from bone marrow-derived mesenchymal stem cells (MSCs), which are defined as multipotent progenitor cells with the capacity to differentiate into lineage-specific cells[44,48,49]. Currently, it is not clear whether circulating MSCs significantly contribute to ECM deposition in the course of liver fibrosis or not, but they most likely represent a population that is distinct from hematopoietic-derived fibrocytes[49].
EMT
EMT is a process during which fully differentiated epithelial cells undergo phenotypic transition to fully differentiated mesenchymal cells. Liver cell culture studies have shown that hepatocytes and cholangiocytes may undergo EMT and acquire mesenchymal features, including FSP-1 expression[50-52]. However, more recent reports provide strong evidence against EMT in the liver as a source of MFs, convincingly arguing for an epithelial origin of ECM-producing cells[52,53].
HSCS IN LIVER FIROSIS
During liver fibrogenesis, parenchymal injury and the resulting inflammatory reaction generate a large panel of signals that stimulate the induction of specific transcription factors and morphogens in quiescent HSCs, thereby initiating the activation and the acquisition of fibrogenic and proinflammatory properties. Sustained activation leads to discrete changes in hepatic stellate cell (HSC) behavior, including proliferation, chemotaxis, fibrogenesis, contractility, retinoid loss and WBC chemoattractant/cytokine release[1]. In these phases there is a release of proinflammatory, profibrogenic and promitogenic stimuli acting in an autocrine and paracrine manner (Figure 3).
Figure 3.
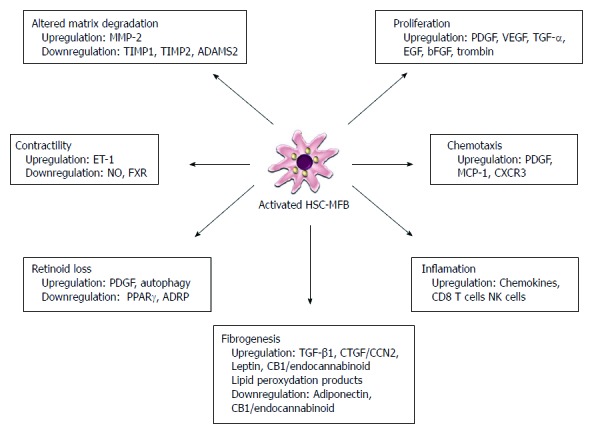
Hepatic myofibroblasts myofibroblasts have multiple functions during liver fibrogenesis. In the activated form, hepatic stellate cells show de novo properties, including increased proliferation, fibrogenesis, contractility, chemotaxis, matrix degradation, retinoid loss and secretion of chemokines. Each of these properties is controlled by the release of many cytokines acting in an autocrine and paracrine manner offering many potential sites for therapeutic intervention. MMP: Matrix metalloproteinase; TIMP: Tissue inhibitor of matrix metalloproteinase; ADAMS2: A disintegrin and metalloproteinase 2; PDGF: Platelet derived growth factor; VEGF: Vascular endothelial growth factor; TGF-α: Transforming growth factor-α; EGF: Epidermal growth factor; bFGF: Basic fibroblast growth factor; TGF-β1: Transforming growth factor-β1; CTGF/CCN2: Connective tissue growth factor; ET-1: Endothelin 1; NO: Nitric oxide; FXR: Farnesoid X receptor; PPARγ: Peroxisome proliferators activated nuclear receptorsγ; ADRP: Adipose differentiation related protein.
ACTIVATION OF HSCS
Activation of HSCs by neighboring cells
In the early stage of injury, all neighboring cell types can contribute to the paracrine stimulation of HSC activation.
Hepatocytes
Hepatocyte apoptosis is a common feature in liver injury. This process is mediated partially by Fas and may also involve TNF-related-apoptosis-inducing ligand (TRAIL)[54-56]. Recent data have shown that the engulfment of the apoptotic bodies of hepatocytes by HSC lines results in a profibrogenic response and activates Kupffer cells[57,58]. A similar profibrogenic response can be observed following disruption of Bcl-xl (an anti-apoptotic mediator) that leads to hepatocyte apoptosis[59,60]. HSC activation by hepatocyte-derived apoptotic bodies is partially mediated by the interaction of hepatocyte DNA with Toll-like receptor 9 (TLR9) expressed in HSCs[61]. Hepatocytes also produce fibrogenic lipid peroxides[62]. Experimental studies have demonstrated that either blockage of hepatocyte apoptosis or selective stimulation of apoptosis in HSCs could be a therapeutic strategy for the prevention of fibrosis[63-66]. However, this approach has not been successful in clinical trials[26].
Liver sinusoidal endothelial cells
In response to injury, sinusoidal endothelial cells contribute to HSC activation, owing to their capacity to produce fibronectin, TGF-β1 and PDGF[67]. Conversely, recent data indicate that restoration of liver sinusoidal endothelial cell differentiation may contribute to fibrosis regression by promoting HSC quiescence[68-70]. It has been proposed that a loss of endothelial fenestration following injury leads to changes in liver sinusoidal endothelial cell differentiation and, consequently, HSC activation[3].
Kupffer cells
Kupffer cells and infiltrating monocytes express a number of chemokine receptors that influence fibrosis progression and resolution[71-74]. Indeed, different macrophage subsets have been described in experimental models; however, their molecular profile is incomplete and additional studies are warranted[74-78]. To date, profibrogenic macrophages have been shown to have high Gr1 (Ly6c) expression and to activate HSCs[74,78]. Additionally, another subset of monocytes (Gr1Io) is vital for fibrosis regression[79,80].
Lymphocytes
Lymphocytes, especially CD4 T-helper lymphocytes, may activate HSCs via cytokine production. Previous experimental models imply that during liver injury Th2 lymphocytes, a subset of T-helper lymphocytes, are more fibrogenic as compared to the Th1 lymphocytes subset[81,82].
Natural killer cells
Recent findings indicate that natural killer (NK) cells inhibit liver fibrosis by directly killing activated HSCs[83-86]. In cases of liver injury, NK cells induce apoptosis of HSCs by IFN-γ. Moreover, IFN-γ not only inhibits HSC activation directly but also amplifies NK cell cytotoxicity against HSCs via upregulation of NKG2D (best defined natural cytotoxicity receptor) and TRAIL expression on NK cells[87-90]. It has been shown that HSCs in the early stages of activation are more prone to be killed by NK cells than quiescent or fully activated HSCs, because they still produce retinoic acid that is important in the induction of NK cell-activating ligands (MICA in humans)[91]. Thus, activation of NK cells could be a novel, therapeutic target to treat liver fibrosis[91,92]. It should be noted that another T cell subset, NKT cells, has diverse effects on liver fibrosis depending on the stage of the disease[91-93].
Leukocytes recruited to the liver during injury produce compounds that modulate HSC behavior. Neutrophils are an important source of reactive oxygen species (ROS) that also produce nitric oxide (NO), which may counteract the effect of superoxide on collagen production[94,95].
Platelets that produce TGF-β1, PDGF and epidermal growth factor (EGF) are also an important source of paracrine stimuli in HSC activation and fibrogenesis[96-98].
Molecular activation of HSCs
ROS: ROS that are generated through lipid peroxidation have the ability to activate HSCs and stimulate the progression of fibrosis[99,100] They can originate from hepatocytes, macrophages, cholangiocytes and inflammatory cells[99,100]. Moreover, ROS can also be produced by HSCs in response to several fibrogenic mediators, such as PDGF, TGF-β leptin and Angiotensin II[101-104]. Although it has been suggested that the loss of antioxidant capacity in activated HSCs amplifies the effects of lipid peroxidation products, more recent studies have indicated that activated HSCs have an increased ROS-detoxifying capacity compared to quiescent HSCs[62,105-107]. It has also been demonstrated that increased glutathione levels and hydrogen peroxide detoxifying enzymes protect HSCs from ROS-induced necrosis and apoptotic cell death, respectively[107]. Because ROS can activate signal transduction pathways and transcription factors, including JNK and NFκB, they also upregulate the expression of fibrosis-associated genes, including COL1A1, COL1A2, MCP1 and TIMP1 in HSCs[102-104]. At the cellular level, ROS are generated via mitochondrial damage, mitochondrial transport chain or via activation of cytochrome P450 (especially cytochrome P450 2E1), xanthine oxidase and NADPH oxidase[108]. It has been demonstrated that, through the induction of oxidative stress, homologs of NADPH oxidase (NOX) might contribute not only to HSC activation but also to the activation of Kupffer cells and macrophages[109]. More recently it has been shown that the phagocytic NADPH oxidase NOX2 is expressed in HSCs and its activation leads to the induction of fibrogenic cascades[110,111]. Angiotensin II-mediated induction of NOX1 was also described as profibrogenic[111,112]. In a recent study, Jiang et al[113] demonstrated that NOX4 plays an important role in ROS production and HSC activation. They proposed that inhibition of NOX4 might be a promising new strategy for translational trials in liver fibrosis. The cytochrome P450 2E1 (CYP2E1) may also contribute to activation of HSCs via the generation of ROS. In the presence of cells that express CYP2E1 (E47 cells), the production of collagen by HSCs is increased[114,115]. Conversely, in the presence of antioxidants or a CYP2E1 inhibitor the increase in collagen production is blocked, suggesting that the CYP2E1 derived ROS are responsible for the increased collagen production[115].
Because ROS constitute a heterogeneous group of species with widely varying chemical reactivity and biological properties, the blockade of oxidative stress as a therapeutic target is still under investigation. Early results demonstrated that the use of an antioxidant mitoquinone might decrease liver inflammation possibly through the induction of the antioxidant transcription factor Nrf2[116]. In a more recent study, chloride channels that are involved in HSC activation by superoxide anion radicals were proposed as a potential target for new anti-fibrotic drugs[117].
Toll-like receptors: Toll-like receptors (TLRs), receptors for microbial products, are present in HSCs and Kupffer cells, introducing a role of immunity in HSC activation and hepatic fibrosis. In chronic liver diseases, increased intestinal permeability results in an enhanced portal inflow of gut-derived microbial products, lipopolysaccharides (LPS), bacterial DNA, peptidoglycan and viral and fungal components[118]. The impact of intestinal decontamination on liver fibrogenesis has been reported. Parallel to this data, mice with a knockout of TLR4 (the LPS receptor), TLR2 and TLR9 were shown to be protected from liver fibrosis[118]. The stimulation of HSCs by LPS or bacterial products through TLR4, TLR9 and TLR2 has been shown to induce a proinflammatory response[118,119]. The activation of HSCs in response to LPS and its receptor TLR4 may elicit a fibrogenic response by downregulating a transmembrane suppressor of TGF-β-1, BAMBI[119-121]. By contrast, it has been indicated that in addition to LPS (exogenous ligand) TLR4 signaling may also be activated by endogenous ligands from cellular compartments that are released and/or increased during tissue injury, including high mobility group box 1 protein (HMGB1)[122,123]. This chromatin-associated, highly conserved nuclear protein has been shown to be upregulated during liver fibrosis. In vitro studies have demonstrated that HMGB1 activates TLR4 signaling in HSCs to enhance their inflammatory phenotype, indicating that TLR4 signaling need not rely solely on gut-derived LPS for activation during liver injury[123]. HMGB1 also has a synergistic effect with TGF-β1 to stimulate fibrogenic protein expression, which is likely to be TLR4-dependent[123]. It has been suggested that inhibition of HMGB1 and TLR4 signaling activity may therefore be important targets of antifibrotic therapy, warranting further investigation by in vitro and in vivo studies[122,123].
Gene regulations in activated HSCs
There are countless changes in gene transcription that may take place after HSC activation. Among the many target genes of transcription factors described in HSCs include: Type 1 collagen, α-SMA, TGF-β-1, TGF-β receptors, MMP-2, TIMPs 1 and 2[124-126]. The transcription factors that activate these downstream targets are Ets-1, Mef2, CREB, Egr-1, Vitamin D receptor, Foxf1, JunD and C/EBPβ[127].
HSCs also express many nuclear receptors, such as the retinoid responsive RxR and RAR, the farnesoid X receptor (FXR), the pregane X receptor (PXR) and peroxisome proliferators-activated nuclear receptorsγ (PPARγ)[128-130]. While RXR an FXP suppress collagen production, PXR is activated by steroids and antibiotics, dimerizes RXR to induce cytochrome p450 and thereby induces fibrosis[128]. By contrast, PPARγ down-regulates HSC activation and reduces collagen production[128-130].
MicroRNAs: Micro RNAs (mi-Rs) regulate posttranscriptional gene repression by decreasing target mRNA levels. Many mi-Rs are expressed in HSCs and control fibrosis progression[131] including mi-R29, mi-R19b and miR 221/222, among others[132-134]. Based on gene array analysis, mi-R29, which is a physiological inhibitor of various ECM proteins, including collagens, is down regulated by TGF-β and LPS in cultured HSCs[132,133]. MiR-19b is an inhibitor of TGF-β signaling and its expression is decreased in patients with advanced fibrosis, while its overexpression in HSCs blocks activation[132]. In contrast, miR-221/222 is upregulated in human livers in parallel with progression of liver fibrosis. Its expression also increases during HSC activation, and its contribution to HSC proliferation has been proposed[134].
DNA methylation and histone modifications: DNA methylation of genes expressed in quiescent HSCs contributes to the maintenance of the quiescent phenotype. During activation, HSCs express DNA-methyl binding proteins (MeCP2). These proteins promote the silencing of antifibrogenic genes and increase the expression of histone methyl transferases, leading to enhanced transcription of collagen, TIMP-1 and TGF-β[135-137].
It is noteworthy that epigenetic changes can also modulate fibrosis susceptibility[136]. In a recent study, offspring from the progeny of male fibrotic rat ancestors are found to be more resistant to liver fibrosis than their counterparts with no previous history of fibrosis[137]. In experimental models, DNA methylation and histone acetylation in the sperm of rats with fibrosis may also take place in the resistance to the wound healing process, leading to hypomethylation of the PPARγ gene, resulting in elevated hepatic expression of this antifibrogenic transcription factor in adult offspring[137].
PROLIFERATION OF HSCS
The most potent mitogen in HSCs is PDGF. Other mitogens that stimulate HSC proliferation are VEGF, thrombin and its receptors, EGF, TGFα and bFGF[3,104]. Downstream pathways in HSCs include PI3 kinase and ERK/MAP kinase, among others[104,138]. PDGF signaling at the cell membrane of HSCs can also be enhanced by a co-receptor, neuropilin-1[139]. In addition to its mitogenic effect, PDGF also stimulates Na+/H+ exchange, providing a potential site for therapeutic intervention by blocking ion transport[140]. Signaling pathways for these mitogens have been clearly identified in HSCs, offering many potential sites for therapeutic intervention[141,142].
CHEMOTAXIS OF HSCS
HSCs can migrate towards many chemokines, including VEGF, PDGF, MCP-1, CXCR4 and CXCR3[3]. For example, CCR5 and its ligand RANTES stimulate the migration of HSCs[143]. Hypoxia is another activator of HSC migration. In hypoxic conditions the motility of HSCs is not only induced by ROS but also by VEGF in an autocrine manner because prolonged hypoxia induces HSCs to produce and secrete VEGF in an HIF-1α-dependent manner[144].
The role of ECM in migratory behavior of HSCs has been previously described. Additionally, cellular fibronectin containing an alternatively spliced domain A (EIIA) has been shown to induce motility of HSCs, supporting the role of ECM in HSC behavior[145].
Interestingly, while adenosine blunts chemotaxis and fixes cells at sites of injury via the loss of actin fibers, enhanced adenosine signaling may also stimulate HSC fibrogenesis[146,147]. Therefore, understanding the dual role of adenosine will be important in the development of antifibrotic agents. Recent epidemiologic studies demonstrated that caffeine exerts its protective effect by inhibiting adenosine signaling in HSCs[148,149].
HSCS IN FIBROGENESIS
Production of the ECM, in particular collagen type I, is a major characteristic of HSCs. The expression of collagen type I in HSCs is regulated posttranscriptionally by multiple stimuli and pathways. Prominent among these is TGF-β, the most profibrogenic cytokine in the liver[150,151]. TGF-β is produced by Kupffer cells, liver sinusoidal endothelial cells, hepatocytes and HSCs and has paracrine/autocrine effects on HSCs[150,151]. It has three major isoforms: TGF-β1, TGF-β2 and TGF-β3. In addition to its role in the stimulation of collagen type I, TGF-β also stimulates the production of other matrix components, including cellular fibronectin and proteoglycans[150,151]. Although none appears to be as potent as TGF-β, a variety of other factors have profibrogenic effects on HSCs, including retinoids and angiotensin II[103,152]. TGF-β1 is stored as an inactivated protein and, when activated, signals via its receptors to Smad proteins, which enhance the transcription of target genes, such as procollagens I and III[150,151]. The response of SMADs in HSCs differs between acute and chronic injury to further favor matrix production[151]. Because TGF-β1 may also contribute to liver homeostasis during regeneration, therapeutic antagonization of TGF-β1 is challenging[153].
Connective tissue growth factor (CTGF/CCN2) is a growth factor protein that is upregulated by hyperglycemia, hyperinsulinemia and alcohol-induced cellular injury[154,155]. While the stimulation of CTGF/CCN2 in hepatocytes is TGF-β dependent, this stimulation in HSCs is independent of TGF-β, highlighting the fact that, in exception to the general rule, cytokine signaling in HSCs is not always autocrine[156].
Adipokines are polypeptides mainly secreted in adipose tissue and, to lesser extent, by stromal cells. In the liver, they not only contribute to the hepatic manifestation of obesity but are increasingly recognized as key mediators of liver fibrogenesis. Leptin, adiponectin and ghrelin are the main adipokines that contribute to liver injury[157-161]. Leptin is an adipogenic hormone that promotes HSC fibrogenesis and activates Kupffer cells, macrophages and endothelial cells to produce TGF-β1[162]. It modulates the HSC phenotype through the leptin receptor (OB-R), which leads to stimulation of the Janus kinase 2 (JAK 2) and signal transducer and activator of transcription 3 (STAT 3) pathways[157]. Leptin also partially suppresses PPARγ, which can reverse HSC activation and maintain senescence[163]. Recently, it has been demonstrated that leptin deficiency may reduce the activity of norepinephrine, thereby reducing fibrogenesis[164]. Reduced activity of norepinephrine leads to decreased activity of NK cells and attenuates the release of profibrogenic cytokines and reduces ECM production[164]. Adiponectin, a counter-regulatory hormone of leptin, inhibits hepatic fibrogenesis both in vivo and in vitro[160,162]. Ghrelin also appears to attenuate hepatocellular damage and fibrosis in experimental studies[161].
Neurochemical and neurotrophic factors also contribute to the fibrogenic function of HSCs. Following liver injury, activated HSCs express specific receptors (CB1 and CB2) that are components of the endocannabinoid system that regulates the fibrogenic cascade[165-168]. Two receptors exert opposing effects; while CB1 stimulation induces fibrogenesis, the stimulation of the CB2 receptor is anti-fibrotic and hepatoprotective[165-167]. The overexpression of these receptors is observed both in experimental models of liver fibrosis and in the livers of patients with chronic liver disease[165,167]. Therefore, efforts for therapeutic strategies are being directed to either antagonize CB1 or agonize CB2. Non-brain penetrant CB1 antagonists have shown promising results in experimental models[168]. Similarly, opioids that contribute to fibrogenesis by stimulating HSCs can be antagonized by naltrexone[169,170]. Serotonin and thyroid hormones are also involved in fibrogenesis, with agonists or antagonists for these mediators already in existence[41,171].
CONTRACTILITY OF HSCS
Activation of HSCs is accompanied by an increase in expression of proteins characteristic of contractile cells[172]. In the process of becoming contractile, HSCs develop an increased expression of the cytoskeletal protein α-smooth muscle actin (α-SMA)[172]. It has also been reported that HSC contraction is mediated by both Ca2+ dependent and Ca2+ independent mechanisms[173,174]. Contractility of HSCs has a multitude of effects in the injured liver, including perisinusoidal constriction and portal hypertension, leading to an increase in portal resistance during liver fibrosis[174]. Contractile HSCs impede portal blood flow by constricting sinusoids and by contracting the cirrhotic liver[174-177]. This contractility is likely associated with multiple different systems, including endothelin-1. Endothelin-1 receptors are expressed in both quiescent and activated HSCs[176]. Nuclear receptor FXR antagonizes endothelin 1[176]. There is a shift in the predominant type of endothelin receptor and increased sensitivity to endothelin-1 after activation of HSCs[178]. The effect of endothelin-1 may also be reversed by locally produced vasodilator substances; particularly, nitric oxide (NO) may counteract the constrictive effects of endothelin-1[179]. Similarly, carbon monoxide also mediates sinusoidal dilatation[179].
RETINOID LOSS OF HSCS
Retinoid is stored as retinyl esters in the form of perinuclear droplets in the cytoplasm of quiescent HSCs. Activation of HSCs is accompanied by the loss of these characteristic droplets. The form of retinoid released outside the cell during activation is retinol, suggesting that there is intracellular hydrolysis of esters prior to export[127]. Several nuclear retinoid receptors have been identified in HSCs. Lecithin retinol acetyl transferase (LRAT) catalyzes the esterification of retinol into retinyl ester in liver[180]. In liver injury models, LRAT-deficient animals exhibit increased fibrogenesis in the liver[181]. In contrast, treatment with retinoid acid decrease activation of HSCs by inhibiting TGF-β[182].
PPARs regulate glucose and lipid metabolism[129]. Their expression decreases with the activation of HSCs[128,130]. In contrast, forced expression of PPARγ in activated HSCs inhibits collagen expression, blocks TGF-β1 signaling and increases cytoplasmic lipid droplets[129].
Adipose differentiation related protein (ADRP), an intracellular lipid storage protein, is present in quiescent HSCs and its expression is reduced during HSC activation. ADRP is induced by retinoid exposure, suggesting that ADRP may have a regulatory role between lipid content and cellular activation through an unknown mechanism[183,184].
Because energy homeostasis is maintained through autophagic digestion of lipid droplets in many cells, it has been hypothesized that autophagy drives HSC activation by digesting lipid droplets, thereby providing energy required for the activation process[185,186]. Recent studies have demonstrated that inhibition of autophagy down-regulates the fibrogenic properties of HSCs, revealing HSC autophagy as a therapeutic target[185-187].
HSCS IN INFLAMMATION AND WBC CHEMOATTRACTION
HSCs may produce chemokines that amplify inflammatory responses by inducing migration of inflammatory cells[141,188]. Additionally, cell surface expression of chemokines by HSCs promotes ICAM-1- and VCAM-1-dependent adhesion and migration of lymphocytes[189]. Therefore, some of these chemokines are attractive therapeutic targets[188]. The interaction of HSCs with immune cells (especially with T cells) promotes or inhibits their maturation[190]. The results from a recent proteomics analysis supports the immunosuppressive role of activated HSCs[191]. It has been suggested that HSCs also have the capacity to interact with bacterial LPS because they express TLRs[94,118,119].
PATHOGENESIS OF FIBROSIS ASSOCIATED WITH VARIOUS ETIOLOGIES
Alcoholic liver disease
The pathogenesis of liver fibrosis in alcoholic liver disease (ALD) is complex and may be cell specific and controlled through feedback mechanisms and cross-talk between neighboring and distant cells. The development of liver fibrosis in alcoholics has been linked to the oxidation of ethanol to the highly reactive compound acetaldehyde. After alcohol consumption, acetaldehyde stimulates type I collagen synthesis and gene transcription in cultured rat and human HSCs through the activation of protein kinase C (PKC)[192]. Acetaldehyde was also shown to increase NFκB (p65) and its binding to the α2(I) collagen promoter as well as to enhance NFκB by a mechanism dependent on H2O2 accumulation[90,193-195]. The activity of cytochrome P450 isoform 2E1 (CYP2E1) is an important source of ROS in alcohol-induced injury. It has been reported that the inhibition of CYP2E1 activity prevented the induction of collagen I gene expression in rat stellate cells overexpressing CYP2E1[196]. Oxidative stress also activates c-Jun N-terminal kinase (JNK), a protein that regulates the secretion of proinflammatory cytokines in cultured HSCs[144]. The results of a recent study indicated that butein inhibited ethanol- and acetaldehyde-induced activation of HSCs at different levels, acting as an antioxidant and inhibitor of ethanol-induced MAPK, TGF-β and NFκB/IκB transduction signaling; therefore, butein is a promising agent for antifibrotic therapies[197].
Alcohol inhibits the anti-fibrogenic effects of NK cells by stimulating TGF-β production by HSCs, inducing suppressors of cytokine signaling (SOCS-1) and ROS in hepatocytes, thereby sustaining HSC activation and reducing HSC apoptosis[90,198]. Recently, it has been suggested that alcohol increases the binding of the early growth response-1 (Egr-1) transcription factor to the TNF-α promoter and enhances macrophage sensitivity to LPS in the progression of liver injury to fibrosis[199]. Recent discoveries have revealed that alcohol inhibits PPARα, suppressing sterol-regulatory element binding protein-1 (SREBP-1), which is involved in fatty acid synthesis, leading to the activation of HSCs and ultimately fibrosis[90,200]. Other recently identified novel molecules and physiological/cell signaling pathways include hedgehog (Hh) signaling, fibrinolysis and involvement of novel cytokines such as osteopontin. Alcohol increases liver progenitor cell accumulation by providing an increase of Hh and Hh ligands in an autocrine manner[201]. Osteopontin (OPN), which is secreted by several cell types in the presence of alcohol, activates NFκB and activator protein 1 (AP-1) as well as several other genes, including urokinase plasminogen activator (uPA), MMPs and TGF-β[202,203]. Moreover, the profibrogenic plasminogen activator inhibitor (PAI-1) was increased in liver cells after alcohol consumption, leading to the inhibition of uPA, plasmin and fibrinolysis, thereby tipping the balance in favor of fibrosis[204].
Non-alcoholic fatty liver diseases and non-alcoholic steatohepatitis
Although the role of HSC activation in non-alcoholic fatty liver disease (NAFLD) has not been completely clarified, several studies have reported increased HSC activation in non-alcoholic steatohepatitis (NASH)[205]. Although the TGF-β signaling pathway plays a major role in the activation of HSCs in liver fibrosis, many other signaling pathways are implicated in liver fibrosis in NAFLD, including the hedgehog (Hh), PI3K/AKT and JAK/STAT signaling pathways[206].
Several studies have demonstrated that insulin resistance is associated with advanced stages of fibrosis in NAFLD[206,207]. Because insulin promotes HSC activation and insulin sensitizers can attenuate hepatic fibrosis in NASH, it has been suggested that insulin resistance plays an important role in NASH-related fibrogenesis[208,209].
It is understood that oxidative stress induces the activation of HSCs in NASH[108]. The role of oxidative stress in fibrogenesis is supported by the finding that antioxidants, such as vitamin E and astaxanthin, can decrease NASH-related fibrogenesis[210].
Recently reported data also indicate that adipokines affect not only lipid metabolism but also inflammatory and fibrotic processes in NAFLD[157] (the adipokines are described in more detail in the section “HSCs in fibrogenesis”). Recent data related to the newly described adipokines visfatin, chemerin and vaspin in NASH fibrogenesis is limited, warranting further studies to better understand their importance in the pathogenesis of NASH[211,212].
It has been hypothesized that various factors might contribute to the development of liver fibrosis in NAFLD, including LPS-derived from gut bacteria. Because LPS presents its effects by binding TLRs and because a recent finding in a murine NAFLD model demonstrated that TLR9 knockout mice demonstrate less steatohepatitis and liver fibrosis than controls, a role for TLRs in the progression of fibrosis of NASH have been proposed[213]. Recently, it has also been suggested that NK cells may play a pivotal role in NAFLD-related liver fibrogenesis. Although the population of hepatic NK cells in NAFLD patients is controversial, it has been shown that activation of the Hh pathway lead to hepatic accumulation of NK cells, resulting in progression of liver fibrosis in NASH[214].
In experimental studies as well as studies in patients with NASH, PPARγ agonists and especially pioglitazone have been shown to diminish liver fibrosis[209,215]. These data support the key role of PPARs in fibrosis in NASH. Among the other nuclear receptor family, liver X receptors (LXRs) play important roles in the regulation of cholesterol absorption, efflux, transport and excretion. In a more recent experimental study LXR ligands were found to suppress the activation of HSCs and the expression of fibrosis related genes[216].
Chronic viral hepatitis
During liver fibrogenesis, hepatotrophic viruses can induce HSC activation through several mechanisms. Immune cell types, especially NK cells, are engaged in the hepatitis B virus (HBV)-related acceleration of fibrosis[217]. It has been demonstrated that hepatitis B virus X protein (HBx) expression in hepatocytes leads to paracrine activation and proliferation of HSCs[218]. Moreover, in patients with chronic HBV, superinfection of hepatitis delta virus (HDV) accelerates the progression of fibrosis. The large isoform of hepatitis delta antigen (LHDAg) can induce liver fibrosis through the regulation of TGF-β-mediated signal transduction. LHDAg synergistically activates HBx protein-mediated TGF-β and AP-1 signaling, enhancing the level of TGF-β-induced PAI-1[219].
It has been found that the biology of activated HSCs is modulated by hepatitis C virus (HCV)-derived proteins in a profibrogenic manner[220]. Recent findings indicate that both oxidative stress and mitochondrial dysfunction are related to HCV pathogenesis. The blockade of oxidative stress as a therapeutic target in patients with HCV hepatitis remains under investigation[221,222]. Recent studies have indicated that hepatic iron accumulation is also correlated with histologic disease severity and with HSC numbers in patients with HCV infection, supporting the assumption that hepatic iron concentration may also influence fibrogenesis[223]. Huang et al[224] demonstrated that specific single nucleotide polymorphisms of TLR4 are related to the rate of progression of fibrosis in patients with HCV hepatitis. It has been suggested that this finding presents a link between a genetic marker and disease pathogenesis.
Although a correlation between HCV viral load and the progression of fibrosis has not been demonstrated in HCV hepatitis, HIV RNA levels predict the fibrogenic progression of chronic hepatitis in HCV/HIV-co-infected individuals[225,226]. In contrast, patients infected with HIV alone do not show significant liver fibrosis, indicating that HIV infection is not profibrogenic per se but rather accelerates the fibrogenic process in the presence of hepatic damage induced by hepatotropic viruses[225-227]. A recent, elegant study by Bruno et al[228] demonstrated that HIV gp120 modulates HSC behavior, including directional cell movement and expression of proinflammatory cytokines. They concluded that these results identify a direct pathway that most likely links HIV infection with liver fibrosis via envelope proteins, presenting new prospective strategies for the management of liver diseases in HCV/HIV-co-infected patients.
CONCLUSION
In conclusion, there have been considerable advances in the understanding of the mechanisms that underlie hepatic fibrogenesis. A critical event in liver fibrogenesis is that the ECM is a dynamic structure, and even advanced fibrosis may be reversible. Multiple interactions between the ECM, HSCs, endothelial cells and immune cells have been identified. The central event in fibrogenesis appears to be the activation of HSCs, which is a complex process, leading to multiple potential sites for therapeutic interventions. Although specific, effective and safe antifibrotic therapies are not currently available for the identification of potential new therapeutic agents, once available, they will mediate the progression of hepatic fibrogenesis.
Footnotes
P- Reviewers: Gong Y, Liu XD, Seki E S- Editor: Ma YJ L- Editor: A E- Editor: Zhang DN
References
- 1.Cohen-Naftaly M, Friedman SL. Current status of novel antifibrotic therapies in patients with chronic liver disease. Therap Adv Gastroenterol. 2011;4:391–417. doi: 10.1177/1756283X11413002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. 2007;117:539–548. doi: 10.1172/JCI30542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Puche JE, Saiman Y, Friedman SL. Hepatic stellate cells and liver fibrosis. Compr Physiol. 2013;3:1473–1492. doi: 10.1002/cphy.c120035. [DOI] [PubMed] [Google Scholar]
- 4.Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011;6:425–456. doi: 10.1146/annurev-pathol-011110-130246. [DOI] [PubMed] [Google Scholar]
- 5.Rockey DC. Antifibrotic therapy in chronic liver disease. Clin Gastroenterol Hepatol. 2005;3:95–107. doi: 10.1016/s1542-3565(04)00445-8. [DOI] [PubMed] [Google Scholar]
- 6.Ellis EL, Mann DA. Clinical evidence for the regression of liver fibrosis. J Hepatol. 2012;56:1171–1180. doi: 10.1016/j.jhep.2011.09.024. [DOI] [PubMed] [Google Scholar]
- 7.McGuire RF, Bissell DM, Boyles J, Roll FJ. Role of extracellular matrix in regulating fenestrations of sinusoidal endothelial cells isolated from normal rat liver. Hepatology. 1992;15:989–997. doi: 10.1002/hep.1840150603. [DOI] [PubMed] [Google Scholar]
- 8.Schuppan D, Ruehl M, Somasundaram R, Hahn EG. Matrix as a modulator of hepatic fibrogenesis. Semin Liver Dis. 2001;21:351–372. doi: 10.1055/s-2001-17556. [DOI] [PubMed] [Google Scholar]
- 9.Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, Pellicoro A, Raschperger E, Betsholtz C, Ruminski PG, et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med. 2013;19:1617–1624. doi: 10.1038/nm.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ray K. Liver: Key role for αv integrins in myofibroblasts in liver fibrosis. Nat Rev Gastroenterol Hepatol. 2014;11:4. doi: 10.1038/nrgastro.2013.227. [DOI] [PubMed] [Google Scholar]
- 11.Patsenker E, Popov Y, Stickel F, Jonczyk A, Goodman SL, Schuppan D. Inhibition of integrin alphavbeta6 on cholangiocytes blocks transforming growth factor-beta activation and retards biliary fibrosis progression. Gastroenterology. 2008;135:660–670. doi: 10.1053/j.gastro.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo J, Friedman SL. Hepatic fibrogenesis. Semin Liver Dis. 2007;27:413–426. doi: 10.1055/s-2007-991517. [DOI] [PubMed] [Google Scholar]
- 13.Das SK, Vasudevan DM. Genesis of hepatic fibrosis and its biochemical markers. Scand J Clin Lab Invest. 2008;68:260–269. doi: 10.1080/00365510701668516. [DOI] [PubMed] [Google Scholar]
- 14.Ramachandran P, Iredale JP. Reversibility of liver fibrosis. Ann Hepatol. 2009;8:283–291. [PubMed] [Google Scholar]
- 15.Fowell AJ, Collins JE, Duncombe DR, Pickering JA, Rosenberg WM, Benyon RC. Silencing tissue inhibitors of metalloproteinases (TIMPs) with short interfering RNA reveals a role for TIMP-1 in hepatic stellate cell proliferation. Biochem Biophys Res Commun. 2011;407:277–282. doi: 10.1016/j.bbrc.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 16.Lee JS, Semela D, Iredale J, Shah VH. Sinusoidal remodeling and angiogenesis: a new function for the liver-specific pericyte? Hepatology. 2007;45:817–825. doi: 10.1002/hep.21564. [DOI] [PubMed] [Google Scholar]
- 17.Rosmorduc O, Housset C. Hypoxia: a link between fibrogenesis, angiogenesis, and carcinogenesis in liver disease. Semin Liver Dis. 2010;30:258–270. doi: 10.1055/s-0030-1255355. [DOI] [PubMed] [Google Scholar]
- 18.Johnson A, DiPietro LA. Apoptosis and angiogenesis: an evolving mechanism for fibrosis. FASEB J. 2013;27:3893–3901. doi: 10.1096/fj.12-214189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wight TN, Potter-Perigo S. The extracellular matrix: an active or passive player in fibrosis? Am J Physiol Gastrointest Liver Physiol. 2011;301:G950–G955. doi: 10.1152/ajpgi.00132.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kisseleva T, Brenner DA. Anti-fibrogenic strategies and the regression of fibrosis. Best Pract Res Clin Gastroenterol. 2011;25:305–317. doi: 10.1016/j.bpg.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tacke F, Weiskirchen R. Update on hepatic stellate cells: pathogenic role in liver fibrosis and novel isolation techniques. Expert Rev Gastroenterol Hepatol. 2012;6:67–80. doi: 10.1586/egh.11.92. [DOI] [PubMed] [Google Scholar]
- 22.Kisseleva T, Brenner DA. Hepatic stellate cells and the reversal of fibrosis. J Gastroenterol Hepatol. 2006;21 Suppl 3:S84–S87. doi: 10.1111/j.1440-1746.2006.04584.x. [DOI] [PubMed] [Google Scholar]
- 23.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lekkerkerker AN, Aarbiou J, van Es T, Janssen RA. Cellular players in lung fibrosis. Curr Pharm Des. 2012;18:4093–4102. doi: 10.2174/138161212802430396. [DOI] [PubMed] [Google Scholar]
- 25.Fallowfield JA, Mizuno M, Kendall TJ, Constandinou CM, Benyon RC, Duffield JS, Iredale JP. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol. 2007;178:5288–5295. doi: 10.4049/jimmunol.178.8.5288. [DOI] [PubMed] [Google Scholar]
- 26.Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest. 2013;123:1887–1901. doi: 10.1172/JCI66028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parola M, Marra F, Pinzani M. Myofibroblast - like cells and liver fibrogenesis: Emerging concepts in a rapidly moving scenario. Mol Aspects Med. 2008;29:58–66. doi: 10.1016/j.mam.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 28.Kisseleva T, Brenner DA. Fibrogenesis of parenchymal organs. Proc Am Thorac Soc. 2008;5:338–342. doi: 10.1513/pats.200711-168DR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalluri R. EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest. 2009;119:1417–1419. doi: 10.1172/JCI39675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi SS, Diehl AM. Epithelial-to-mesenchymal transitions in the liver. Hepatology. 2009;50:2007–2013. doi: 10.1002/hep.23196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 32.Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 2008;19:2282–2287. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Asahina K. Hepatic stellate cell progenitor cells. J Gastroenterol Hepatol. 2012;27 Suppl 2:80–84. doi: 10.1111/j.1440-1746.2011.07001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szatmari I, Iacovino M, Kyba M. The retinoid signaling pathway inhibits hematopoiesis and uncouples from the Hox genes during hematopoietic development. Stem Cells. 2010;28:1518–1529. doi: 10.1002/stem.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Costa G, Kouskoff V, Lacaud G. Origin of blood cells and HSC production in the embryo. Trends Immunol. 2012;33:215–223. doi: 10.1016/j.it.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 36.McDonald LT, LaRue AC. Hematopoietic stem cell derived carcinoma-associated fibroblasts: a novel origin. Int J Clin Exp Pathol. 2012;5:863–873. [PMC free article] [PubMed] [Google Scholar]
- 37.Krivtsov AV, Figueroa ME, Sinha AU, Stubbs MC, Feng Z, Valk PJ, Delwel R, Döhner K, Bullinger L, Kung AL, et al. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia. 2013;27:852–860. doi: 10.1038/leu.2012.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vishnubalaji R, Al-Nbaheen M, Kadalmani B, Aldahmash A, Ramesh T. Skin-derived multipotent stromal cells--an archrival for mesenchymal stem cells. Cell Tissue Res. 2012;350:1–12. doi: 10.1007/s00441-012-1471-z. [DOI] [PubMed] [Google Scholar]
- 39.Geerts A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin Liver Dis. 2001;21:311–335. doi: 10.1055/s-2001-17550. [DOI] [PubMed] [Google Scholar]
- 40.Sachs BD, Baillie GS, McCall JR, Passino MA, Schachtrup C, Wallace DA, Dunlop AJ, MacKenzie KF, Klussmann E, Lynch MJ, et al. p75 neurotrophin receptor regulates tissue fibrosis through inhibition of plasminogen activation via a PDE4/cAMP/PKA pathway. J Cell Biol. 2007;177:1119–1132. doi: 10.1083/jcb.200701040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kendall TJ, Hennedige S, Aucott RL, Hartland SN, Vernon MA, Benyon RC, Iredale JP. p75 Neurotrophin receptor signaling regulates hepatic myofibroblast proliferation and apoptosis in recovery from rodent liver fibrosis. Hepatology. 2009;49:901–910. doi: 10.1002/hep.22701. [DOI] [PubMed] [Google Scholar]
- 42.Senoo H, Kojima N, Sato M. Vitamin A-storing cells (stellate cells) Vitam Horm. 2007;75:131–159. doi: 10.1016/S0083-6729(06)75006-3. [DOI] [PubMed] [Google Scholar]
- 43.Guyot C, Lepreux S, Combe C, Doudnikoff E, Bioulac-Sage P, Balabaud C, Desmoulière A. Hepatic fibrosis and cirrhosis: the (myo)fibroblastic cell subpopulations involved. Int J Biochem Cell Biol. 2006;38:135–151. doi: 10.1016/j.biocel.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 44.Yovchev MI, Zhang J, Neufeld DS, Grozdanov PN, Dabeva MD. Thymus cell antigen-1-expressing cells in the oval cell compartment. Hepatology. 2009;50:601–611. doi: 10.1002/hep.23012. [DOI] [PubMed] [Google Scholar]
- 45.Bosselut N, Housset C, Marcelo P, Rey C, Burmester T, Vinh J, Vaubourdolle M, Cadoret A, Baudin B. Distinct proteomic features of two fibrogenic liver cell populations: hepatic stellate cells and portal myofibroblasts. Proteomics. 2010;10:1017–1028. doi: 10.1002/pmic.200900257. [DOI] [PubMed] [Google Scholar]
- 46.Dranoff JA, Wells RG. Portal fibroblasts: Underappreciated mediators of biliary fibrosis. Hepatology. 2010;51:1438–1444. doi: 10.1002/hep.23405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quan TE, Cowper S, Wu SP, Bockenstedt LK, Bucala R. Circulating fibrocytes: collagen-secreting cells of the peripheral blood. Int J Biochem Cell Biol. 2004;36:598–606. doi: 10.1016/j.biocel.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 48.Strieter RM, Keeley EC, Burdick MD, Mehrad B. The role of circulating mesenchymal progenitor cells, fibrocytes, in promoting pulmonary fibrosis. Trans Am Clin Climatol Assoc. 2009;120:49–59. [PMC free article] [PubMed] [Google Scholar]
- 49.Kisseleva T, Uchinami H, Feirt N, Quintana-Bustamante O, Segovia JC, Schwabe RF, Brenner DA. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J Hepatol. 2006;45:429–438. doi: 10.1016/j.jhep.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 50.Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H, Kalluri R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007;282:23337–23347. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]
- 51.Pinzani M. Epithelial-mesenchymal transition in chronic liver disease: fibrogenesis or escape from death? J Hepatol. 2011;55:459–465. doi: 10.1016/j.jhep.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 52.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taura K, Miura K, Iwaisako K, Osterreicher CH, Kodama Y, Penz-Osterreicher M, Brenner DA. Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology. 2010;51:1027–1036. doi: 10.1002/hep.23368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Canbay A, Friedman S, Gores GJ. Apoptosis: the nexus of liver injury and fibrosis. Hepatology. 2004;39:273–278. doi: 10.1002/hep.20051. [DOI] [PubMed] [Google Scholar]
- 55.Mehal W, Imaeda A. Cell death and fibrogenesis. Semin Liver Dis. 2010;30:226–231. doi: 10.1055/s-0030-1255352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sánchez-Valle V, Chávez-Tapia NC, Uribe M, Méndez-Sánchez N. Role of oxidative stress and molecular changes in liver fibrosis: a review. Curr Med Chem. 2012;19:4850–4860. doi: 10.2174/092986712803341520. [DOI] [PubMed] [Google Scholar]
- 57.Canbay A, Taimr P, Torok N, Higuchi H, Friedman S, Gores GJ. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab Invest. 2003;83:655–663. doi: 10.1097/01.lab.0000069036.63405.5c. [DOI] [PubMed] [Google Scholar]
- 58.Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, Bronk SF, Gores GJ. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology. 2003;38:1188–1198. doi: 10.1053/jhep.2003.50472. [DOI] [PubMed] [Google Scholar]
- 59.Takehara T, Tatsumi T, Suzuki T, Rucker EB, Hennighausen L, Jinushi M, Miyagi T, Kanazawa Y, Hayashi N. Hepatocyte-specific disruption of Bcl-xL leads to continuous hepatocyte apoptosis and liver fibrotic responses. Gastroenterology. 2004;127:1189–1197. doi: 10.1053/j.gastro.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 60.Ghavami S, Hashemi M, Kadkhoda K, Alavian SM, Bay GH, Los M. Apoptosis in liver diseases--detection and therapeutic applications. Med Sci Monit. 2005;11:RA337–RA345. [PubMed] [Google Scholar]
- 61.Guicciardi ME, Gores GJ. Apoptosis as a mechanism for liver disease progression. Semin Liver Dis. 2010;30:402–410. doi: 10.1055/s-0030-1267540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Novo E, Marra F, Zamara E, Valfrè di Bonzo L, Caligiuri A, Cannito S, Antonaci C, Colombatto S, Pinzani M, Parola M. Dose dependent and divergent effects of superoxide anion on cell death, proliferation, and migration of activated human hepatic stellate cells. Gut. 2006;55:90–97. doi: 10.1136/gut.2005.069633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wright MC, Issa R, Smart DE, Trim N, Murray GI, Primrose JN, Arthur MJ, Iredale JP, Mann DA. Gliotoxin stimulates the apoptosis of human and rat hepatic stellate cells and enhances the resolution of liver fibrosis in rats. Gastroenterology. 2001;121:685–698. doi: 10.1053/gast.2001.27188. [DOI] [PubMed] [Google Scholar]
- 64.Taimr P, Higuchi H, Kocova E, Rippe RA, Friedman S, Gores GJ. Activated stellate cells express the TRAIL receptor-2/death receptor-5 and undergo TRAIL-mediated apoptosis. Hepatology. 2003;37:87–95. doi: 10.1053/jhep.2003.50002. [DOI] [PubMed] [Google Scholar]
- 65.Anan A, Baskin-Bey ES, Bronk SF, Werneburg NW, Shah VH, Gores GJ. Proteasome inhibition induces hepatic stellate cell apoptosis. Hepatology. 2006;43:335–344. doi: 10.1002/hep.21036. [DOI] [PubMed] [Google Scholar]
- 66.Pockros PJ, Schiff ER, Shiffman ML, McHutchison JG, Gish RG, Afdhal NH, Makhviladze M, Huyghe M, Hecht D, Oltersdorf T, et al. Oral IDN-6556, an antiapoptotic caspase inhibitor, may lower aminotransferase activity in patients with chronic hepatitis C. Hepatology. 2007;46:324–329. doi: 10.1002/hep.21664. [DOI] [PubMed] [Google Scholar]
- 67.March S, Hui EE, Underhill GH, Khetani S, Bhatia SN. Microenvironmental regulation of the sinusoidal endothelial cell phenotype in vitro. Hepatology. 2009;50:920–928. doi: 10.1002/hep.23085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Witek RP, Yang L, Liu R, Jung Y, Omenetti A, Syn WK, Choi SS, Cheong Y, Fearing CM, Agboola KM, et al. Liver cell-derived microparticles activate hedgehog signaling and alter gene expression in hepatic endothelial cells. Gastroenterology. 2009;136:320–330.e2. doi: 10.1053/j.gastro.2008.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xie G, Wang X, Wang L, Wang L, Atkinson RD, Kanel GC, Gaarde WA, Deleve LD. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology. 2012;142:918–927.e6. doi: 10.1053/j.gastro.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ding BS, Cao Z, Lis R, Nolan DJ, Guo P, Simons M, Penfold ME, Shido K, Rabbany SY, Rafii S. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature. 2014;505:97–102. doi: 10.1038/nature12681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006;26:1175–1186. doi: 10.1111/j.1478-3231.2006.01342.x. [DOI] [PubMed] [Google Scholar]
- 72.Fischer R, Cariers A, Reinehr R, Häussinger D. Caspase 9-dependent killing of hepatic stellate cells by activated Kupffer cells. Gastroenterology. 2002;123:845–861. doi: 10.1053/gast.2002.35384. [DOI] [PubMed] [Google Scholar]
- 73.Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, Jang MK, Guenther ND, Mederacke I, Friedman R, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58:1461–1473. doi: 10.1002/hep.26429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, Hartland SN, Snowdon VK, Cappon A, Gordon-Walker TT, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci USA. 2012;109:E3186–E3195. doi: 10.1073/pnas.1119964109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chu PS, Nakamoto N, Ebinuma H, Usui S, Saeki K, Matsumoto A, Mikami Y, Sugiyama K, Tomita K, Kanai T, et al. C-C motif chemokine receptor 9 positive macrophages activate hepatic stellate cells and promote liver fibrosis in mice. Hepatology. 2013;58:337–350. doi: 10.1002/hep.26351. [DOI] [PubMed] [Google Scholar]
- 76.Heymann F, Hammerich L, Storch D, Bartneck M, Huss S, Rüsseler V, Gassler N, Lira SA, Luedde T, Trautwein C, et al. Hepatic macrophage migration and differentiation critical for liver fibrosis is mediated by the chemokine receptor C-C motif chemokine receptor 8 in mice. Hepatology. 2012;55:898–909. doi: 10.1002/hep.24764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mitchell C, Couton D, Couty JP, Anson M, Crain AM, Bizet V, Rénia L, Pol S, Mallet V, Gilgenkrantz H. Dual role of CCR2 in the constitution and the resolution of liver fibrosis in mice. Am J Pathol. 2009;174:1766–1775. doi: 10.2353/ajpath.2009.080632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Seki E, De Minicis S, Gwak GY, Kluwe J, Inokuchi S, Bursill CA, Llovet JM, Brenner DA, Schwabe RF. CCR1 and CCR5 promote hepatic fibrosis in mice. J Clin Invest. 2009;119:1858–1870. doi: 10.1172/JCI37444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aoyama T, Inokuchi S, Brenner DA, Seki E. CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice. Hepatology. 2010;52:1390–1400. doi: 10.1002/hep.23795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Karlmark KR, Zimmermann HW, Roderburg C, Gassler N, Wasmuth HE, Luedde T, Trautwein C, Tacke F. The fractalkine receptor CX3CR1 protects against liver fibrosis by controlling differentiation and survival of infiltrating hepatic monocytes. Hepatology. 2010;52:1769–1782. doi: 10.1002/hep.23894. [DOI] [PubMed] [Google Scholar]
- 81.Marra F, Aleffi S, Galastri S, Provenzano A. Mononuclear cells in liver fibrosis. Semin Immunopathol. 2009;31:345–358. doi: 10.1007/s00281-009-0169-0. [DOI] [PubMed] [Google Scholar]
- 82.Li JT, Liao ZX, Ping J, Xu D, Wang H. Molecular mechanism of hepatic stellate cell activation and antifibrotic therapeutic strategies. J Gastroenterol. 2008;43:419–428. doi: 10.1007/s00535-008-2180-y. [DOI] [PubMed] [Google Scholar]
- 83.Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006;130:435–452. doi: 10.1053/j.gastro.2005.10.055. [DOI] [PubMed] [Google Scholar]
- 84.Melhem A, Muhanna N, Bishara A, Alvarez CE, Ilan Y, Bishara T, Horani A, Nassar M, Friedman SL, Safadi R. Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J Hepatol. 2006;45:60–71. doi: 10.1016/j.jhep.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 85.Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Muhanna N, Doron S, Wald O, Horani A, Eid A, Pappo O, Friedman SL, Safadi R. Activation of hepatic stellate cells after phagocytosis of lymphocytes: A novel pathway of fibrogenesis. Hepatology. 2008;48:963–977. doi: 10.1002/hep.22413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jeong WI, Park O, Radaeva S, Gao B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology. 2006;44:1441–1451. doi: 10.1002/hep.21419. [DOI] [PubMed] [Google Scholar]
- 88.Rockey DC, Chung JJ. Interferon gamma inhibits lipocyte activation and extracellular matrix mRNA expression during experimental liver injury: implications for treatment of hepatic fibrosis. J Investig Med. 1994;42:660–670. [PubMed] [Google Scholar]
- 89.Baroni GS, D’Ambrosio L, Curto P, Casini A, Mancini R, Jezequel AM, Benedetti A. Interferon gamma decreases hepatic stellate cell activation and extracellular matrix deposition in rat liver fibrosis. Hepatology. 1996;23:1189–1199. doi: 10.1002/hep.510230538. [DOI] [PubMed] [Google Scholar]
- 90.Jeong WI, Park O, Gao B. Abrogation of the antifibrotic effects of natural killer cells/interferon-gamma contributes to alcohol acceleration of liver fibrosis. Gastroenterology. 2008;134:248–258. doi: 10.1053/j.gastro.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gao B, Radaeva S, Park O. Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol. 2009;86:513–528. doi: 10.1189/jlb.0309135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gao B, Radaeva S, Jeong WI. Activation of natural killer cells inhibits liver fibrosis: a novel strategy to treat liver fibrosis. Expert Rev Gastroenterol Hepatol. 2007;1:173–180. doi: 10.1586/17474124.1.1.173. [DOI] [PubMed] [Google Scholar]
- 93.Rehermann B. Pathogenesis of chronic viral hepatitis: differential roles of T cells and NK cells. Nat Med. 2013;19:859–868. doi: 10.1038/nm.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yi HS, Jeong WI. Interaction of hepatic stellate cells with diverse types of immune cells: foe or friend? J Gastroenterol Hepatol. 2013;28 Suppl 1:99–104. doi: 10.1111/jgh.12017. [DOI] [PubMed] [Google Scholar]
- 95.Svegliati-Baroni G, Saccomanno S, van Goor H, Jansen P, Benedetti A, Moshage H. Involvement of reactive oxygen species and nitric oxide radicals in activation and proliferation of rat hepatic stellate cells. Liver. 2001;21:1–12. doi: 10.1034/j.1600-0676.2001.210101.x. [DOI] [PubMed] [Google Scholar]
- 96.Kodama T, Takehara T, Hikita H, Shimizu S, Li W, Miyagi T, Hosui A, Tatsumi T, Ishida H, Tadokoro S, et al. Thrombocytopenia exacerbates cholestasis-induced liver fibrosis in mice. Gastroenterology. 2010;138:2487–2498, 2498.e1-7. doi: 10.1053/j.gastro.2010.02.054. [DOI] [PubMed] [Google Scholar]
- 97.Ikeda N, Murata S, Maruyama T, Tamura T, Nozaki R, Kawasaki T, Fukunaga K, Oda T, Sasaki R, Homma M, et al. Platelet-derived adenosine 5’-triphosphate suppresses activation of human hepatic stellate cell: In vitro study. Hepatol Res. 2012;42:91–102. doi: 10.1111/j.1872-034X.2011.00893.x. [DOI] [PubMed] [Google Scholar]
- 98.Takahashi K, Murata S, Fukunaga K, Ohkohchi N. Human platelets inhibit liver fibrosis in severe combined immunodeficiency mice. World J Gastroenterol. 2013;19:5250–5260. doi: 10.3748/wjg.v19.i32.5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jaeschke H. Mechanisms of Liver Injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1083–G1088. doi: 10.1152/ajpgi.00568.2005. [DOI] [PubMed] [Google Scholar]
- 100.Ghatak S, Biswas A, Dhali GK, Chowdhury A, Boyer JL, Santra A. Oxidative stress and hepatic stellate cell activation are key events in arsenic induced liver fibrosis in mice. Toxicol Appl Pharmacol. 2011;251:59–69. doi: 10.1016/j.taap.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Aleffi S, Navari N, Delogu W, Galastri S, Novo E, Rombouts K, Pinzani M, Parola M, Marra F. Mammalian target of rapamycin mediates the angiogenic effects of leptin in human hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2011;301:G210–G219. doi: 10.1152/ajpgi.00047.2010. [DOI] [PubMed] [Google Scholar]
- 102.Zhang F, Ni C, Kong D, Zhang X, Zhu X, Chen L, Lu Y, Zheng S. Ligustrazine attenuates oxidative stress-induced activation of hepatic stellate cells by interrupting platelet-derived growth factor-β receptor-mediated ERK and p38 pathways. Toxicol Appl Pharmacol. 2012;265:51–60. doi: 10.1016/j.taap.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 103.Moreno-Alvarez P, Sosa-Garrocho M, Briones-Orta MA, González-Espinosa C, Medina-Tamayo J, Molina-Jijón E, Pedraza-Chaverri J, Macías-Silva M. Angiotensin II increases mRNA levels of all TGF-beta isoforms in quiescent and activated rat hepatic stellate cells. Cell Biol Int. 2010;34:969–978. doi: 10.1042/CBI20090074. [DOI] [PubMed] [Google Scholar]
- 104.Shah R, Reyes-Gordillo K, Arellanes-Robledo J, Lechuga CG, Hernández-Nazara Z, Cotty A, Rojkind M, Lakshman MR. TGF-β1 up-regulates the expression of PDGF-β receptor mRNA and induces a delayed PI3K-, AKT-, and p70(S6K) -dependent proliferative response in activated hepatic stellate cells. Alcohol Clin Exp Res. 2013;37:1838–1848. doi: 10.1111/acer.12167. [DOI] [PubMed] [Google Scholar]
- 105.Thirunavukkarasu C, Watkins S, Harvey SA, Gandhi CR. Superoxide-induced apoptosis of activated rat hepatic stellate cells. J Hepatol. 2004;41:567–575. doi: 10.1016/j.jhep.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 106.Dunning S, Hannivoort RA, de Boer JF, Buist-Homan M, Faber KN, Moshage H. Superoxide anions and hydrogen peroxide inhibit proliferation of activated rat stellate cells and induce different modes of cell death. Liver Int. 2009;29:922–932. doi: 10.1111/j.1478-3231.2009.02004.x. [DOI] [PubMed] [Google Scholar]
- 107.Dunning S, Ur Rehman A, Tiebosch MH, Hannivoort RA, Haijer FW, Woudenberg J, van den Heuvel FA, Buist-Homan M, Faber KN, Moshage H. Glutathione and antioxidant enzymes serve complementary roles in protecting activated hepatic stellate cells against hydrogen peroxide-induced cell death. Biochim Biophys Acta. 2013;1832:2027–2034. doi: 10.1016/j.bbadis.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 108.Koek GH, Liedorp PR, Bast A. The role of oxidative stress in non-alcoholic steatohepatitis. Clin Chim Acta. 2011;412:1297–1305. doi: 10.1016/j.cca.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 109.De Minicis S, Brenner DA. NOX in liver fibrosis. Arch Biochem Biophys. 2007;462:266–272. doi: 10.1016/j.abb.2007.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jiang JX, Venugopal S, Serizawa N, Chen X, Scott F, Li Y, Adamson R, Devaraj S, Shah V, Gershwin ME, et al. Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology. 2010;139:1375–1384. doi: 10.1053/j.gastro.2010.05.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Paik YH, Iwaisako K, Seki E, Inokuchi S, Schnabl B, Osterreicher CH, Kisseleva T, Brenner DA. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology. 2011;53:1730–1741. doi: 10.1002/hep.24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cui W, Matsuno K, Iwata K, Ibi M, Matsumoto M, Zhang J, Zhu K, Katsuyama M, Torok NJ, Yabe-Nishimura C. NOX1/nicotinamide adenine dinucleotide phosphate, reduced form (NADPH) oxidase promotes proliferation of stellate cells and aggravates liver fibrosis induced by bile duct ligation. Hepatology. 2011;54:949–958. doi: 10.1002/hep.24465. [DOI] [PubMed] [Google Scholar]
- 113.Jiang JX, Chen X, Serizawa N, Szyndralewiez C, Page P, Schröder K, Brandes RP, Devaraj S, Török NJ. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic Biol Med. 2012;53:289–296. doi: 10.1016/j.freeradbiomed.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nieto N, Friedman SL, Cederbaum AI. Cytochrome P450 2E1-derived reactive oxygen species mediate paracrine stimulation of collagen I protein synthesis by hepatic stellate cells. J Biol Chem. 2002;277:9853–9864. doi: 10.1074/jbc.M110506200. [DOI] [PubMed] [Google Scholar]
- 115.Liu T, Wang P, Cong M, Xu Y, Jia J, You H. The CYP2E1 inhibitor DDC up-regulates MMP-1 expression in hepatic stellate cells via an ERK1/2- and Akt-dependent mechanism. Biosci Rep. 2013;33:pii: e00041. doi: 10.1042/BSR20130033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, Lockhart MM, Frampton CM, Taylor KM, Smith RA, Murphy MP. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010;30:1019–1026. doi: 10.1111/j.1478-3231.2010.02250.x. [DOI] [PubMed] [Google Scholar]
- 117.den Hartog GJ, Qi S, van Tilburg JH, Koek GH, Bast A. Superoxide anion radicals activate hepatic stellate cells after entry through chloride channels: a new target in liver fibrosis. Eur J Pharmacol. 2014;724:140–144. doi: 10.1016/j.ejphar.2013.12.033. [DOI] [PubMed] [Google Scholar]
- 118.Huebener P, Schwabe RF. Regulation of wound healing and organ fibrosis by toll-like receptors. Biochim Biophys Acta. 2013;1832:1005–1017. doi: 10.1016/j.bbadis.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 120.Li YS, Ni SY, Meng Y, Shi XL, Zhao XW, Luo HH, Li X. Angiotensin II facilitates fibrogenic effect of TGF-β1 through enhancing the down-regulation of BAMBI caused by LPS: a new pro-fibrotic mechanism of angiotensin II. PLoS One. 2013;8:e76289. doi: 10.1371/journal.pone.0076289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liu C, Chen X, Yang L, Kisseleva T, Brenner DA, Seki E. Transcriptional Repression of the Transforming Growth Factor β (TGF-β) Pseudoreceptor BMP and Activin Membrane-bound Inhibitor (BAMBI) by Nuclear Factor κB (NF-κB) p50 Enhances TGF-β Signaling in Hepatic Stellate Cells. J Biol Chem. 2014;289:7082–7091. doi: 10.1074/jbc.M113.543769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li J, Wang FP, She WM, Yang CQ, Li L, Tu CT, Wang JY, Jiang W. Enhanced high-mobility group box 1 (HMGB1) modulates regulatory T cells (Treg)/T helper 17 (Th17) balance via toll-like receptor (TLR)-4-interleukin (IL)-6 pathway in patients with chronic hepatitis B. J Viral Hepat. 2014;21:129–140. doi: 10.1111/jvh.12152. [DOI] [PubMed] [Google Scholar]
- 123.Wang FP, Li L, Li J, Wang JY, Wang LY, Jiang W. High mobility group box-1 promotes the proliferation and migration of hepatic stellate cells via TLR4-dependent signal pathways of PI3K/Akt and JNK. PLoS One. 2013;8:e64373. doi: 10.1371/journal.pone.0064373. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 124.Rippe RA, Brenner DA. From quiescence to activation: Gene regulation in hepatic stellate cells. Gastroenterology. 2004;127:1260–1262. doi: 10.1053/j.gastro.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 125.Mann J, Oakley F, Akiboye F, Elsharkawy A, Thorne AW, Mann DA. Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: implications for wound healing and fibrogenesis. Cell Death Differ. 2007;14:275–285. doi: 10.1038/sj.cdd.4401979. [DOI] [PubMed] [Google Scholar]
- 126.Tsukamoto H, She H, Hazra S, Cheng J, Miyahara T. Anti-adipogenic regulation underlies hepatic stellate cell transdifferentiation. J Gastroenterol Hepatol. 2006;21 Suppl 3:S102–S105. doi: 10.1111/j.1440-1746.2006.04573.x. [DOI] [PubMed] [Google Scholar]
- 127.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lee UE, Friedman SL. Mechanisms of hepatic fibrogenesis. Best Pract Res Clin Gastroenterol. 2011;25:195–206. doi: 10.1016/j.bpg.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhang F, Kong D, Lu Y, Zheng S. Peroxisome proliferator-activated receptor-γ as a therapeutic target for hepatic fibrosis: from bench to bedside. Cell Mol Life Sci. 2013;70:259–276. doi: 10.1007/s00018-012-1046-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sharvit E, Abramovitch S, Reif S, Bruck R. Amplified inhibition of stellate cell activation pathways by PPAR-γ, RAR and RXR agonists. PLoS One. 2013;8:e76541. doi: 10.1371/journal.pone.0076541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov. 2010;9:775–789. doi: 10.1038/nrd3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lakner AM, Steuerwald NM, Walling TL, Ghosh S, Li T, McKillop IH, Russo MW, Bonkovsky HL, Schrum LW. Inhibitory effects of microRNA 19b in hepatic stellate cell-mediated fibrogenesis. Hepatology. 2012;56:300–310. doi: 10.1002/hep.25613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Roderburg C, Urban GW, Bettermann K, Vucur M, Zimmermann H, Schmidt S, Janssen J, Koppe C, Knolle P, Castoldi M, et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology. 2011;53:209–218. doi: 10.1002/hep.23922. [DOI] [PubMed] [Google Scholar]
- 134.Ogawa T, Enomoto M, Fujii H, Sekiya Y, Yoshizato K, Ikeda K, Kawada N. MicroRNA-221/222 upregulation indicates the activation of stellate cells and the progression of liver fibrosis. Gut. 2012;61:1600–1609. doi: 10.1136/gutjnl-2011-300717. [DOI] [PubMed] [Google Scholar]
- 135.Bian EB, Huang C, Wang H, Wu BM, Zhang L, Lv XW, Li J. DNA methylation: new therapeutic implications for hepatic fibrosis. Cell Signal. 2013;25:355–358. doi: 10.1016/j.cellsig.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 136.Tsukamoto H, Zhu NL, Asahina K, Mann DA, Mann J. Epigenetic cell fate regulation of hepatic stellate cells. Hepatol Res. 2011;41:675–682. doi: 10.1111/j.1872-034X.2011.00804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zeybel M, Hardy T, Wong YK, Mathers JC, Fox CR, Gackowska A, Oakley F, Burt AD, Wilson CL, Anstee QM, et al. Multigenerational epigenetic adaptation of the hepatic wound-healing response. Nat Med. 2012;18:1369–1377. doi: 10.1038/nm.2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lechuga CG, Hernández-Nazara ZH, Hernández E, Bustamante M, Desierto G, Cotty A, Dharker N, Choe M, Rojkind M. PI3K is involved in PDGF-beta receptor upregulation post-PDGF-BB treatment in mouse HSC. Am J Physiol Gastrointest Liver Physiol. 2006;291:G1051–G1061. doi: 10.1152/ajpgi.00058.2005. [DOI] [PubMed] [Google Scholar]
- 139.Cao S, Yaqoob U, Das A, Shergill U, Jagavelu K, Huebert RC, Routray C, Abdelmoneim S, Vasdev M, Leof E, et al. Neuropilin-1 promotes cirrhosis of the rodent and human liver by enhancing PDGF/TGF-beta signaling in hepatic stellate cells. J Clin Invest. 2010;120:2379–2394. doi: 10.1172/JCI41203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Di Sario A, Bendia E, Taffetani S, Marzioni M, Candelaresi C, Pigini P, Schindler U, Kleemann HW, Trozzi L, Macarri G, et al. Selective Na+/H+ exchange inhibition by cariporide reduces liver fibrosis in the rat. Hepatology. 2003;37:256–266. doi: 10.1053/jhep.2003.50028. [DOI] [PubMed] [Google Scholar]
- 141.Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. 2013;59:583–594. doi: 10.1016/j.jhep.2013.03.033. [DOI] [PubMed] [Google Scholar]
- 142.Kim SY, Cho BH, Kim UH. CD38-mediated Ca2+ signaling contributes to angiotensin II-induced activation of hepatic stellate cells: attenuation of hepatic fibrosis by CD38 ablation. J Biol Chem. 2010;285:576–582. doi: 10.1074/jbc.M109.076216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Affò S, Bataller R. RANTES antagonism: a promising approach to treat chronic liver diseases. J Hepatol. 2011;55:936–938. doi: 10.1016/j.jhep.2011.04.023. [DOI] [PubMed] [Google Scholar]
- 144.Novo E, Povero D, Busletta C, Paternostro C, di Bonzo LV, Cannito S, Compagnone A, Bandino A, Marra F, Colombatto S, et al. The biphasic nature of hypoxia-induced directional migration of activated human hepatic stellate cells. J Pathol. 2012;226:588–597. doi: 10.1002/path.3005. [DOI] [PubMed] [Google Scholar]
- 145.Olsen AL, Sackey BK, Marcinkiewicz C, Boettiger D, Wells RG. Fibronectin extra domain-A promotes hepatic stellate cell motility but not differentiation into myofibroblasts. Gastroenterology. 2012;142:928–937.e3. doi: 10.1053/j.gastro.2011.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hashmi AZ, Hakim W, Kruglov EA, Watanabe A, Watkins W, Dranoff JA, Mehal WZ. Adenosine inhibits cytosolic calcium signals and chemotaxis in hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2007;292:G395–G401. doi: 10.1152/ajpgi.00208.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sohail MA, Hashmi AZ, Hakim W, Watanabe A, Zipprich A, Groszmann RJ, Dranoff JA, Torok NJ, Mehal WZ. Adenosine induces loss of actin stress fibers and inhibits contraction in hepatic stellate cells via Rho inhibition. Hepatology. 2009;49:185–194. doi: 10.1002/hep.22589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ruhl CE, Everhart JE. Coffee and tea consumption are associated with a lower incidence of chronic liver disease in the United States. Gastroenterology. 2005;129:1928–1936. doi: 10.1053/j.gastro.2005.08.056. [DOI] [PubMed] [Google Scholar]
- 149.Shim SG, Jun DW, Kim EK, Saeed WK, Lee KN, Lee HL, Lee OY, Choi HS, Yoon BC. Caffeine attenuates liver fibrosis via defective adhesion of hepatic stellate cells in cirrhotic model. J Gastroenterol Hepatol. 2013;28:1877–1884. doi: 10.1111/jgh.12317. [DOI] [PubMed] [Google Scholar]
- 150.Inagaki Y, Okazaki I. Emerging insights into Transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut. 2007;56:284–292. doi: 10.1136/gut.2005.088690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yoshida K, Matsuzaki K. Differential Regulation of TGF-β/Smad Signaling in Hepatic Stellate Cells between Acute and Chronic Liver Injuries. Front Physiol. 2012;3:53. doi: 10.3389/fphys.2012.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lee YS, Jeong WI. Retinoic acids and hepatic stellate cells in liver disease. J Gastroenterol Hepatol. 2012;27 Suppl 2:75–79. doi: 10.1111/j.1440-1746.2011.07007.x. [DOI] [PubMed] [Google Scholar]
- 153.Yang L, Pang Y, Moses HL. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31:220–227. doi: 10.1016/j.it.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chen L, Charrier AL, Leask A, French SW, Brigstock DR. Ethanol-stimulated differentiated functions of human or mouse hepatic stellate cells are mediated by connective tissue growth factor. J Hepatol. 2011;55:399–406. doi: 10.1016/j.jhep.2010.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hora C, Negro F, Leandro G, Oneta CM, Rubbia-Brandt L, Muellhaupt B, Helbling B, Malinverni R, Gonvers JJ, Dufour JF. Connective tissue growth factor, steatosis and fibrosis in patients with chronic hepatitis C. Liver Int. 2008;28:370–376. doi: 10.1111/j.1478-3231.2007.01608.x. [DOI] [PubMed] [Google Scholar]
- 156.Gressner OA, Lahme B, Demirci I, Gressner AM, Weiskirchen R. Differential effects of TGF-beta on connective tissue growth factor (CTGF/CCN2) expression in hepatic stellate cells and hepatocytes. J Hepatol. 2007;47:699–710. doi: 10.1016/j.jhep.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 157.Marra F, Bertolani C. Adipokines in liver diseases. Hepatology. 2009;50:957–969. doi: 10.1002/hep.23046. [DOI] [PubMed] [Google Scholar]
- 158.Savvidou S, Hytiroglou P, Orfanou-Koumerkeridou H, Panderis A, Frantzoulis P, Goulis J. Low serum adiponectin levels are predictive of advanced hepatic fibrosis in patients with NAFLD. J Clin Gastroenterol. 2009;43:765–772. doi: 10.1097/MCG.0b013e31819e9048. [DOI] [PubMed] [Google Scholar]
- 159.Choi SS, Syn WK, Karaca GF, Omenetti A, Moylan CA, Witek RP, Agboola KM, Jung Y, Michelotti GA, Diehl AM. Leptin promotes the myofibroblastic phenotype in hepatic stellate cells by activating the hedgehog pathway. J Biol Chem. 2010;285:36551–36560. doi: 10.1074/jbc.M110.168542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Korah TE, El-Sayed S, Elshafie MK, Hammoda GE, Safan MA. Significance of serum leptin and adiponectin levels in Egyptian patients with chronic hepatitis C virus associated hepatic steatosis and fibrosis. World J Hepatol. 2013;5:74–81. doi: 10.4254/wjh.v5.i2.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Moreno M, Chaves JF, Sancho-Bru P, Ramalho F, Ramalho LN, Mansego ML, Ivorra C, Dominguez M, Conde L, Millán C, et al. Ghrelin attenuates hepatocellular injury and liver fibrogenesis in rodents and influences fibrosis progression in humans. Hepatology. 2010;51:974–985. doi: 10.1002/hep.23421. [DOI] [PubMed] [Google Scholar]
- 162.Marra F. Leptin and liver tissue repair: do rodent models provide the answers? J Hepatol. 2007;46:12–18. doi: 10.1016/j.jhep.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 163.Zhou Y, Jia X, Wang G, Wang X, Liu J. PI-3 K/AKT and ERK signaling pathways mediate leptin-induced inhibition of PPARgamma gene expression in primary rat hepatic stellate cells. Mol Cell Biochem. 2009;325:131–139. doi: 10.1007/s11010-009-0027-3. [DOI] [PubMed] [Google Scholar]
- 164.Li Z, Oben JA, Yang S, Lin H, Stafford EA, Soloski MJ, Thomas SA, Diehl AM. Norepinephrine regulates hepatic innate immune system in leptin-deficient mice with nonalcoholic steatohepatitis. Hepatology. 2004;40:434–441. doi: 10.1002/hep.20320. [DOI] [PubMed] [Google Scholar]
- 165.Mallat A, Teixeira-Clerc F, Lotersztajn S. Cannabinoid signaling and liver therapeutics. J Hepatol. 2013;59:891–896. doi: 10.1016/j.jhep.2013.03.032. [DOI] [PubMed] [Google Scholar]
- 166.Trebicka J, Racz I, Siegmund SV, Cara E, Granzow M, Schierwagen R, Klein S, Wojtalla A, Hennenberg M, Huss S, et al. Role of cannabinoid receptors in alcoholic hepatic injury: steatosis and fibrogenesis are increased in CB2 receptor-deficient mice and decreased in CB1 receptor knockouts. Liver Int. 2011;31:860–870. doi: 10.1111/j.1478-3231.2011.02496.x. [DOI] [PubMed] [Google Scholar]
- 167.Giannone FA, Baldassarre M, Domenicali M, Zaccherini G, Trevisani F, Bernardi M, Caraceni P. Reversal of liver fibrosis by the antagonism of endocannabinoid CB1 receptor in a rat model of CCl(4)-induced advanced cirrhosis. Lab Invest. 2012;92:384–395. doi: 10.1038/labinvest.2011.191. [DOI] [PubMed] [Google Scholar]
- 168.Tam J, Vemuri VK, Liu J, Bátkai S, Mukhopadhyay B, Godlewski G, Osei-Hyiaman D, Ohnuma S, Ambudkar SV, Pickel J, et al. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J Clin Invest. 2010;120:2953–2966. doi: 10.1172/JCI42551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ebrahimkhani MR, Kiani S, Oakley F, Kendall T, Shariftabrizi A, Tavangar SM, Moezi L, Payabvash S, Karoon A, Hoseininik H, et al. Naltrexone, an opioid receptor antagonist, attenuates liver fibrosis in bile duct ligated rats. Gut. 2006;55:1606–1616. doi: 10.1136/gut.2005.076778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.De Minicis S, Candelaresi C, Marzioni M, Saccomano S, Roskams T, Casini A, Risaliti A, Salzano R, Cautero N, di Francesco F, et al. Role of endogenous opioids in modulating HSC activity in vitro and liver fibrosis in vivo. Gut. 2008;57:352–364. doi: 10.1136/gut.2007.120303. [DOI] [PubMed] [Google Scholar]
- 171.Zvibel I, Atias D, Phillips A, Halpern Z, Oren R. Thyroid hormones induce activation of rat hepatic stellate cells through increased expression of p75 neurotrophin receptor and direct activation of Rho. Lab Invest. 2010;90:674–684. doi: 10.1038/labinvest.2010.48. [DOI] [PubMed] [Google Scholar]
- 172.Shi Z, Rockey DC. Interferon-gamma-mediated inhibition of serum response factor-dependent smooth muscle-specific gene expression. J Biol Chem. 2010;285:32415–32424. doi: 10.1074/jbc.M110.164863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Iizuka M, Murata T, Hori M, Ozaki H. Increased contractility of hepatic stellate cells in cirrhosis is mediated by enhanced Ca2+-dependent and Ca2+-sensitization pathways. Am J Physiol Gastrointest Liver Physiol. 2011;300:G1010–G1021. doi: 10.1152/ajpgi.00350.2010. [DOI] [PubMed] [Google Scholar]
- 174.Xu H, Zhou Y, Lu C, Ping J, Xu LM. Salvianolic acid B lowers portal pressure in cirrhotic rats and attenuates contraction of rat hepatic stellate cells by inhibiting RhoA signaling pathway. Lab Invest. 2012;92:1738–1748. doi: 10.1038/labinvest.2012.113. [DOI] [PubMed] [Google Scholar]
- 175.Rockey DC. Hepatic blood flow regulation by stellate cells in normal and injured liver. Semin Liver Dis. 2001;21:337–349. doi: 10.1055/s-2001-17551. [DOI] [PubMed] [Google Scholar]
- 176.Li J, Kuruba R, Wilson A, Gao X, Zhang Y, Li S. Inhibition of endothelin-1-mediated contraction of hepatic stellate cells by FXR ligand. PLoS One. 2010;5:e13955. doi: 10.1371/journal.pone.0013955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.He C, Miao X, Li J, Qi H. Angiotensin II induces endothelin-1 expression in human hepatic stellate cells. Dig Dis Sci. 2013;58:2542–2549. doi: 10.1007/s10620-013-2685-y. [DOI] [PubMed] [Google Scholar]
- 178.Khimji AK, Rockey DC. Endothelin and hepatic wound healing. Pharmacol Res. 2011;63:512–518. doi: 10.1016/j.phrs.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Michalski Ł, Kleniewska P, Piechota-Polańczyk A, Gorąca A. The role of endothelin-1 and its receptor blockers on the liver function. Gen Physiol Biophys. 2012;31:383–388. doi: 10.4149/gpb_2012_046. [DOI] [PubMed] [Google Scholar]
- 180.Kida Y, Xia Z, Zheng S, Mordwinkin NM, Louie SG, Zheng SG, Feng M, Shi H, Duan Z, Han YP. Interleukin-1 as an injury signal mobilizes retinyl esters in hepatic stellate cells through down regulation of lecithin retinol acyltransferase. PLoS One. 2011;6:e26644. doi: 10.1371/journal.pone.0026644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.He H, Mennone A, Boyer JL, Cai SY. Combination of retinoic acid and ursodeoxycholic acid attenuates liver injury in bile duct-ligated rats and human hepatic cells. Hepatology. 2011;53:548–557. doi: 10.1002/hep.24047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kluwe J, Wongsiriroj N, Troeger JS, Gwak GY, Dapito DH, Pradere JP, Jiang H, Siddiqi M, Piantedosi R, O’Byrne SM, et al. Absence of hepatic stellate cell retinoid lipid droplets does not enhance hepatic fibrosis but decreases hepatic carcinogenesis. Gut. 2011;60:1260–1268. doi: 10.1136/gut.2010.209551. [DOI] [PubMed] [Google Scholar]
- 183.Takahashi K, Sasabe N, Ohshima K, Kitazato K, Kato R, Masuda Y, Tsurumaki M, Obama T, Okudaira S, Aoki J, et al. Glucagon regulates intracellular distribution of adipose differentiation-related protein during triacylglycerol accumulation in the liver. J Lipid Res. 2010;51:2571–2580. doi: 10.1194/jlr.M004648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lee TF, Mak KM, Rackovsky O, Lin YL, Kwong AJ, Loke JC, Friedman SL. Downregulation of hepatic stellate cell activation by retinol and palmitate mediated by adipose differentiation-related protein (ADRP) J Cell Physiol. 2010;223:648–657. doi: 10.1002/jcp.22063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hernández-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, Czaja MJ, Friedman SL. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142:938–946. doi: 10.1053/j.gastro.2011.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Thoen LF, Guimarães EL, Dollé L, Mannaerts I, Najimi M, Sokal E, van Grunsven LA. A role for autophagy during hepatic stellate cell activation. J Hepatol. 2011;55:1353–1360. doi: 10.1016/j.jhep.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 187.Hernández-Gea V, Hilscher M, Rozenfeld R, Lim MP, Nieto N, Werner S, Devi LA, Friedman SL. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J Hepatol. 2013;59:98–104. doi: 10.1016/j.jhep.2013.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Harvey SA, Dangi A, Tandon A, Gandhi CR. The transcriptomic response of rat hepatic stellate cells to endotoxin: implications for hepatic inflammation and immune regulation. PLoS One. 2013;8:e82159. doi: 10.1371/journal.pone.0082159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Holt AP, Haughton EL, Lalor PF, Filer A, Buckley CD, Adams DH. Liver myofibroblasts regulate infiltration and positioning of lymphocytes in human liver. Gastroenterology. 2009;136:705–714. doi: 10.1053/j.gastro.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 190.Schildberg FA, Wojtalla A, Siegmund SV, Endl E, Diehl L, Abdullah Z, Kurts C, Knolle PA. Murine hepatic stellate cells veto CD8 T cell activation by a CD54-dependent mechanism. Hepatology. 2011;54:262–272. doi: 10.1002/hep.24352. [DOI] [PubMed] [Google Scholar]
- 191.Ji J, Yu F, Ji Q, Li Z, Wang K, Zhang J, Lu J, Chen L, E Q, Zeng Y, et al. Comparative proteomic analysis of rat hepatic stellate cell activation: a comprehensive view and suppressed immune response. Hepatology. 2012;56:332–349. doi: 10.1002/hep.25650. [DOI] [PubMed] [Google Scholar]
- 192.Svegliati-Baroni G, Ridolfi F, Di Sario A, Saccomanno S, Bendia E, Benedetti A, Greenwel P. Intracellular signaling pathways involved in acetaldehyde-induced collagen and fibronectin gene expression in human hepatic stellate cells. Hepatology. 2001;33:1130–1140. doi: 10.1053/jhep.2001.23788. [DOI] [PubMed] [Google Scholar]
- 193.Novitskiy G, Ravi R, Potter JJ, Rennie-Tankersley L, Wang L, Mezey E. Effects of acetaldehyde and TNF alpha on the inhibitory kappa B-alpha protein and nuclear factor kappa B activation in hepatic stellate cells. Alcohol Alcohol. 2005;40:96–101. doi: 10.1093/alcalc/agh116. [DOI] [PubMed] [Google Scholar]
- 194.Novitskiy G, Potter JJ, Rennie-Tankersley L, Mezey E. Identification of a novel NF-kappaB-binding site with regulation of the murine alpha2(I) collagen promoter. J Biol Chem. 2004;279:15639–15644. doi: 10.1074/jbc.M311499200. [DOI] [PubMed] [Google Scholar]
- 195.Svegliati-Baroni G, Inagaki Y, Rincon-Sanchez AR, Else C, Saccomanno S, Benedetti A, Ramirez F, Rojkind M. Early response of alpha2(I) collagen to acetaldehyde in human hepatic stellate cells is TGF-beta independent. Hepatology. 2005;42:343–352. doi: 10.1002/hep.20798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lee HI, McGregor RA, Choi MS, Seo KI, Jung UJ, Yeo J, Kim MJ, Lee MK. Low doses of curcumin protect alcohol-induced liver damage by modulation of the alcohol metabolic pathway, CYP2E1 and AMPK. Life Sci. 2013;93:693–699. doi: 10.1016/j.lfs.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 197.Szuster-Ciesielska A, Mizerska-Dudka M, Daniluk J, Kandefer-Szerszeń M. Butein inhibits ethanol-induced activation of liver stellate cells through TGF-β, NFκB, p38, and JNK signaling pathways and inhibition of oxidative stress. J Gastroenterol. 2013;48:222–237. doi: 10.1007/s00535-012-0619-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Vidali M, Stewart SF, Albano E. Interplay between oxidative stress and immunity in the progression of alcohol-mediated liver injury. Trends Mol Med. 2008;14:63–71. doi: 10.1016/j.molmed.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 199.McMullen MR, Pritchard MT, Wang Q, Millward CA, Croniger CM, Nagy LE. Early growth response-1 transcription factor is essential for ethanol-induced fatty liver injury in mice. Gastroenterology. 2005;128:2066–2076. doi: 10.1053/j.gastro.2005.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Purohit V, Gao B, Song BJ. Molecular mechanisms of alcoholic fatty liver. Alcohol Clin Exp Res. 2009;33:191–205. doi: 10.1111/j.1530-0277.2008.00827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Syn WK, Jung Y, Omenetti A, Abdelmalek M, Guy CD, Yang L, Wang J, Witek RP, Fearing CM, Pereira TA, et al. Hedgehog-mediated epithelial-to-mesenchymal transition and fibrogenic repair in nonalcoholic fatty liver disease. Gastroenterology. 2009;137:1478–1488.e8. doi: 10.1053/j.gastro.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Bellahcène A, Castronovo V, Ogbureke KU, Fisher LW, Fedarko NS. Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): multifunctional proteins in cancer. Nat Rev Cancer. 2008;8:212–226. doi: 10.1038/nrc2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Das R, Philip S, Mahabeleshwar GH, Bulbule A, Kundu GC. Osteopontin: it’s role in regulation of cell motility and nuclear factor kappa B-mediated urokinase type plasminogen activator expression. IUBMB Life. 2005;57:441–447. doi: 10.1080/15216540500159424. [DOI] [PubMed] [Google Scholar]
- 204.Arteel GE. New role of plasminogen activator inhibitor-1 in alcohol-induced liver injury. J Gastroenterol Hepatol. 2008;23 Suppl 1:S54–S59. doi: 10.1111/j.1440-1746.2007.05285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kaji K, Yoshiji H, Kitade M, Ikenaka Y, Noguchi R, Shirai Y, Aihara Y, Namisaki T, Yoshii J, Yanase K, et al. Combination treatment of angiotensin II type I receptor blocker and new oral iron chelator attenuates progression of nonalcoholic steatohepatitis in rats. Am J Physiol Gastrointest Liver Physiol. 2011;300:G1094–G1104. doi: 10.1152/ajpgi.00365.2010. [DOI] [PubMed] [Google Scholar]
- 206.Bian Z, Ma X. Liver fibrogenesis in non-alcoholic steatohepatitis. Front Physiol. 2012;3:248. doi: 10.3389/fphys.2012.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Krawczyk M, Bonfrate L, Portincasa P. Nonalcoholic fatty liver disease. Best Pract Res Clin Gastroenterol. 2010;24:695–708. doi: 10.1016/j.bpg.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 208.Bugianesi E, Manzini P, D’Antico S, Vanni E, Longo F, Leone N, Massarenti P, Piga A, Marchesini G, Rizzetto M. Relative contribution of iron burden, HFE mutations, and insulin resistance to fibrosis in nonalcoholic fatty liver. Hepatology. 2004;39:179–187. doi: 10.1002/hep.20023. [DOI] [PubMed] [Google Scholar]
- 209.Aithal GP, Thomas JA, Kaye PV, Lawson A, Ryder SD, Spendlove I, Austin AS, Freeman JG, Morgan L, Webber J. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology. 2008;135:1176–1184. doi: 10.1053/j.gastro.2008.06.047. [DOI] [PubMed] [Google Scholar]
- 210.McCarty MF. Full-spectrum antioxidant therapy featuring astaxanthin coupled with lipoprivic strategies and salsalate for management of non-alcoholic fatty liver disease. Med Hypotheses. 2011;77:550–556. doi: 10.1016/j.mehy.2011.06.029. [DOI] [PubMed] [Google Scholar]
- 211.Kukla M, Ciupińska-Kajor M, Kajor M, Wyleżoł M, Żwirska-Korczala K, Hartleb M, Berdowska A, Mazur W. Liver visfatin expression in morbidly obese patients with nonalcoholic fatty liver disease undergoing bariatric surgery. Pol J Pathol. 2010;61:147–153. [PubMed] [Google Scholar]
- 212.Yilmaz Y, Yonal O, Kurt R, Alahdab YO, Eren F, Ozdogan O, Celikel CA, Imeryuz N, Kalayci C, Avsar E. Serum levels of omentin, chemerin and adipsin in patients with biopsy-proven nonalcoholic fatty liver disease. Scand J Gastroenterol. 2011;46:91–97. doi: 10.3109/00365521.2010.516452. [DOI] [PubMed] [Google Scholar]
- 213.Miura K, Seki E, Ohnishi H, Brenner DA. Role of toll-like receptors and their downstream molecules in the development of nonalcoholic Fatty liver disease. Gastroenterol Res Pract. 2010;2010:362847. doi: 10.1155/2010/362847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Syn WK, Oo YH, Pereira TA, Karaca GF, Jung Y, Omenetti A, Witek RP, Choi SS, Guy CD, Fearing CM, et al. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology. 2010;51:1998–2007. doi: 10.1002/hep.23599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Nakagami H, Shimamura M, Miyake T, Shimosato T, Minobe N, Moritani T, Kiomy Osako M, Nakagami F, Koriyama H, Kyutoku M, et al. Nifedipine prevents hepatic fibrosis in a non-alcoholic steatohepatitis model induced by an L-methionine-and choline-deficient diet. Mol Med Rep. 2012;5:37–40. doi: 10.3892/mmr.2011.594. [DOI] [PubMed] [Google Scholar]
- 216.Mallat A, Lotersztajn S. The liver X receptor in hepatic stellate cells: a novel antifibrogenic target? J Hepatol. 2011;55:1452–1454. doi: 10.1016/j.jhep.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 217.Jin Z, Sun R, Wei H, Gao X, Chen Y, Tian Z. Accelerated liver fibrosis in hepatitis B virus transgenic mice: involvement of natural killer T cells. Hepatology. 2011;53:219–229. doi: 10.1002/hep.23983. [DOI] [PubMed] [Google Scholar]
- 218.Martín-Vílchez S, Sanz-Cameno P, Rodríguez-Muñoz Y, Majano PL, Molina-Jiménez F, López-Cabrera M, Moreno-Otero R, Lara-Pezzi E. The hepatitis B virus X protein induces paracrine activation of human hepatic stellate cells. Hepatology. 2008;47:1872–1883. doi: 10.1002/hep.22265. [DOI] [PubMed] [Google Scholar]
- 219.Choi SH, Jeong SH, Hwang SB. Large hepatitis delta antigen modulates transforming growth factor-beta signaling cascades: implication of hepatitis delta virus-induced liver fibrosis. Gastroenterology. 2007;132:343–357. doi: 10.1053/j.gastro.2006.10.038. [DOI] [PubMed] [Google Scholar]
- 220.Chouteau P, Defer N, Florimond A, Caldéraro J, Higgs M, Gaudin A, Mérour E, Dhumeaux D, Lerat H, Pawlotsky JM. Hepatitis C virus (HCV) protein expression enhances hepatic fibrosis in HCV transgenic mice exposed to a fibrogenic agent. J Hepatol. 2012;57:499–507. doi: 10.1016/j.jhep.2012.04.019. [DOI] [PubMed] [Google Scholar]
- 221.Paracha UZ, Fatima K, Alqahtani M, Chaudhary A, Abuzenadah A, Damanhouri G, Qadri I. Oxidative stress and hepatitis C virus. Virol J. 2013;10:251. doi: 10.1186/1743-422X-10-251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Arciello M, Gori M, Balsano C. Mitochondrial dysfunctions and altered metals homeostasis: new weapons to counteract HCV-related oxidative stress. Oxid Med Cell Longev. 2013;2013:971024. doi: 10.1155/2013/971024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Hino K, Nishina S, Hara Y. Iron metabolic disorder in chronic hepatitis C: mechanisms and relevance to hepatocarcinogenesis. J Gastroenterol Hepatol. 2013;28 Suppl 4:93–98. doi: 10.1111/jgh.12243. [DOI] [PubMed] [Google Scholar]
- 224.Huang H, Shiffman ML, Friedman S, Venkatesh R, Bzowej N, Abar OT, Rowland CM, Catanese JJ, Leong DU, Sninsky JJ, et al. A 7 gene signature identifies the risk of developing cirrhosis in patients with chronic hepatitis C. Hepatology. 2007;46:297–306. doi: 10.1002/hep.21695. [DOI] [PubMed] [Google Scholar]
- 225.Qurishi N, Kreuzberg C, Lüchters G, Effenberger W, Kupfer B, Sauerbruch T, Rockstroh JK, Spengler U. Effect of antiretroviral therapy on liver-related mortality in patients with HIV and hepatitis C virus coinfection. Lancet. 2003;362:1708–1713. doi: 10.1016/S0140-6736(03)14844-1. [DOI] [PubMed] [Google Scholar]
- 226.Sitia G, De Bona A, Bagaglio S, Galli L, Paties CT, Uberti-Foppa C, Guidotti LG, Lazzarin A, Morsica G. Naive HIV/HCV-coinfected patients have higher intrahepatic pro-inflammatory cytokines than coinfected patients treated with antiretroviral therapy. Antivir Ther. 2006;11:385–389. [PubMed] [Google Scholar]
- 227.Saracino A, Bruno G, Scudeller L, Punzi G, Lagioia A, Ladisa N, Monno L, Angarano G. Does HIV-1 co-receptor tropism correlate with fibrosis progression in HIV/HCV co-infected patients? J Clin Virol. 2014;59:167–171. doi: 10.1016/j.jcv.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 228.Bruno R, Galastri S, Sacchi P, Cima S, Caligiuri A, DeFranco R, Milani S, Gessani S, Fantuzzi L, Liotta F, et al. gp120 modulates the biology of human hepatic stellate cells: a link between HIV infection and liver fibrogenesis. Gut. 2010;59:513–520. doi: 10.1136/gut.2008.163287. [DOI] [PubMed] [Google Scholar]