

Abstract
Autophagy is a mechanism involved in cellular homeostasis under basal and stressed conditions delivering cytoplasmic content to the lysosomes for degradation to macronutrients. The potential role of autophagy in disease is increasingly recognised and investigated in the last decade. Nowadays it is commonly accepted that autophagy plays a role in the hepatic lipid metabolism. Hence, dysfunction of autophagy may be an underlying cause of non-alcoholic fatty liver disease. However, controversy of the exact role of autophagy in the lipid metabolism exists: some publications report a lipolytic function of autophagy, whereas others claim a lipogenic function. This review aims to give an update of the present knowledge on autophagy in the hepatic lipid metabolism, hepatic insulin resistance, steatohepatitis and hepatic fibrogenesis.
Keywords: Non-alcoholic fatty liver disease, Non-alcoholic fatty liver disease, Steatohepatitis, Non-alcoholic steatohepatitis, Autophagy, Lipophagy, Lipid metabolism
Core tip: Autophagy is a mechanism involved in cellular homeostasis. In this review the current knowledge on the role of autophagy in non-alcoholic fatty liver disease (NAFLD) is summarised, with emphasis on the current controversy on the lipolytic vs lipogenic function in hepatic lipid metabolism. Furthermore the role of autophagy in the pathophysiology of insulin resistance, hepatocellular injury and fibrogenesis is reviewed to better understand its importance in NAFLD.
INTRODUCTION
The term autophagy has been introduced by De Duve et al[1] over forty years ago to define a process of vacuolisation for the transport of intracellular material to the lysosomes for degradation. The knowledge and number of autophagy-related publications increased exponentially in the last decade, as the importance of autophagy in (patho)physiology became recognised. Indeed, autophagy is progressively acknowledged as an important regulator of intracellular homeostasis. Dysfunction of this process has been linked with cardiovascular, respiratory, neurodegenerative and metabolic diseases and with cancer[2,3].
Non-alcoholic fatty liver disease (NAFLD) is characterised by macrovesicular fat accumulation in more than 5% of the hepatocytes in the absence of known causes of secondary steatosis[4,5]. This accumulation ranges from scarce to panacinar steatosis and usually starts in Rapaport’s zone 3[6]. It is important to distinguish non-alcoholic fatty liver (NAFL, also known as simple steatosis) from non-alcoholic steatohepatitis (NASH), which is diagnosed when macrovesicular steatosis is accompanied by both hepatocyte ballooning degeneration and lobular inflammation[5,6]. Simple steatosis has a low risk for the development of advanced disease, while NASH is associated with an increased risk of hepatic and non-hepatic co-morbidities and mortality. NAFLD is epidemiologically linked with obesity and diabetes, and is currently considered as the hepatic manifestation of the metabolic syndrome. Given that the prevalence of these metabolic disorders rises, the prevalence of NAFLD and hence its clinical impact, is rising too[4,7].
A growing body of evidence indicates that autophagy and lipid metabolism are correlated. Dysfunctional autophagy may therefore contribute to the pathogenesis of NAFLD. However, controversies still exist and the exact role of autophagy in the hepatic lipid metabolism is not entirely elucidated yet. This review aims at summarising current knowledge on autophagy in NAFLD.
AUTOPHAGY
Autophagy is derived from the Greek “auto” and “phagos” and literally means “self-eating”. Basal autophagy serves as a housekeeper in the continuous turnover of cellular contents, thereby removing damaged or dysfunctional cellular contents and supplying substrates for energy production. Autophagy can be induced in response to oxidative or metabolic stress[8,9]. Starvation is commonly used to induce autophagy in research settings. Moreover, liver tissue and hepatocytes are frequently used in research and as such were involved in many major discoveries[9].
There are three types of autophagy identified in mammalian cells: macroautophagy, chaperone-mediated autophagy (CMA) and microautophagy[2,10,11]. In macroautophagy, cytoplasmic material (e.g., organelles or protein aggregates) is sequestrated in a double membrane structure, the autophagosome (Figure 1). This process initiates the formation of a phagophore (also known as isolation membrane), which subsequently lengthens to create an autophagosome. The autophagosome fuses with a lysosome to form an autolysosome where its content will be degraded. When a small portion of cytoplasm is engulfed directly by the lysosome, the term microautophagy is used. In CMA, proteins that contain a special targeting motif, recognised by heat shock cognate protein 70 (HSC70) and co-chaperones, will be selectively delivered to lysosomes where they are internalised via a lysosomal-associated membrane protein 2A (LAMP2A).
Figure 1.
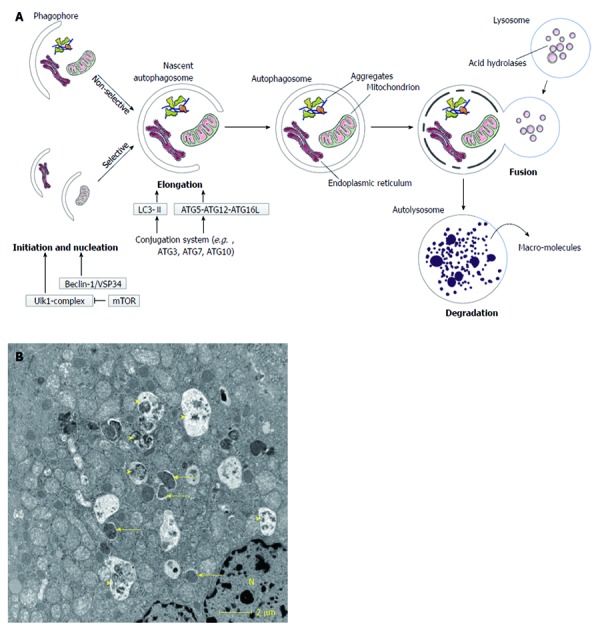
Macroautophagy. A: Schematic overview of macroautophagy. Macroautophagy starts with the formation of a double-layered membrane, the phagophore (isolation membrane). Phagophore formation is regulated by the ULK1 complex (initiation), which is under control of the mammalian target of rapamycin (mTOR) complex, and the beclin-1/VSP34 interacting complex (nucleation). Two major ubiquitine-like conjugated complexes take care of the elongation of the double membrane: light chain 3 (LC3)-II and ATG5-ATG12-ATG16L1. ATG7 is one of the proteins necessary for formation of both elongation complexes. When an autophagosome is formed, it will fuse with a lysosome. The inner membrane of the autophagosome and the sequestered cytoplasm will be degraded and macromolecules can subsequently be (re-)used. Macroautophagy can be non-selective (random uptake of intracellular material) or selective [uptake of specific cargo, e.g., mitochondria, endoplasmic reticulum (ER)]; B: Transmission electron microsocopy (TEM)-image of a normal mouse liver fasted overnight. The arrows indicate autophagosomes, the arrowheads indicate autolysosomes. N: Nucleus.
Among the three types of autophagy, macroautophagy (hereafter called autophagy) is considered to play the most important role in pathophysiology and is well studied in recent years. Even though autophagy was initially believed to be a non-selective bulky degradation pathway, selective forms such as “mitophagy” (selective autophagy of mitochondria), “peroxiphagy” (peroxisomes), “ribophagy” (ribosomes) or “xenophagy” (invading microbes) are also acknowledged[2,3,8,12].
The formation of autophagosomes is a dynamic and highly regulated process (Figure 1). It is regulated at the molecular level by autophagy related (Atg) genes. These genes were originally identified in yeast, but many orthologues in higher eukaryotes have been found[2]. A central regulator in autophagy is the mammalian target of rapamycin (mTOR)[13]. This protein complex inhibits the initiation of autophagosome formation by phosphorylating UNC51-like kinase 1 (ULK1). The class I phosphatidylinositol 3-kinase (PI3K)/AKT pathway stimulates mTOR in response to growth factors, such as insulin. However, under conditions of low energy status the AMP/ATP ratio increases, leading to adenosine 5’-monophosphate-activated protein kinase (AMPK) activation and mTOR inhibition, thereby activating autophagy[2,13]. The beclin-1/VSP34 (a class III PI3K)-interacting complex mediates nucleation of the phagophore[2,13]. Two ubiquitin-like conjugated complexes take care of elongation of the formed phagophore into an autophagosome: the ATG5-ATG12-ATG16L1 complex and light chain 3-II (LC3-II). An E1-like protein, ATG7, is a necessary mediator of both conjugation processes, hence an interesting target for the study of autophagy[14]. LC3 (also known as MAP1LC3) is the major mammalian orthologue of ATG8 and also one of the key regulators in autophagosome formation[3]. The active conjugated form of LC3, LC3-II, is frequently used as a marker for autophagy in studies[15]. For further extensive review of autophagy regulation, see references[11,16].
AUTOPHAGY IN LIPID METABOLISM
Singh et al[17] were the first to convincingly correlate autophagy with the lipid metabolism. They considered it as a novel selective pathway in lipid breakdown and called it ‘lipophagy’, even though the first clues pointing towards a potential role of autophagy in lipid metabolism were already seen a couple of decades earlier[18]. In contrast, Shibata et al[19] claimed that autophagy was necessary for the genesis of lipid droplets (LDs) rather than being involved in the breakdown of LDs. Currently, several papers with supporting evidence for both theories have been published. In this section, some common findings will be discussed, followed by reviewing both the opposing theories and the contextual alterations of autophagy in the lipid metabolism.
Common findings in autophagy and lipid metabolism
Despite the contradictory results in the recent literature, some common findings supporting the relationship between autophagy and the lipid metabolism in the liver deserve to be mentioned.
First, a close relationship between LDs and LC3 has been demonstrated. A notable portion of LC3-positive structures, as demonstrated by immunofluorescence microscopy, co-localised with markers for LDs in liver tissue[19] and in cell lines[20-22]. Likewise, immunohistochemistry revealed positive LC3B dots localised on the surface of LDs[23]. Co-localisation could also be confirmed by LC3 immunogold staining transmission electron microscopy (TEM) and suggests a LD-regulating function of autophagy[17,19,20]. This co-localisation was not influenced by inhibition of autophagosome-lysosome fusion or knockout of autophagy, suggesting that conjugation to the active form of LC3 (LC3-II) occurs on the surface of LDs and not only on autophagosomes[17].
Secondly, steatosis is most of the time present in acinar zone 3[6]. In parallel with this histological finding, immunohistochemical staining for autophagy (staining of LC3) is more localised around the central veins[23,24]. Zonal distribution of autophagy is also postulated from a theoretical point of view, based on findings in glutamine metabolism[25]. It is assumed that low rates of autophagy occur in periportal areas and constitutive high levels of autophagy pericentrally in well-nourished conditions. This assumption serves as a potential explanation for the pattern of steatosis in the liver.
Thirdly, LDs have shown to be associated with lysosomes. Immunofluorescence microscopy reveals increased co-localisation of LDs with lysosomal markers such as LAMP1[17] or lysotracker[22] in fat-loaden cells. Co-localisation decreases after inhibiting autophagosome formation pharmacologically or by knockdown techniques[17].
Autophagy as a lipolytic mechanism
Considering autophagy as a lipolytic mechanism is an attractive theory, because it helps explaining the ability of the liver to mobilise free fatty acids (FFA) rapidly, if needed, taking into account that hepatocytes have relatively low concentrations of cytosolic lipases[26].
Pharmacological inhibition, silencing or knockdown of autophagy (by targeting ATG5) in hepatocytes results in an increased hepatocyte triglyceride (TG) level and accumulation of LDs when cultured in the presence of an exogenous or endogenous lipid stimulus[17]. It was shown that this increase was due to impaired lipolysis (to fuel β-oxidation) and not to increased TG synthesis. Additionally, the opposite happened after pharmacological induction of autophagy: lipid stores in hepatocytes decreased. The development of hepatocyte-specific autophagy-deficient mice (by targeting Atg7) could confirm the in vitro results. Indeed, these mice develop hepatomegaly with increased TG and cholesterol content, compared to their wild type countermates[17]. Autophagy-deficient and -competent mice that were starved for 24 h showed an increased presence of TG in their livers (the so-called fasting-induced steatosis). In wild type mice this accumulation was less pronounced. Moreover, TEM demonstrated an increase of lysosomes and lipid-containing autophago(lyso)somes, supporting lipolysis. These findings were even more pronounced with prolonged fasting.
Obese mice (either genetically or dietary) show reduced ATG7 protein levels (although the mRNA expression is comparable) as well as decreased levels of autophagy[27]. Autophagy induction via liver-specific overexpression of Atg7 in ob/ob mice improves the metabolic state and reduces the steatosis significantly. These findings further support a lipolytic function of autophagy. Unfortunately, the effects of induced autophagy on lipid metabolism were only reported in ob/ob mice and not in high fat diet (HFD)-fed animals.
When blocking autophagy pharmacologically, a decrease in the oxidation of FFA and in the VLDL production was observed, whilst stimulation of autophagy resulted in opposite effects[28,29]. In vivo results also showed a change in the distribution of lysosomal lipases (LAL) towards the autophagosome fraction after starvation, suggesting an increase in the autophagy-mediated lysosomal lipolysis[28].
Defects in forkhead box class O (FOXO) transcription factors are linked to steatosis and dyslipidaemia[30]. Mice with a liver-specific triple knockout of FOXO1/3/4 (LTKO) demonstrate steatosis and hypertriglyceridemia[31]. Reduced autophagy in these mice confirm FOXO1 mediated regulation of the key autophagy genes[32]. Of note, Atg14 is regulated by the FOXO transcription factors 1 and 3. Knockdown of hepatic ATG14 increases hepatic and serum TG, whereas overexpression in HFD-fed animals decreases steatosis. In LTKO mice, overexpression of Atg14 was able to counteract lipid disturbances including steatosis[31]. In contrast with these experiments, an increase rather than a decrease of FOXO1 levels was described in a small cohort of patients with NASH[33].
One of the latest contributions in the knowledge of autophagy regulation is the discovery of transcription factor EB (TFEB), which seems to be a master regulator of autophagy[34]. TFEB is involved in the lipid metabolism as its overexpression inhibits and its suppression induces steatosis, respectively[35]. The effects on the lipid metabolism were mediated by the stimulation of the peroxisome proliferator-activated receptor γ coactivator 1 α-peroxisome proliferator-activated receptor α (pgc-1α-PPARα) pathway as well as by autophagy. Overexpression of TFEB could not counteract steatosis caused by the disruption of autophagy, implying dependency of TFEB function on the autophagy mechanism[35].
It is generally known that antiretroviral therapy can induce steatosis. In an in vitro study, it was shown that the thymidine analogues zidovudine and stavudine inhibit the autophagic flux of hepatocytes in a dose-dependent manner, thereby inducing the accumulation of lipids and mitochondrial dysfunction[36]. Though not yet used for the treatment of steatosis, glucagon like peptide-1 (GLP-1) analogues were able to reduce endoplasmic reticulum (ER) stress and fat accumulation in vitro and in vivo by the activation of autophagy[37]. Carbamazepine and rapamycin induce autophagy and show to be effective in reducing steatosis in models of alcoholic and non-alcoholic fatty liver disease[38]. Very recently, caffeine was shown to induce autophagy with increased lipid clearance[22]. Other possible mechanisms parallel to changes in autophagy that might explain the observed effects are formally not excluded, however, are less likely.
Data in humans are scarce. Studies in human liver tissue are limited partly by experimental restrictions, e.g., the impossibility to use specific pharmacologic interventions or to perform consecutive biopsies. Some currently used markers, such as LC3, do not allow good identification of autophagosomal structures unless the target protein is overexpressed[23]. Moreover, immunohistochemical markers are a snapshot of a dynamic state and are not able to discern between increased autophagy versus decreased degradation of autophagosomes[15,23]. Nonetheless, a small immunohistochemical study on post-mortem liver tissue demonstrated decreased LC3 and increased p62 staining with an increased degree of steatosis, suggesting decreased autophagy in more severe steatosis[24]. Likewise, patients with proven NAFLD demonstrate increased numbers of autophagic vesicles and p62 accumulation on their liver biopsy[39].
Finally, two clinical observations in patients with NAFLD deserve attention. First, hypothyroidism was found to be more frequent in patients with NAFLD[40-42]. Thyroid hormone (T3) is a known regulator of the basal metabolism and acts on different mechanisms. Recently, T3 was shown to be a powerful inducer of autophagy in vitro and in vivo, and autophagy accounted for a crucial portion of T3 stimulated β-oxidation[21]. As a result, autophagy may provide the explanation for this association.
Secondly, patients with NAFL have an increased prevalence of hypovitaminosis D[43]. Vitamin D prevents the development of steatosis, whereas knockout of its receptor (VDR) promotes steatosis[44]. Others have shown that vitamin D acts as a potent inducer of autophagy[13]. Indirectly, these two facts together are in line with lipophagy, but hitherto the hypothesis that these effects are directly mediated via autophagy has not been investigated.
Autophagy as a lipogenic mechanism
In fasting conditions, the body’s energy supply is maintained by adaptive mechanisms. As insulin levels decrease, lipolysis in the adipose tissue (AT) is no longer inhibited and FFA are released in the serum. These FFA are captured by the liver and either used for formation of ketone bodies, or temporary stored as TG in LDs[19,45]. In rodents this mechanism may cause substantial accumulation of TG in the liver, known as fasting-induced steatosis. C57Bl/6 mice showed to be very prone to develop steatosis in fasting conditions[45]. Also in humans, the liver fat content increases on imaging (with 1H-magnetic resonance spectroscopy) after 36h fasting[46].
Mice with a hepatocyte-specific autophagy deficiency do not show fasting-induced steatosis as compared to wild type animals. The remaining LDs are much smaller in number and size and the total TG content in the liver is lower. This lack of fasting-induced steatosis was first seen in very young mice (twenty-two days old), but was confirmed in eight to twelve week old mice[19,23,47]. Autophagy is therefore implicated in LD formation and growth. The co-localisation of LC3 (necessary for autophagosome formation) with LDs in starved wild type mice further supports these findings[19].
Subsequent in vitro research confirmed these findings in different cell lines (including hepatocytes)[20]. Cells subjected to knockdown of LC3 form less LDs and have less TG content compared to their controls. Neither FFA uptake, nor TG synthesis or TG breakdown are influenced in LC3 knockdown cells, suggesting an impaired ability to preserve synthesised TG within these cells[20].
Very recently, an improved metabolic profile was observed in both hepatocyte- or skeletal muscle-specific autophagy-deficient mice[48]. Adult mice with a deficiency in hepatic autophagy[47,48] and fed a control diet show reduced fasting-induced steatosis. Moreover, lipid accumulation did not develop[48] or increase[47] after feeding a HFD. Gene expression of proteins involved in fatty acid and TG synthesis was lower compared to control littermates. On the other hand, gene expression of proteins involved in β-oxidation and TG secretion was also reduced[47,48]. It is not clear whether these findings are epiphenomena or directly involved in preventing steatosis. The “mitokine” fibroblast growth factor 21 (FGF21), induced by mitochondrial stress, was held responsible as a central mediator of the metabolic alterations[48].
Decreased autophagy has been reported in dietary and genetic models of obesity, whereas overexpression of Atg7 had beneficial metabolic effects[27] as discussed above. Nevertheless, suppressing ATG7 expression in lean mice increases hepatic glycogen content, but fails to alter lipid accumulation in the liver (as well as TG or FFA in serum)[27]. This study is therefore non-conclusive about a lipolytic or lipogenic function of autophagy.
Contextual variability of autophagy in lipid metabolism
Besides the duality in autophagy as a lipolytic or lipogenic process, there are also differences described depending on its context. In most cases these differences were found in experiments supporting autophagy as a lipolytic mechanism.
Based on in vitro experiments, basal autophagy is supposed to be a more important pathway for lipid metabolism than induced autophagy[17]. Hepatocytes in culture did not demonstrate signs of induced autophagy in response to lipid stimuli. Moreover, in contrast with endogenous lipid load, these hepatocytes were unable to adjust the autophagic flow to a sudden increase in the external lipid load. In line with this in vitro finding, the external lipid load by prolonged HFD decreases the efficiency of autophagy[17,27,29,37]. Intriguingly, a biphasic time course is observed with an increase in the autophagic flux and mRNA of autophagic markers after two weeks HFD and a decrease afterwards (ten weeks HFD)[29]. Recent data (only presented as an abstract) suggest that autophagy decreases on short term HFD (three days) and normalises after long term HFD (ten weeks)[49]. Instead of a decrease, eight weeks of a diet high in fat load generates an increase in autophagy[50]. Paradoxically, a further increase of autophagy by a compound found in garlic decreases the lipid content, possibly by autophagy-independent mechanisms that have to be further elucidated. All together, it seems that alterations of autophagy are dynamic in states of overnutrition.
The term lipotoxicity covers all detrimental effects of fatty acids on the cellular integrity[51,52]. Hence, it is not surprising that lipids as such can influence autophagy. Short chain fatty acids are able to induce autophagy in vitro[53]. Unsaturated fatty acids (e.g., oleic acid) stimulate autophagy and protect against apoptosis, whilst saturated fatty acids (e.g., palmitic acid) inhibit autophagy and promote apoptosis[37,54,55].
Not only fatty acids, but also the lipid composition of membranes or vesicular compartments can influence autophagic behaviour. A long exposure to high lipid concentrations alters the membrane composition and diminishes the fusion capacity of autophagosomes and lysosomes[56]. This may explain the altered autophagy after prolonged fatty diets. Attenuation of CMA was also observed after lipid challenge[10]. However, some authors did not observe an attenuated fusion capacity in ob/ob mice Instead they report a decrease in clearance of autophagosomes due to a disturbed acidification of lysosomal compartments[57] and/or down-regulated cathepsin expression[39,57].
Variation of autophagic behaviour has also been reported with respect to tissue type. For example, autophagy is indispensable for adipogenesis and transdifferentiation of white AT[58,59], contradicting a potential lipolytic function in AT. These findings are in contrast with findings of autophagy in the liver, which mainly claim a lipolytic function of autophagy (as discussed above). In addition, an increased level of autophagy in AT of patients with metabolic syndrome or type 2 diabetes mellitus is observed[60-62].
Discrepancies and hypotheses
A clear-cut explanation for the aforementioned discrepancies in autophagy and lipid metabolism is currently lacking. Several hypotheses, however, have been put forward.
Because autophagy declines with age[63], some concerns were raised about the age of the laboratory animals[64]. Sometimes, juvenile rather than adult mice were used in experiments[19], which may be less dependent on autophagy. However, additional experiments with younger and older mice provide similar results[19,23,47,48], suggesting that the observed differences cannot be explained solely by age.
Secondly, small variances in the mouse strains might be responsible for different outcomes in in vivo experiments[48]. This explanation, however, is even so not likely to offer a solution. Most of the experiments were performed on a C57Bl/6 background, which is an inbred strain. Moreover, tracing back the cited resources of the hepatocyte-specific Atg7 knockout mice that were used, leads to the same origin of the mice: Atg7 flox mice (used by[17,23,65-67]) were created by Komatsu et al[14] and albumin-Cre mice (used by[17,23,48,66]) were created by the group of Magnuson[68]. Furthermore, in vitro experiments on hepatocyte cell lines also showed conflicting results[17,20,36,37].
Thirdly, many different methods can be used to examine lipid accumulation and autophagy. Liver steatosis can be induced by fasting, by genetic and/or by dietary alterations. Different genetic modifications or pharmacological interferences can also alter autophagy. Whereas papers claiming a lipolytic role use a wide range of methods (e.g., in vitro methods, pharmacological interference, genetic modifications) (see 3.2), articles claiming the opposite mainly use in vivo knockout models and fasting-induced steatosis (see 3.3). However, one of the caveats in knockout models is the potential of influencing developmental stages, e.g., transdifferentiation of white adipocytes requires autophagy[58,59]. This implies that the observed differences in autophagic lipid handling might be due to the method of inducing fat accumulation or an altered maturation of hepatocytes.
Fourthly, distinction is made between basal and induced autophagy. Basal autophagy is supposed to be the most important type of autophagy in the pathogenesis of NAFLD[17,69]. However, most studies are performed after total blockage of autophagy, making it difficult to discern between the two. A Bcl-2 knock-in model is able to selectively block stimulus-induced (i.e., by exercise or starvation) autophagy[70]. These mice have an impaired glucose-uptake during exercise and an impairment of the exercise-induced protection against glucose-intolerance and increased serum lipid levels caused by HFD. The liver and pancreas morphology did not alter after HFD, supporting the importance of basal autophagy in the lipid metabolism. Further elucidating the role of basal vs induced autophagy, including the relationship towards the lipid source, may provide a possible explanation for the divergent findings in the lipid metabolism.
Moreover, when studying autophagy, it is sometimes difficult to distinguish whether the observed effects are a secondary/adaptive process or directly casused by autophagy[59,71]. Furthermore, autophagy not only influences lipid metabolism, other organelles and cellular systems are also influenced by dysfunctional autophagy and may partly explain observed differences in the liver metabolism (e.g., ER stress and dysfunctional mitochondria[44,72,73]). Disturbance of very low density lipoprotein (VLDL) production might also be implicated, as autophagy can degrade apoB, a necessary protein for the VLDL formation[71]. There even may be non-autophagic (and autophagy-independent) functions of ATG-proteins[26,27,74] in the lipid metabolism.
Inversely, the lipid metabolism is not solely dependent on autophagy alone. Cytosolic lipases still account for a substantial part of the lipolysis[51]. For example, after total blockage of lipolysis by diethylumbelliferyl phosphate (DEUP), the cellular TG content increases more than after blocking autophagy alone[17]. Additionaly, if LD-formation is autophagy-dependent, small LD-like bodies are observed on TEM in autophagy-deficient cells, suggesting that the lumenally-sorted LD production (i.e., LDs formed out of the ER) is not affected[19].
Finally, while the resolution limit of microscopical techniques does not allow to visualise the smallest LDs in living cells[75], it is possible that the observed effects of autophagy only reflect LD modulation after the formation of LDs. Autophagy might be a dynamically active process which controls LD size and the amount of lipotoxic FFA in the cytoplasm. The behaviour of autophagy will be context-dependent and drives the main outcome of autophagy in the lipid metabolism[51]. In this view, lipolysis and lipogenesis are no longer mutually exclusive and in fact co-exist.
AUTOPHAGY AND INSULIN RESISTANCE
The liver plays a central role in the glucose metabolism and an impaired insulin signalling is an important feature of NAFLD[4]. Autophagy substantially contributes to maintain the glucose homeostasis and is strictly regulated by insulin[76]. Similarly to the lipid metabolism, the exact interactions between the action of insulin and autophagy are not entirely clarified yet. An overview of the current knowledge is given in Figure 2.
Figure 2.
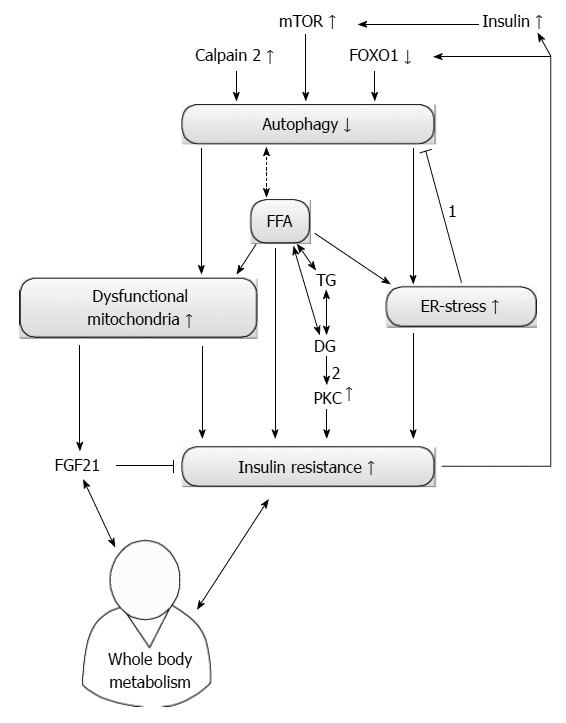
Current knowledge of autophagy and insulin resistance. Autophagy and insulin resistance (IR) seem to influence each other reciprocally. On one hand, an increased level of calpain 2 reduces autophagy and increases IR by increasing endoplasmic reticulum (ER) stress. The amount of dysfunctional mitochondria will also increase and contribute to IR. On the other hand, secondary hyperinsulinism due to IR can decrease autophagy if the insulin sensitivity remains present. Furthermore, autophagy can also be suppressed via an IR-mediated reduction in forkhead box class O 1 (FOXO1). Free fatty acids (FFA) can induce IR directly or by increasing ER stress. Correct stereoisomers of diacylglycerol (DG) can induce protein kinase c (PKC) dependent IR. Controversy exists on how autophagy influences the level of FFA and if subsequent correct stereoisomers of DG can be formed. Dysfunctional mitochondria can increase the level of fibroblast growth factor 21 (FGF21), which is able to reduce IR. FGF21 and IR interact with whole body metabolism. Arrows indicate a consequence of a certain alteration, bar headed arrows denote an inhibition. Double-headed arrows present a reciprocal influence. The dashed and double-headed arrow denotes the uncertain relation between FFA and autophagy. 1: ER stress actually increases autophagy; 2: Only right stereoisomers induce PKC. mTOR: Mammalian target of rapamycin; TG: Triglycerides.
Insulin stimulates mTOR via the class I PI3K /AKT pathway, and thus inhibits autophagy[2,16]. In case of a normal insulin sensitivity of the liver, insulin-dependent stimulation of this pathway is a possible mechanism of reduced hepatocellular autophagy in hyperinsulinemic states. Furthermore, an alternative pathway was found, which could explain the reduced autophagy in case of insulin resistance (IR)[32]. In HFD-induced IR, a downregulation of autophagy was noticed due to a reduced FOXO1-mediated expression of key autophagy genes. The authors suggested that IR was a consequence of the reduced clearance of dysfunctional/aged mitochondria via mitophagy, as oxidative stress and altered mitochondrial integrity (and mass) are related to IR[32]. The impact of other dysfunctional organelles on IR was not investigated, but could also contribute to these findings.
Reduced autophagy was also linked to IR in a study with HFD and ob/ob mice[27]. In contrast to the aforementioned study, IR was not the cause but the result of decreased autophagy. Knockdown of autophagy in lean mice induced severe IR, while overexpression of Atg7 in obese mice improved the insulin sensitivity and glucose tolerance, decreased the hepatic glucose production and decreased steatosis. An increased level of the calcium-dependent protease calpain 2, which can cleave several autophagy-related proteins, was observed and was held responsible for the decreased protein level of ATG7. The subsequent increase of cellular stress, with emphasis on ER stress, might be the mechanism behind IR[27].
Hepatic IR appears to be the result of ER stress-mediated processes[72], therefore ER stress secondary to decreased autophagy might be a plausible underlying mechanism for IR. Intracellular saturated fatty acids can also contribute to IR by an increase in ER stress, but also via an ER-stress independent mechanism[72]. On the other hand, ER stress stimulates autophagy[16,77], thus autophagy potentially acts as an escape mechanism to prevent cell injury and IR in particular.
Recently, Kim et al[48] described a novel endocrine and metabolic function of autophagy. Defective mitophagy causes cellular stress, inducing a stress response regulated by activating transcription factor 4 (ATF4), which promotes the expression of FGF21. FGF21 has several beneficial metabolic effects in lean and HFD-fed animals including an improved glucose tolerance and insulin sensitivity[48,78]. In human subjects, however, a positive correlation was observed between plasma FGF21 levels, IR[79] and steatosis[80]. This apparent paradox might be explained by the resistance to FGF21 (as with increasing levels of FGF21 less IR is actually expected, based on experimental data) or can either be explained by an adaptive increase of FGF21 after establishment of IR. In this study, mitochondrial dysfunction is no longer seen as detrimental, but rather as beneficial by reducing the fasting-induced steatosis and improving the glucose metabolism.
Finally, inhibiting and stimulating effects of protein kinase C (PKC), an important effector enzyme in several signal transduction cascades, on autophagy have been described[81,82]. Several isoforms of PKC are known and many of them can be activated by diacylglycerol (DG). Insulin resistance is also linked to PKC[83]. As DG is an intermediate as well as a product of lipolysis, these findings may indicate an additional crosslink between IR and autophagy. Not all DG is able to activate PKC due to differences in stereoisomers. DG produced by lipolysis does not show bioactivity, and therefore potential crosstalk is less likely[51,83]. On the other hand, active stereoisomers of DG (i.e., 1,2-diacyl-glycerol) can be generated in lipid synthesis and interference with insulin signalling can still occur.
AUTOPHAGY AND HEPATOCELLULAR INJURY
In some patients, steatosis leads to cellular injury and inflammation (NASH) with a subsequent risk for progression to cirrhosis and eventually for hepatocellular carcinoma (HCC) in a subset of patients[4]. Because damaged organelles are removed by autophagy, dysfunction of autophagy likely will result in cellular injury. In line with this hypothesis, stimulation of autophagy could indeed reduce liver injury in animal models of ethanol-induced steatohepatitis[38,84]. Carbamazepine-induced autophagy also demonstrated a tendency to reduce cell injury in a mouse model of NAFLD[38].
A potential role exists for mitophagy since mitochondrial dysfunction leads to the production of reactive oxygen species (ROS), causes oxidative stress and is involved in the pathogenesis of NASH[73]. Many of the aforementioned studies do report damaged mitochondria when autophagy is defective[14,23,36,48,54,66]. In one study defective autophagy was accompanied by an increased production of ROS[36]. Knockdown of autophagy makes hepatocytes more susceptible to cell death caused by menadione-induced oxidative stress[69]. This type of cell death is caspase-dependent and activated via the mitochondrial death pathway due to c-JUN N-terminal kinase (JNK)/c-JUN overactivation. CMA is upregulated as a compensatory mechanism, but fails to overcome the induced oxidative stress. Furthermore, CMA as such also provides protection to menadione-induced cell death, but through a different mechanism[69].
Comparable to menadione, TNF-induced hepatic injury also causes increased cell death, JNK/c-JUN overactivation and activation of the mitochondrial death pathway in hepatocyte-specific autophagy-deficient mice. However, primary mitochondrial dysfunction followed by oxidative stress or impaired energy homeostasis is not responsible for cell injury in this model[47,65].
The protein p62/SQSTM1 selectively guides proteinaceous aggregates to autophagosomes and accumulates in autophagy-deficient cells. Presence of this protein contributes significantly to hepatocellular injury caused by autophagy deficiency, as double knockouts of autophagy (Atg7) and p62 (DKO) diminish hepatocyte injury compared to autophagy knockout (Atg7) only[67]. Nevertheless, p62 is believed to be a beneficial adaptive response to promote formation of relatively harmless aggregates. Toxic intermediates are formed during the aggregate formation and are considered responsible for cell injury. Furthermore, p62 also aids nuclear translocation of the beneficial transcription factor NF-E2 related factor 2 (NRF2), which induces transcription of various detoxifying enzymes[67]. Of note, despite the alleviation of cellular injury by DKO, a complete abolishment of the cellular injury comparable to control levels cannot be achieved as turnover of disturbed organelles is still not corrected.
Autophagy is linked to the inflammatory cytokines in adipocytes. When p62 is knocked out in adipocytes, an increased invasion of macrophages and production of pro-inflammatory cytokines in AT is observed[85]. Moreover, knockout of p62 in AT also causes obesity and glucose intolerance, whereas knockout of p62 in hepatocytes does not. Whether these inflammatory changes also occurred in liver tissue was not investigated. A direct inhibition of autophagy in human or mice adipocytes increases the production of pro-inflammatory cytokines[62] as was described in other tissues as well[86].
The role of autophagy in tumourigenesis is dual and depends on the stage of tumour development. Autophagy acts as a tumour suppressor in normal tissue and prevents the development of malignant neoplasia. Once a tumour is developed, autophagy drives survival of tumour cells by supplying nutrients[2]. An extensive discussion of the role(s) of autophagy in liver tumour development can be found in ref.[9].
AUTOPHAGY IN LIVER FIBROSIS
The knowledge on autophagy in fibrogenesis is scarce, but available evidence suggests an elementary role in different fibrogenic cells[87]. Autophagy seems to provide nutrients to fuel the processes involved in the activation of these cells.
Hepatic stellate cells (HSCs) are considered major fibrogenic cells in the liver. A central observation in the transdifferentiation from a quiescent state to active myofibroblasts is the depletion of lipid stores, in HSC typically mainly composed of vitamin A[88].
Autophagy in HSC activation was first evidenced in 2011[89]. In this study, an increased autophagic flux was observed during HSC activation. Pharmacological inhibition of autophagy inhibited the activation of HSCs. Autophagy also interfered with the LD metabolism after stimulation by platelet derived growth factor BB (PDGF-BB), a known mitogen of HSCs, as shown by co-localisation of LC3B with LDs. Interestingly, autophagy seems to affect only larger LDs and co-localisation became absent once HSCs were activated.
The importance of autophagy in HSC activation was confirmed in another study using pharmacological and genetic tools to inhibit autophagy in vivo and in vitro[90]. The results showed an energy-supplying role (via delivery of FFA out of LDs for β-oxidation) of autophagy needed for the transdifferentiation of HSCs. Administration of oleic acid in autophagy-deficient stellate cells could partly restore the activation, but did not augment fibrogenesis in autophagy-competent cells.
In line with the aforementioned results, one may expect an increase in fibrosis when inducing autophagy. Paradoxically, reduced hepatic fibrosis is observed after administration of rapamycin, a known potent inducer of autophagy. The involvement of autophagy was, however, not specifically tested in these studies[91-93]. Rapamycin is known to have a complex mode of action. Hence, studies with other autophagy-inducers may clarify whether the rapamycin-induced reduction in hepatic fibrosis is due to autophagy-related mechanisms.
FUTURE PERSPECTIVES
Further research is clearly needed to elucidate the exact role of autophagy in NAFLD. Fortunately, the increasing research interest in NAFLD and the growing awareness of autophagy as a pathophysiological mechanism will most likely result in new discoveries in the next decade.
The major issue to be resolved is the apparent paradoxical behaviour of autophagy in the lipid metabolism as discussed in this review. At this moment, evidence towards a pro-lipolytic function in the liver is more solid and outnumbers the evidence against this theory. However, the evidence for a lipogenic role cannot be ignored. It has to be addressed whether the two roles might co-exist or whether one role dominates in the hepatocyte, and how their balance and/or function is regulated exactly.
Contextual differences were noted, but not fully understood. Detailed knowledge of basal and induced autophagy may resolve a part of this puzzle. It would be very interesting if future research can more specifically and separately investigate the role of basal and induced autophagy in liver metabolism. Additionally, the relationship with other cellular observations such as dysfunctional mitochondria should be clarified.
Most studies perform experiments on one specific cell type (i.e., hepatocytes), although the liver is composed of many different cell types (e.g., endothelial cells, HSCs and Kupffer cells[94]). More extensive research on whole liver tissue may be useful as several cell types are also involved in the development of steatohepatitis[4,88] or may influence each other. Moreover, potential zonal differences of autophagy in liver tissue should be investigated and correlated with the already known different functions of the acinar liver zones[25].
The liver is not a “cavalier seul” in the metabolism, but one of the central players of the whole body metabolism. The role of autophagy is site-specific. For example, in AT stronger evidence exists for a lipogenic action of autophagy[58,59] contrary to the results in hepatocytes. In order to understand the complex role of autophagy, other tissues and other metabolic pathways, including the glucose and protein metabolism, must be incorporated in the research projects.
Ultimately, if knowledge on autophagy in NAFLD increases, therapeutic interventions can be developed and tested. Systemic therapy will potentially be hampered if context- and tissue-dependent behaviour of autophagy appears to play an important role in the pathogenesis of the metabolic syndrome. Several clinical trials are already ongoing, almost exclusively in oncological settings (www.clinicaltrials.gov). Investigators should be stimulated to include repetitive evaluation of the metabolic parameters in their study as secondary objectives.
CONCLUSION
Autophagy is an important factor in the lipid metabolism, but its exact role has not yet been fully clarified and appears to be context- and tissue-specific. Increasing knowledge on its exact role in the complex pathophysiology of metabolic disturbances and NAFLD might make autophagy a target for treatment of the metabolic syndrome or NAFLD. We should, however, always keep in mind that altering a key cellular process such as autophagy might lead to a better metabolic state, but that this not automatically equals a better general health.
Footnotes
Supported by Fund for Scientific Research (FWO) - Flanders (11J9513N, G007412N, G044312N, 1802514N), to Kwanten WJ, Martinet W and Francque SM
P- Reviewers: Elisaf MS, Imatoh T, Yang YF S- Editor: Wen LL L- Editor: A E- Editor: Liu XM
References
- 1.De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–492. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 2.Martinet W, Agostinis P, Vanhoecke B, Dewaele M, De Meyer GRY. Autophagy in disease: a double-edged sword with therapeutic potential. Clin Sci (Lond) 2009;116:697–712. doi: 10.1042/CS20080508. [DOI] [PubMed] [Google Scholar]
- 3.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 4.Torres DM, Williams CD, Harrison SA. Features, diagnosis, and treatment of nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2012;10:837–858. doi: 10.1016/j.cgh.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 5.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ. The diagnosis and management of non-alcoholic fatty liver disease: practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012;55:2005–2023. doi: 10.1002/hep.25762. [DOI] [PubMed] [Google Scholar]
- 6.Kleiner DE, Brunt EM. Nonalcoholic fatty liver disease: pathologic patterns and biopsy evaluation in clinical research. Semin Liver Dis. 2012;32:3–13. doi: 10.1055/s-0032-1306421. [DOI] [PubMed] [Google Scholar]
- 7.Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274–285. doi: 10.1111/j.1365-2036.2011.04724.x. [DOI] [PubMed] [Google Scholar]
- 8.Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344–1348. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Czaja MJ, Ding WX, Donohue TM, Friedman SL, Kim JS, Komatsu M, Lemasters JJ, Lemoine A, Lin JD, Ou JH, et al. Functions of autophagy in normal and diseased liver. Autophagy. 2013;9:1131–1158. doi: 10.4161/auto.25063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodriguez-Navarro JA, Kaushik S, Koga H, Dall’Armi C, Shui G, Wenk MR, Di Paolo G, Cuervo AM. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc Natl Acad Sci USA. 2012;109:E705–E714. doi: 10.1073/pnas.1113036109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 12.Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012;393:547–564. doi: 10.1515/hsz-2012-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 17.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Debeer LJ, Thomas J, De Schepper PJ, Mannaerts GP. Lysosomal triacylglycerol lipase and lipolysis in isolated rat hepatocytes. J Biol Chem. 1979;254:8841–8846. [PubMed] [Google Scholar]
- 19.Shibata M, Yoshimura K, Furuya N, Koike M, Ueno T, Komatsu M, Arai H, Tanaka K, Kominami E, Uchiyama Y. The MAP1-LC3 conjugation system is involved in lipid droplet formation. Biochem Biophys Res Commun. 2009;382:419–423. doi: 10.1016/j.bbrc.2009.03.039. [DOI] [PubMed] [Google Scholar]
- 20.Shibata M, Yoshimura K, Tamura H, Ueno T, Nishimura T, Inoue T, Sasaki M, Koike M, Arai H, Kominami E, et al. LC3, a microtubule-associated protein1A/B light chain3, is involved in cytoplasmic lipid droplet formation. Biochem Biophys Res Commun. 2010;393:274–279. doi: 10.1016/j.bbrc.2010.01.121. [DOI] [PubMed] [Google Scholar]
- 21.Sinha RA, You SH, Zhou J, Siddique MM, Bay BH, Zhu X, Privalsky ML, Cheng SY, Stevens RD, Summers SA, et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J Clin Invest. 2012;122:2428–2438. doi: 10.1172/JCI60580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sinha RA, Farah BL, Singh BK, Siddique MM, Li Y, Wu Y, Ilkayeva OR, Gooding J, Ching J, Zhou J, et al. Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology. 2014;59:1366–1380. doi: 10.1002/hep.26667. [DOI] [PubMed] [Google Scholar]
- 23.Martinet W, Schrijvers DM, Timmermans JP, Bult H, De Meyer GRY. Immunohistochemical analysis of macroautophagy: recommendations and limitations. Autophagy. 2013;9:386–402. doi: 10.4161/auto.22968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kashima J, Shintani-Ishida K, Nakajima M, Maeda H, Unuma K, Uchiyama Y, Yoshida KI. Immunohistochemical study of the autophagy marker microtubule-associated protein 1 light chain 3 in normal and steatotic human livers. Hepatol Res. 2013:Epub ahead of print. doi: 10.1111/hepr.12183. [DOI] [PubMed] [Google Scholar]
- 25.Gebhardt R, Coffer PJ. Hepatic autophagy is differentially regulated in periportal and pericentral zones - a general mechanism relevant for other tissues? Cell Commun Signal. 2013;11:21. doi: 10.1186/1478-811X-11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zechner R, Madeo F. Cell biology: Another way to get rid of fat. Nature. 2009;458:1118–1119. doi: 10.1038/4581118a. [DOI] [PubMed] [Google Scholar]
- 27.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skop V, Cahová M, Papáčková Z, Páleníčková E, Daňková H, Baranowski M, Zabielski P, Zdychová J, Zídková J, Kazdová L. Autophagy-lysosomal pathway is involved in lipid degradation in rat liver. Physiol Res. 2012;61:287–297. doi: 10.33549/physiolres.932285. [DOI] [PubMed] [Google Scholar]
- 29.Papáčková Z, Daňková H, Páleníčková E, Kazdová L, Cahová M. Effect of short- and long-term high-fat feeding on autophagy flux and lysosomal activity in rat liver. Physiol Res. 2012;61 Suppl 2:S67–S76. doi: 10.33549/physiolres.932394. [DOI] [PubMed] [Google Scholar]
- 30.Tikhanovich I, Cox J, Weinman SA. Forkhead box class O transcription factors in liver function and disease. J Gastroenterol Hepatol. 2013;28 Suppl 1:125–131. doi: 10.1111/jgh.12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiong X, Tao R, DePinho RA, Dong XC. The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J Biol Chem. 2012;287:39107–39114. doi: 10.1074/jbc.M112.412569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu HY, Han J, Cao SY, Hong T, Zhuo D, Shi J, Liu Z, Cao W. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J Biol Chem. 2009;284:31484–31492. doi: 10.1074/jbc.M109.033936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valenti L, Rametta R, Dongiovanni P, Maggioni M, Fracanzani AL, Zappa M, Lattuada E, Roviaro G, Fargion S. Increased expression and activity of the transcription factor FOXO1 in nonalcoholic steatohepatitis. Diabetes. 2008;57:1355–1362. doi: 10.2337/db07-0714. [DOI] [PubMed] [Google Scholar]
- 34.Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14:283–296. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013;15:647–658. doi: 10.1038/ncb2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stankov MV, Panayotova-Dimitrova D, Leverkus M, Vondran FW, Bauerfeind R, Binz A, Behrens GM. Autophagy inhibition due to thymidine analogues as novel mechanism leading to hepatocyte dysfunction and lipid accumulation. AIDS. 2012;26:1995–2006. doi: 10.1097/QAD.0b013e32835804f9. [DOI] [PubMed] [Google Scholar]
- 37.Sharma S, Mells JE, Fu PP, Saxena NK, Anania FA. GLP-1 analogs reduce hepatocyte steatosis and improve survival by enhancing the unfolded protein response and promoting macroautophagy. PLoS One. 2011;6:e25269. doi: 10.1371/journal.pone.0025269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin CW, Zhang H, Li M, Xiong X, Chen X, Chen X, Dong XC, Yin XM. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol. 2013;58:993–999. doi: 10.1016/j.jhep.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fukuo Y, Yamashina S, Sonoue H, Arakawa A, Nakadera E, Aoyama T, Uchiyama A, Kon K, Ikejima K, Watanabe S. Abnormality of autophagic function and cathepsin expression in the liver from patients with non-alcoholic fatty liver disease. Hepatol Res. 2013:Epub ahead of print. doi: 10.1111/hepr.12282. [DOI] [PubMed] [Google Scholar]
- 40.Liangpunsakul S, Chalasani N. Is hypothyroidism a risk factor for non-alcoholic steatohepatitis? J Clin Gastroenterol. 2003;37:340–343. doi: 10.1097/00004836-200310000-00014. [DOI] [PubMed] [Google Scholar]
- 41.Chung GE, Kim D, Kim W, Yim JY, Park MJ, Kim YJ, Yoon JH, Lee HS. Non-alcoholic fatty liver disease across the spectrum of hypothyroidism. J Hepatol. 2012;57:150–156. doi: 10.1016/j.jhep.2012.02.027. [DOI] [PubMed] [Google Scholar]
- 42.Pagadala MR, Zein CO, Dasarathy S, Yerian LM, Lopez R, McCullough AJ. Prevalence of hypothyroidism in nonalcoholic fatty liver disease. Dig Dis Sci. 2012;57:528–534. doi: 10.1007/s10620-011-2006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eliades M, Spyrou E, Agrawal N, Lazo M, Brancati FL, Potter JJ, Koteish AA, Clark JM, Guallar E, Hernaez R. Meta-analysis: vitamin D and non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2013;38:246–254. doi: 10.1111/apt.12377. [DOI] [PubMed] [Google Scholar]
- 44.Bechmann LP, Hannivoort RA, Gerken G, Hotamisligil GS, Trauner M, Canbay A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol. 2012;56:952–964. doi: 10.1016/j.jhep.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 45.Guan HP, Goldstein JL, Brown MS, Liang G. Accelerated fatty acid oxidation in muscle averts fasting-induced hepatic steatosis in SJL/J mice. J Biol Chem. 2009;284:24644–24652. doi: 10.1074/jbc.M109.034397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gan SK, Watts GF. Is adipose tissue lipolysis always an adaptive response to starvation?: implications for non-alcoholic fatty liver disease. Clin Sci (Lond) 2008;114:543–545. doi: 10.1042/CS20070461. [DOI] [PubMed] [Google Scholar]
- 47.Ma D, Molusky MM, Song J, Hu CR, Fang F, Rui C, Mathew AV, Pennathur S, Liu F, Cheng JX, et al. Autophagy deficiency by hepatic FIP200 deletion uncouples steatosis from liver injury in NAFLD. Mol Endocrinol. 2013;27:1643–1654. doi: 10.1210/me.2013-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim do H, Hur KY, Kim HK, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med. 2013;19:83–92. doi: 10.1038/nm.3014. [DOI] [PubMed] [Google Scholar]
- 49.Zhang H, Chen X, Yin XM. The dynamics of liver autophagy status during steatosis development. (abstract 1440) Hepatology. 2012;56(Suppl S1):869A. [Google Scholar]
- 50.Xiao J, Guo R, Fung ML, Liong EC, Chang RC, Ching YP, Tipoe GL. Garlic-Derived S-Allylmercaptocysteine Ameliorates Nonalcoholic Fatty Liver Disease in a Rat Model through Inhibition of Apoptosis and Enhancing Autophagy. Evid Based Complement Alternat Med. 2013;2013:642920. doi: 10.1155/2013/642920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zechner R, Zimmermann R, Eichmann TO, Kohlwein SD, Haemmerle G, Lass A, Madeo F. FAT SIGNALS--lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012;15:279–291. doi: 10.1016/j.cmet.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fuchs M, Sanyal AJ. Lipotoxicity in NASH. J Hepatol. 2012;56:291–293. doi: 10.1016/j.jhep.2011.05.019. [DOI] [PubMed] [Google Scholar]
- 53.Tang Y, Chen Y, Jiang H, Nie D. Short-chain fatty acids induced autophagy serves as an adaptive strategy for retarding mitochondria-mediated apoptotic cell death. Cell Death Differ. 2011;18:602–618. doi: 10.1038/cdd.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mei S, Ni HM, Manley S, Bockus A, Kassel KM, Luyendyk JP, Copple BL, Ding WX. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J Pharmacol Exp Ther. 2011;339:487–498. doi: 10.1124/jpet.111.184341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tanaka S, Hikita H, Aono S, Kawaguchi T, Mukai K, Shimizu S, Kodama T, Li W, Miyagi T, Kanto T, et al. Differential roles of palmatic acid and oleic acid in regulation of apoptosis and autophagy in non-alcoholic fatty liver disease. (abstract 1399) Hepatology. 2012;56(Suppl S1):850A–851A. [Google Scholar]
- 56.Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010;24:3052–3065. doi: 10.1096/fj.09-144519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Inami Y, Yamashina S, Izumi K, Ueno T, Tanida I, Ikejima K, Watanabe S. Hepatic steatosis inhibits autophagic proteolysis via impairment of autophagosomal acidification and cathepsin expression. Biochem Biophys Res Commun. 2011;412:618–625. doi: 10.1016/j.bbrc.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 58.Zhang Y, Zeng X, Jin S. Autophagy in adipose tissue biology. Pharmacol Res. 2012;66:505–512. doi: 10.1016/j.phrs.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 59.Maixner N, Kovsan J, Harman-Boehm I, Blüher M, Bashan N, Rudich A. Autophagy in adipose tissue. Obes Facts. 2012;5:710–721. doi: 10.1159/000343983. [DOI] [PubMed] [Google Scholar]
- 60.Kovsan J, Blüher M, Tarnovscki T, Klöting N, Kirshtein B, Madar L, Shai I, Golan R, Harman-Boehm I, Schön MR, et al. Altered autophagy in human adipose tissues in obesity. J Clin Endocrinol Metab. 2011;96:E268–E277. doi: 10.1210/jc.2010-1681. [DOI] [PubMed] [Google Scholar]
- 61.Ost A, Svensson K, Ruishalme I, Brännmark C, Franck N, Krook H, Sandström P, Kjolhede P, Strålfors P. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol Med. 2010;16:235–246. doi: 10.2119/molmed.2010.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jansen HJ, van Essen P, Koenen T, Joosten LA, Netea MG, Tack CJ, Stienstra R. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology. 2012;153:5866–5874. doi: 10.1210/en.2012-1625. [DOI] [PubMed] [Google Scholar]
- 63.Cuervo AM, Bergamini E, Brunk UT, Dröge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy. 2005;1:131–140. doi: 10.4161/auto.1.3.2017. [DOI] [PubMed] [Google Scholar]
- 64.Singh R, Cuervo AM. Lipophagy: connecting autophagy and lipid metabolism. Int J Cell Biol. 2012;2012:282041. doi: 10.1155/2012/282041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Amir M, Zhao E, Fontana L, Rosenberg H, Tanaka K, Gao G, Czaja MJ. Inhibition of hepatocyte autophagy increases tumor necrosis factor-dependent liver injury by promoting caspase-8 activation. Cell Death Differ. 2013;20:878–887. doi: 10.1038/cdd.2013.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 68.Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274:305–315. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- 69.Wang Y, Singh R, Xiang Y, Czaja MJ. Macroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology. 2010;52:266–277. doi: 10.1002/hep.23645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481:511–515. doi: 10.1038/nature10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Christian P, Sacco J, Adeli K. Autophagy: Emerging roles in lipid homeostasis and metabolic control. Biochim Biophys Acta. 2013;1831:819–824. doi: 10.1016/j.bbalip.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 72.Flamment M, Hajduch E, Ferré P, Foufelle F. New insights into ER stress-induced insulin resistance. Trends Endocrinol Metab. 2012;23:381–390. doi: 10.1016/j.tem.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 73.Farrell GC, van Rooyen D, Gan L, Chitturi S. NASH is an Inflammatory Disorder: Pathogenic, Prognostic and Therapeutic Implications. Gut Liver. 2012;6:149–171. doi: 10.5009/gnl.2012.6.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Subramani S, Malhotra V. Non-autophagic roles of autophagy-related proteins. EMBO Rep. 2013;14:143–151. doi: 10.1038/embor.2012.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fujimoto T, Ohsaki Y, Cheng J, Suzuki M, Shinohara Y. Lipid droplets: a classic organelle with new outfits. Histochem Cell Biol. 2008;130:263–279. doi: 10.1007/s00418-008-0449-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ezaki J, Matsumoto N, Takeda-Ezaki M, Komatsu M, Takahashi K, Hiraoka Y, Taka H, Fujimura T, Takehana K, Yoshida M, et al. Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy. 2011;7:727–736. doi: 10.4161/auto.7.7.15371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Knævelsrud H, Simonsen A. Lipids in autophagy: constituents, signaling molecules and cargo with relevance to disease. Biochim Biophys Acta. 2012;1821:1133–1145. doi: 10.1016/j.bbalip.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 78.Camporez JP, Jornayvaz FR, Petersen MC, Pesta D, Guigni BA, Serr J, Zhang D, Kahn M, Samuel VT, Jurczak MJ, et al. Cellular mechanisms by which FGF21 improves insulin sensitivity in male mice. Endocrinology. 2013;154:3099–3109. doi: 10.1210/en.2013-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chavez AO, Molina-Carrion M, Abdul-Ghani MA, Folli F, Defronzo RA, Tripathy D. Circulating fibroblast growth factor-21 is elevated in impaired glucose tolerance and type 2 diabetes and correlates with muscle and hepatic insulin resistance. Diabetes Care. 2009;32:1542–1546. doi: 10.2337/dc09-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yan H, Xia M, Chang X, Xu Q, Bian H, Zeng M, Rao S, Yao X, Tu Y, Jia W, et al. Circulating fibroblast growth factor 21 levels are closely associated with hepatic fat content: a cross-sectional study. PLoS One. 2011;6:e24895. doi: 10.1371/journal.pone.0024895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jiang H, Cheng D, Liu W, Peng J, Feng J. Protein kinase C inhibits autophagy and phosphorylates LC3. Biochem Biophys Res Commun. 2010;395:471–476. doi: 10.1016/j.bbrc.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Patergnani S, Marchi S, Rimessi A, Bonora M, Giorgi C, Mehta KD, Pinton P. PRKCB/protein kinase C, beta and the mitochondrial axis as key regulators of autophagy. Autophagy. 2013;9:1367–1385. doi: 10.4161/auto.25239. [DOI] [PubMed] [Google Scholar]
- 83.Schmitz-Peiffer C. The tail wagging the dog--regulation of lipid metabolism by protein kinase C. FEBS J. 2013;280:5371–5383. doi: 10.1111/febs.12285. [DOI] [PubMed] [Google Scholar]
- 84.Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, Lu B, Stolz DB, Clemens DL, Yin XM. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–1752. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Müller TD, Lee SJ, Jastroch M, Kabra D, Stemmer K, Aichler M, Abplanalp B, Ananthakrishnan G, Bhardwaj N, Collins S, et al. p62 links β-adrenergic input to mitochondrial function and thermogenesis. J Clin Invest. 2013;123:469–478. doi: 10.1172/JCI64209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Harris J. Autophagy and cytokines. Cytokine. 2011;56:140–144. doi: 10.1016/j.cyto.2011.08.022. [DOI] [PubMed] [Google Scholar]
- 87.Hilscher M, Hernandez-Gea V, Friedman SL. Autophagy and mesenchymal cell fibrogenesis. Biochim Biophys Acta. 2012;1831:972–978. doi: 10.1016/j.bbadis.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Thoen LF, Guimarães EL, Dollé L, Mannaerts I, Najimi M, Sokal E, van Grunsven LA. A role for autophagy during hepatic stellate cell activation. J Hepatol. 2011;55:1353–1360. doi: 10.1016/j.jhep.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 90.Hernández-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, Czaja MJ, Friedman SL. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142:938–946. doi: 10.1053/j.gastro.2011.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bridle KR, Popa C, Morgan ML, Sobbe AL, Clouston AD, Fletcher LM, Crawford DH. Rapamycin inhibits hepatic fibrosis in rats by attenuating multiple profibrogenic pathways. Liver Transpl. 2009;15:1315–1324. doi: 10.1002/lt.21804. [DOI] [PubMed] [Google Scholar]
- 92.Neef M, Ledermann M, Saegesser H, Schneider V, Reichen J. Low-dose oral rapamycin treatment reduces fibrogenesis, improves liver function, and prolongs survival in rats with established liver cirrhosis. J Hepatol. 2006;45:786–796. doi: 10.1016/j.jhep.2006.07.030. [DOI] [PubMed] [Google Scholar]
- 93.Zhu J, Wu J, Frizell E, Liu SL, Bashey R, Rubin R, Norton P, Zern MA. Rapamycin inhibits hepatic stellate cell proliferation in vitro and limits fibrogenesis in an in vivo model of liver fibrosis. Gastroenterology. 1999;117:1198–1204. doi: 10.1016/s0016-5085(99)70406-3. [DOI] [PubMed] [Google Scholar]
- 94.Baratta JL, Ngo A, Lopez B, Kasabwalla N, Longmuir KJ, Robertson RT. Cellular organization of normal mouse liver: a histological, quantitative immunocytochemical, and fine structural analysis. Histochem Cell Biol. 2009;131:713–726. doi: 10.1007/s00418-009-0577-1. [DOI] [PMC free article] [PubMed] [Google Scholar]