

Abstract
Cronkhite-Canada syndrome (CCS) is a rare nonfamilial polyposis syndrome characterized by epithelial disturbances in the gastrointestinal tract and skin. The aim of this study was to investigate the clinical features and potential therapies for CCS. Six patients with CCS admitted from December 1992 to July 2008 to Peking Union Medical College Hospital were evaluated. All patients had clinical manifestation of nonhereditary gastrointestinal polyposis with diarrhea, skin hyperpigmentation, alopecia, and nail dystrophy. Fecal occult blood was positive in all six cases. Serum hemoglobin, potassium, calcium and protein were below the normal range in two cases. Anti-Saccharomyces cerevisiae and antinuclear antibodies were present in three cases. Multiple polyps were found in all patients by gastroscopy and colonoscopy, with only one in the esophagus. Histologically, there were hyperplastic polyps in five cases, tubular adenoma in three, and juvenile polyp in one with chronic inflammation and mucosal edema. Comprehensive treatment led by corticosteroids can result in partial remission of clinical symptoms, and long-term follow-up is necessary.
Keywords: Cronkhite-Canada syndrome, Clinical feature, Etiology, Therapeutics
Core tip: Cronkhite-Canada syndrome (CCS) is a rare noncongenital gastrointestinal polyposis syndrome, characterized by skin hyperpigmentation, hair loss and nail atrophy, associated with high morbidity. This case report summarizes the characteristics of six CCS patients, and reviews the literature. Comprehensive treatment led by corticosteroids can improve prognosis, and long-term follow-up is necessary.
INTRODUCTION
Cronkhite-Canada syndrome (CCS) is a rare noncongenital gastrointestinal polyposis syndrome, characterized by ectodermal dysplasia, skin hyperpigmentation, hair loss and nail atrophy. Since CCS was first reported in 1955[1], more than 400 cases have been reported worldwide. CCS has high morbidity but its etiology is still unknown and there is no consistently recommended treatment. In order to gain a detailed understanding of this rare disease, we reviewed the literature and reported on six CCS patients.
CASE REPORT
Data collection
We collected data from the patients with CCS admitted to Peking Union Medical College Hospital from December 1992 to July 2008, who were diagnosed by history, physical examination, endoscopic findings of gastrointestinal polyposis, and histology, including diffuse polyposis of the gastrointestinal tract; ectodermal dysplasia, cutaneous hyperpigmentation, dystrophic changes of fingernails, and alopecia; diarrhea, weight loss, and abdominal pain. All six patients met the diagnostic criteria, with adult onset and no family history of polyposis.
Patients with Peutz-Jeghers syndrome, juvenile polyps, familial polyposis of the colon, Gardner syndrome, Turcot syndrome; and Menetrier’s disease were excluded.
General information
The six cases comprised three male and three female patients aged 24-69 years (mean: 54.67 years).
Clinical manifestations and physical examination
Three patients had used hair dye or topical pharmaceuticals, and two had intestinal ascariasis. The time between onset and diagnosis was 2-7 mo (mean: 4 mo). The most common symptoms were diarrhea (n = 5) and anorexia (n = 6, including 1 each with hypogeusia or dysgeusia) with different degrees of weight loss. Diarrhea often occurred several times per day, sometimes > 10 times. Patients also suffered hair loss (n = 6), nail changes (n = 6), and skin hyperpigmentation (n = 4) (Figure 1). Hair loss occurred on the scalp, eyebrows, eyelashes, axilla, pubic areas, and limbs. Scalp hair was the most predominant type of hair loss reported. Nail changes were described as thinning, splitting, and onycholysis. Skin hyperpigmentation was mainly manifest as brownish patches with a clear boundary with colored spots occurring on the limbs, face, body, palms, and soles of the feet. Two patients had lower limb edema.
Figure 1.
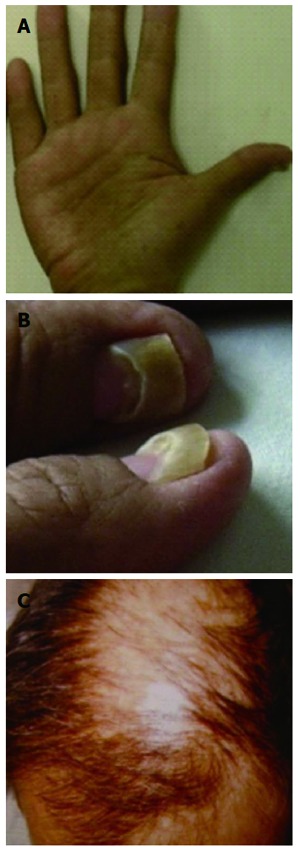
Clinical presence, cutaneous symptoms. A: Hyperpigmentation of palm; B: Onychodystrophy; C: Alopecia.
Laboratory examination
Fecal occult blood was positive in all six cases; maw worm eggs were positive in the feces in two cases; and indistinguishable bacilli were cultivated in the feces from one case. Laboratory data indicated that all kidney and liver functions were normal. Serum hemoglobin, potassium, calcium and protein were below the normal range in two cases, and eosinophilic cells and IgE were elevated in one case. Circulatory anti-Saccharomyces cerevisiae antibody (ASCA) was positive in two cases, antinuclear antibody (ANA) was present in one case, and thyroid function decreased in one case.
Imaging and pathology
Multiple polyps were found in all patients by gastroscopy and colonoscopy, with only one in the esophagus, varying in size from 2 to 40 mm (Figures 2 and 3). Endoscopy results revealed diffuse sessile or pedunculated polyps with either a smooth or rough surface. Radiology of the gastrointestinal tract showed polyposis in the small bowel in two cases. Histology of these polyps showed hyperplastic polyps in five cases, tubular adenoma in three, and juvenile polyp in one with chronic inflammation and mucosal edema.
Figure 2.
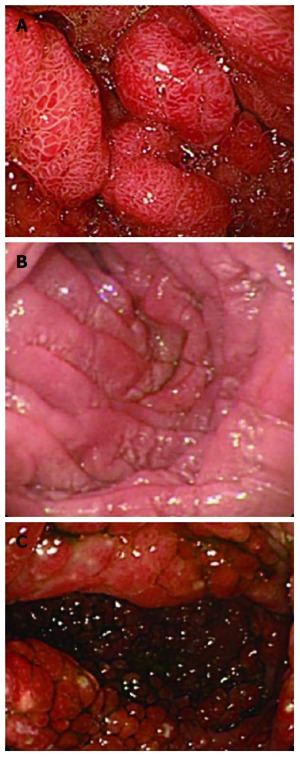
Endoscopy results. A: Numerous polyps in the stomach; B: Mucosal edema in the descendant duodenum; C: Multiple polyps in the colon.
Figure 3.
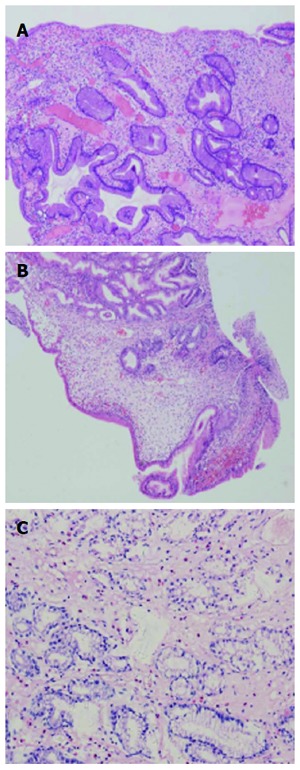
Histology of the biopsy (A), hyperplastic polyps (B) and juvenile polyps (C). The mucosa showed cystically dilated glands in the edematous stroma with eosinophilia and lymphocyte infiltrated.
Treatment
All patients underwent comprehensive therapy, including glucocorticoids, nutritional support, antibiotics, and polypectomy. Five patients underwent a 2-wk to 1-mo course of prednisone at a dose of 1 mg/kg per day before reducing the dose if necessary. Within 2-8 wk after initiation of treatment, symptoms including nail atrophy, anorexia, diarrhea, and skin pigmentation gradually resolved. When prednisone was discontinued or decreased in dose, diarrhea relapsed in two patients but retreatment resulted in remission. In the course of therapy, endoscopy showed that polyps were unchanged in two cases, aggravated in two, and relapsed after remission in two. Polyps of 5-40 mm were resected by endoscopic polypectomy.
Four patients were treated with parenteral nutrition, such as replacement and supplement therapy with blood, albumin, vitamins, amino acids, and lipids with clear improvements. Three patients were treated with antibiotics (albendazole and sulfasalazine). No patient underwent surgery (Table 1).
Table 1.
Therapy and prognosis for all six patients
| Patient | Therapy | Prognosis |
| M/65 | Prednisone, PPI, antibiotics, 5-ASA, Lactobacillus, and nutrition nutritional therapy | Diarrhea, skin hyperpigmentation, atrophic nail remission |
| F/54 | Prednisone, nutritional therapy, and antacid drugs | Diarrhea, appetite, atrophic nail remission Gastrointestinal polyps unchanged 1 mo later |
| M/54 | Prednisone, antibiotics, nutritional therapy, antacid drugs, and polypectomy | Diarrhea, abdominal pain remission Gastrointestinal polyps unchanged 1 mo later |
| F/69 | Prednisone, nutritional therapy, and antacid drugs | Diarrhea, skin hyperpigmentation, atrophic nail remission, gastrointestinal polyps reduced 1 yr later |
| F/24 | Albendazole , antibiotics and sulfasalazine | Diarrhea, appetite remission, atrophic nail, and alopecia aggravated |
| M/62 | Prednisone, albendazole, antibiotics, sulfasalazine and polypectomy | Diarrhea, atrophic nail and polyps resolved 8 wk later Prednisone was discontinued after 3 yr, and all symptoms relapsed |
5-ASA: 5-aminosalicylic acid.
DISCUSSION
CCS is a rare nonfamilial polyposis syndrome characterized by epithelial disturbances both in the gastrointestinal tract and epidermis. At present, the pathogenesis of CCS is unknown, but the following factors may be relevant. The first is immune abnormalities: many studies have shown that patients with CCS are positive for ANA[2]. All six cases in the present study had positive immune parameters; two were positive for ASCA and one was positive for ANA. Hormonal therapy was effective for all three patients, especially in a significantly shorter time for the ANA positive patient. For patients with CCS, their skin and nail changes were similar to the appearance of the ectoderm in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) syndrome. The efficacy of corticosteroids provides the strongest evidence of an inflammatory cause of CCS[3,4]. All the above signs indicated that autoimmunity might play an important role in CCS. The second factor is infection. Two of the present cases had combined infection with two or more pathogens, but the relationship of these pathogens with diarrhea and CCS was difficult to determine. It was suggested the Hp might be a causative factor[5]. The third factor is allergies; after stopping the use of inducers (hair dye and topical medications), IgE and eosinophils were decreased and symptoms improved.
CCS is reported worldwide, with 75% of cases in Japan. The average age of onset is 60 years and the male to female ratio is 3:2. With a relatively acute onset, it typically takes 3 mo to 1 year from onset to diagnosis[6]. There are no clear diagnostic tools for CCS. Diagnosis is based on history, physical examination, endoscopic findings of gastrointestinal polyposis, and histology. The most common clinical manifestations are diarrhea, gustatory loss, weight loss, hair loss, nail atrophy, skin hyperpigmentation, edema, anemia, and glossitis[7]. The condition of the present group of patients was generally consistent with that reported previously. Goto[6] has divided this disease into five types according to its onset. Type I: diarrhea as initial symptom (35.4%); Type II: abnormal gustatory sense as initial symptom (40.9%); Type III : dominated by dry mouth (6.4%); Type IV: initial symptoms include hair loss and nail atrophy (9.1%); and Type V: initial loss of appetite and general malaise, followed by nail atrophy, hair loss and abnormal gustatory sense but no diarrhea (8.2%). In our group of patients, Type I dominated, and other types were relatively rare. One case was Type II, which was mainly characterized by abnormal salt and sweet taste, accompanied by tongue atrophy.
CCS polyps are distributed through the whole digestive tract, being common in the stomach and colon, less common in the small intestine and rectum, and uncommon in the esophagus[3]. In most circumstances, the polyps are found simultaneously in the stomach and intestines. Polyps usually are nodular or irregular in shape, differently sized, and diffusely distributed. The pathology of CCS is not specific. There are four histological types: hyperplastic polyps, tubular adenomas, juvenile polyps, and inflammatory. The present group of patients met the characteristics above. Cancer arising from polyps is an important cause of death, and 12.5% of patients with polyps develop cancer[8], thus close monitoring or removal of polyps is important.
There were some limitations to our study. It was a retrospective study. CCS is a rare disease, so there are no prospective randomized controlled studies. From our experience and review of the literature, comprehensive treatment, mainly with glucocorticoids, is the most effective option at present. Currently, comprehensive treatment includes corticosteroids, polyp electrocision, and enteral/parenteral nutrition, and the recent literature suggests that it could improve prognosis and quality of life[4,9-11]. The currently recommended high-dose hormonal therapy (prednisone ≥ 40 mg/d) was initiated[4], with a treatment course of 6-12 mo, with the longest maintenance period of up to 4 years. In most circumstances, a slow reduction of dosage is suggested. In 26 Chinese cases of CCS, 8 were treated with hormonal and nutritional therapy, seven of which showed varying degrees of improvement[9]. In our six patients, four achieved general remission after hormonal and nutritional therapy. However, the symptoms of CCS have different hormonal responses. In our group of patients, diarrhea and abdominal pain were more easily corrected in five cases, while the improvement of ectodermal symptoms was relatively slow. Hormones played a limited role in gastrointestinal tract polyps. Two cases achieved no improvement in polyps after treatment, one became worse, and one had recurrence after 3 years of full remission. Fossati et al[12] have reported that after 4 years of hormonal maintenance therapy, one patient had improved clinical symptoms, including ectodermal changes, but endoscopic review did not show improvement in polyps. At present, polyps are mainly subject to endoscopic resection to prevent cancerous development. Treatment is still challenged by repeated occurrence of CSS. Two of our six patients experienced recurrence at one-year follow-up. It was reported that immunosuppressants are useful in maintaining remission[3]. However, further clinical observation is needed.
Salazosulfapyridine and 5-aminosalicylic acid are used in patients with gastrointestinal symptoms. Nutritional support, antibiotics, histamine receptor antagonists, cromolyn sodium and surgery can be used to improve symptoms such as diarrhea, weight loss, and ectodermal symptoms[5]. Successful treatment by immunosuppressants for steroid-resistant CCS has also been recently reported[13].
In conclusion, CCS is a rare disease with major symptoms of gastrointestinal polyps, diarrhea, skin hyperpigmentation, and hair/finger (toe) nail atrophy. Comprehensive treatment led by hormonal therapy can lead to partial or full remission of clinical symptoms. Long-term follow-up is necessary for further results.
COMMENTS
Case characteristics
Cronkhite-Canada syndrome (CCS) is a rare nonfamilial polyposis syndrome characterized by epithelial disturbances in the gastrointestinal tract and skin.
Clinical diagnosis
CCS is diagnosed by nonfamilial adenomatous polyposis (diffuse polyposis of the gastrointestinal tract); ectodermal dysplasia (cutaneous hyperpigmentation, dystrophic changes of fingernails, and alopecia); diarrhea, weight loss, and abdominal pain.
Differential diagnosis
Peutz-Jeghers syndrome, juvenile polyps, familial polyposis of the colon, Gardner syndrome, Turcot syndrome, and Menetrier’s disease.
Laboratory diagnosis
Fecal occult blood was positive in all six cases. Serum hemoglobin, potassium, calcium and protein were below the normal range in two cases. Circulatory anti-Saccharomyces cerevisiae antibody and antinuclear antibody were present in three cases.
Imaging diagnosis
Multiple polyps were found in all patients by gastroscopy and colonoscopy, with only one in the esophagus.
Pathological diagnosis
Histological assessment showed hyperplastic polyps in five cases, tubular adenoma in three, and juvenile polyp in one with chronic inflammation and mucosal edema.
Treatment
All patients underwent comprehensive therapy, including glucocorticoids, nutritional support, antibiotics, and polypectomy.
Related reports
There is no prospective randomized controlled study. From their experience and review of the literature, comprehensive treatment mainly with glucocorticoids is the most effective treatment to date.
Term explanation
Comprehensive treatment includes corticosteroids, polyp electrocision, and enteral/parenteral nutrition etc.
Experiences and lessons
CCS has high morbidity, but detailed understanding and comprehensive treatment could improve prognosis and quality of life.
Peer review
This article describes the clinical features, pathophysiology and potential therapy of CCS.
Footnotes
P- Reviewers: Amornyotin S, Chokshi RJ, Gassler N, Lakatos PL, Shen HN S- Editor: Ma YJ L- Editor: A E- Editor: Zhang DN
References
- 1.Cronkhite LW, Canada WJ. Generalized gastrointestinal polyposis; an unusual syndrome of polyposis, pigmentation, alopecia and onychotrophia. N Engl J Med. 1955;252:1011–1015. doi: 10.1056/NEJM195506162522401. [DOI] [PubMed] [Google Scholar]
- 2.Murata I, Yoshikawa I, Endo M, Tai M, Toyoda C, Abe S, Hirano Y, Otsuki M. Cronkhite-Canada syndrome: report of two cases. J Gastroenterol. 2000;35:706–711. doi: 10.1007/s005350070051. [DOI] [PubMed] [Google Scholar]
- 3.Sweetser S, Ahlquist DA, Osborn NK, Sanderson SO, Smyrk TC, Chari ST, Boardman LA. Clinicopathologic features and treatment outcomes in Cronkhite-Canada syndrome: support for autoimmunity. Dig Dis Sci. 2012;57:496–502. doi: 10.1007/s10620-011-1874-9. [DOI] [PubMed] [Google Scholar]
- 4.Liu YX, Wu D, Fei GJ, Sun G, Qian JM, Cai JF, Fan XH, Li YJ, Luo Y. Clinical and pathological features of 12 Cronkhite Canada syndrome patients. Jichu Yixue Linchuang. 2012;32:1349–1352. [Google Scholar]
- 5.She Q, Jiang JX, Si XM, Tian XY, Shi RH, Zhang GX. A severe course of Cronkhite-Canada syndrome and the review of clinical features and therapy in 49 Chinese patients. Turk J Gastroenterol. 2013;24:277–285. doi: 10.4318/tjg.2013.0527. [DOI] [PubMed] [Google Scholar]
- 6.Goto A. Cronkhite-Canada syndrome: epidemiological study of 110 cases reported in Japan. Nihon Geka Hokan. 1995;64:3–14. [PubMed] [Google Scholar]
- 7.Daniel ES, Ludwig SL, Lewin KJ, Ruprecht RM, Rajacich GM, Schwabe AD. The Cronkhite-Canada Syndrome. An analysis of clinical and pathologic features and therapy in 55 patients. Medicine (Baltimore) 1982;61:293–309. [PubMed] [Google Scholar]
- 8.Nakatsubo N, Wakasa R, Kiyosaki K, Matsui K, Konishi F. Cronkhite-Canada syndrome associated with carcinoma of the sigmoid colon: report of a case. Surg Today. 1997;27:345–348. doi: 10.1007/BF00941810. [DOI] [PubMed] [Google Scholar]
- 9.Wu XR, Wang L, Wang Q, Shi SS, Guo WD. Cronkhite-Canada syndrome and review. Zhonghua Neike Zazhi. 2005;44:387–388. [Google Scholar]
- 10.Ward E, Wolfsen HC, Ng C. Medical management of Cronkhite-Canada syndrome. South Med J. 2002;95:272–274. [PubMed] [Google Scholar]
- 11.Ward EM, Wolfsen HC. Pharmacological management of Cronkhite-Canada syndrome. Expert Opin Pharmacother. 2003;4:385–389. doi: 10.1517/14656566.4.3.385. [DOI] [PubMed] [Google Scholar]
- 12.Fossati M, Lombardi F, Cattaneo C, Borsa C, Colombo F. Cronkhite-Canada syndrome. A case of sustained partial-remission. Dig Liver Dis. 2001;33:785–787. doi: 10.1016/s1590-8658(01)80696-4. [DOI] [PubMed] [Google Scholar]
- 13.Ohmiya N, Nakamura M, Yamamura T, Yamada K, Nagura A, Yoshimura T, Hirooka Y, Matsumoto T, Hirata I, Goto H. Steroid-resistant Cronkhite-Canada syndrome successfully treated by cyclosporine and azathioprine. J Clin Gastroenterol. 2014;48:463–464. doi: 10.1097/MCG.0000000000000009. [DOI] [PubMed] [Google Scholar]