

Abstract
Dozens of human diseases and health traits are significantly more common among individuals carrying particular human leukocyte antigens (HLA) alleles. The underlying mechanism of this phenomenon, commonly referred to as “HLA-disease association”, has been the subject of a decades-long debate. The prevailing hypotheses implicate an auto-aggressive immune response due to aberrant presentation of self-, self-mimicking-, or altered self-antigens by HLA molecules. However, the identity of such putative antigens remains elusive in the vast majority of HLA-associated diseases. Moreover, antigen presentation-based hypotheses are difficult to reconcile with epidemiologic data and functional characteristics of HLA molecules. To provide better answers to these inconsistencies an alternative theory involving allele-based, antigen presentation-independent mechanism is proposed here. Recent research findings in rheumatoid arthritis, an emblematic HLA-associated disease, lend support to the proposed theory.
“Two roads diverged in a wood, and I -
I took the one less traveled by,
And that has made all the difference.”
~ Robert Frost (1874–1963)
Introduction
Glycoproteins encoded by genes of the major histocompatibility complex (MHC) – known in humans as HLA (human leukocyte antigens) – specialize in presentation of short peptides to T lymphocytes and play a key role in the body’s immune defense. However, it has long been observed that some allelic variants of certain HLA genes appear to betray their assigned duty and, paradoxically, facilitate certain diseases, many of which involve immune-mediated tissue damage. How does the MHC, a critical immune-policing mechanism become a wrongdoer is unclear. Here, I briefly review some of the prevailing hypotheses concerning the mechanism of MHC-disease association, and discuss an emerging new paradigm.
HLA Molecules
HLA is a large genetic region that encompasses over 200 highly polymorphic genes that encode for class I and class II antigens, as well as other immune system proteins. Class-I and class-II HLA gene products are expressed on the cell surface. Their best-characterized role is to present antigenic peptides to T lymphocytes. In order to selectively target invading microorganisms and other foreign antigens, the immune system must discriminate between self and non-self. To that end, a process called “MHC restriction” (Zinkernagel & Doherty, 1997) has evolved. This restriction is accomplished by an intricate thymic selection process, which eliminates self-reactive T cell clones, while preserving those that recognize foreign peptide antigens presented by self-MHC. At the end of this selection, distinct CD8 and CD4 T lymphocyte clonal populations are produced with restricted recognition of peptides presented only by self class I or class II MHC molecules, respectively. Class I molecules specialize in presentation of endogenous antigens to cytotoxic T lymphocytes, while class II molecules present exogenous antigens to helper T lymphocytes.
Many of the key principles concerning antigen presentation, including MHC restriction, had been reasonably well defined by the mid-1970s, but the structural basis of antigen presentation by MHC molecules had remained elusive. A series of X-ray crystallography studies published between the late-1980s and early-1990s (Bjorkman et al., 1987a; Bjorkman et al., 1987b; Brown et al., 1988; Brown et al., 1993) catapulted the field forward. In a seminal study, Bjorkman et al. resolved the tri-dimensional structure of a human class I HLA molecule, HLA-A2, (Bjorkman et al., 1987a; Bjorkman et al., 1987b). That study revealed that the α1 and α2 domains of the HLA class I heavy chain fold to make up an oval-shaped groove that accommodates 8–10 amino acid-long peptides. The peptide-binding groove is flanked by two parallel α helical structures on its sides. Interestingly, despite a substantial evolutionary distance between class I and class II HLA genes, and the fact that in class I HLA molecules the peptide-binding groove is coded by a single gene, whereas in class II molecules it is formed jointly by the products of two distinct genes, the tri-dimensional conformations of class I and class II molecules are remarkably similar (Brown et al., 1993).
HLA-Disease Association
Population studies carried out over the last several decades have identified a long list of human diseases that are significantly more common among individuals that carry particular HLA alleles. For example, more than 90% of Caucasian patients with ankylosing spondylitis carry particular class I HLA alleles (e.g. HLA-B*27:02, HLA-B*27:05) (Reveille, 2006). Narcolepsy, a brain disorder characterized by sleep abnormality and falling attacks (cataplexy), is an illustrative example of HLA class II-associated disease, in which 90–100% of Caucasian patients carry the DQB1*06:02 allele (Mignot et al., 1994). In type I diabetes mellitus, more than 90% of patients carry either HLA-DRB1*03/DQB1*02:01, or HLA-DRB1*04/DQB1*03:02 gene haplotypes, compared to only 40% of controls (Erlich et al., 2008). Rheumatoid arthritis (RA) is another emblematic HLA class II-associated disease (reviewed in Holoshitz, 2010). Approximately 90% of Caucasian seropositive RA patients carry one or two HLA-DRB1 alleles (e.g. DRB1*04:01, DRB1*04:04, DRB1*04:05, DRB1*01:01, and a few others) that code for a sequence motif in the DRβ chain called “shared epitope” (SE).
The mechanism underlying HLA-disease association is unclear. This question has captivated the imagination of immunologists for decades, and generated many hypotheses, which fall into two general categories: 1) Those that blame ‘mistaken identity’, in which an HLA allele appears to associate with the disease, although the actual culprit belongs to a different locus in the haplotype, or associates through linkage disequilibrium, and 2) Those that implicate immune reactivity to self-antigens due to aberrant T cell repertoire selection (Nepom & Kwok, 1998; Ridgway & Fathman, 1998), immune cross-reactivity with foreign antigens (Oldstone, 1998) or immune attack on “altered self” antigens (Yin et al., 2013).
HLA haplotypes – combinations of genes at adjacent loci that tend to be inherited together – can indeed explain some misinterpreted associations, as exemplified by narcolepsy, where an apparent association with HLA-DRB1*15 was later found to be actually attributed to an HLA-DQ allele in the haplotype, HLA-DQB1*0602 (Ellis et al., 1997). Linkage disequilibrium means a non-random association of alleles at two or more loci. This phenomenon has been implicated in hereditary hemochromatosis, where an apparent association with HLA-A alleles was later determined to be actually due to two point mutations in a non-classical class I HLA gene, HFE, that are found in linkage disequilibrium with HLA-A3 and HLA-A29 alleles (Ajioka et al., 1997; Cardoso et al., 2002). However, compelling as these examples may be, both haplotype-based association and linkage disequilibrium are rather uncommon events in HLA-disease association. In the majority of diseases, the data support a role for the associated HLA alleles themselves. Importantly, neither haplotype-based association nor linkage disequilibrium identifies a bona fide mechanistic basis for HLA-disease association; they merely direct the blame at another gene. As such, these explanations do not provide testable hypotheses to explain the pathogenic mechanism of HLA molecules in disease etiology.
The three other hypotheses listed in the second category above implicate, in one way or another, antigen presentation by HLA molecules. They postulate that the basis of HLA-disease association is immune response to putative self or foreign antigens, either in their native or modified form. Given the known role of MHC molecules in antigen presentation, these hypotheses have resonated well among immunologists. Unfortunately however, despite its plausibility, presentation of specific antigens as a mechanism of HLA-disease association is difficult to reconcile with a considerable body of scientific data.
First, inconsistent with antigen-specific immune recognition, the cause-effect fidelity between HLA alleles and particular diseases is often challenged. For example HLA-DRB1*04:01 best known for its association with RA, associates with type-1 diabetes as well (Tait et al., 1995). Similarly, HLA-DQB1*06:02 increases disease risk in both narcolepsy (Mignot et al., 1994) and multiple sclerosis (Serjeantson et al., 1992); HLA-DQB1*03:02 associates with both type-1 diabetes (Sabbah et al., 1999) and celiac disease (Setty et al., 2008). Thus, an individual HLA allele can promiscuously associate with distinct diseases that do not share pathogeneses, target tissues or putative antigens. Second, contrary to what could be expected from an antigen specific-based mechanism, allele-associated diseases can demonstrate species non-specificity. For example, HLA-DRB1*04:01, which associates with human RA, confers susceptibility to inflammatory arthritis in mice as well (Taneja et al., 2007), and the SE, a sequence motif found in the vast majority of RA patients, is also associated with susceptibility to inflammatory arthritis in dogs (Ollier et al., 2001). Such ‘trans-species susceptibility’ is difficult to reconcile with MHC-restricted antigen presentation. Third, lymphocyte clonality, commonly expected in antigen-specific immune responses, has not been yet convincingly demonstrated in HLA-associated diseases, and in the majority of such diseases the identity of the putative target antigen is unknown, despite decades-long research effort. Fourth, significant HLA-allele-based associations have been observed in conditions that do not involve antigen recognition, or any immune-based pathogenesis. For example, the most significant class II HLA-disease association documented to date has been found in narcolepsy (Nishino et al., 2010), a neurotransmitter-mediated brain disorder that is not known to involve antigen presentation. Moreover, MHC associations have been shown to exist with certain traits, such as cognition (Shepherd et al., 2004), that do not involve any known immune basis, let alone antigen presentation. Finally, antigen presentation-based hypotheses cannot easily explain allele-dose impact on disease severity, or offer a plausible explanation for allele-dose effects on concordance rates in monozygotic twins (Jawaheer et al., 1994).
Thus, HLA-restricted antigen presentation, a common thread in most of the prevailing hypotheses addressing HLA-disease association, appears to be inharmonious with many observations concerning the biology, epidemiology and evolution of HLA molecules.
The MHC Cusp Theory – A Road Less Travelled By
Seeking better answers to the question of HLA-disease association, we have recently put forward a heterodox, MHC Cusp theory (de Almeida & Holoshitz, 2011). At the focus of this theory is a polymorphic region on HLA molecules whose tri-dimensional cusp-like shape appears to have been preserved in the entire MHC family. The theory proposes that the MHC codes for allele-specific ligands in the cusp region, which interact with non-MHC receptors and activate various pathways. Aberrations in those pathways could cause MHC-associated diseases.
The rationale behind the MHC Cusp theory is based on structural and functional evidence. As discussed above, the crystal structures of class I and class II HLA molecules demonstrate remarkable tri-dimensional conservation despite their considerable evolutionary distance. One of the most conspicuous features in the MHC fold is a prominent cusp-shaped prominence in the α2 domain of class I HLA molecules, and its tri-dimensionally-equivalent β1 domain in class II HLA molecules. The cusp in both molecules involves allele-diversity regions. Similarly shaped structures have been preserved in the entire MHC gene product family, irrespective of whether or not they can present antigens (Rudolph et al., 2006). Importantly, cusp regions in several MHC molecules have been found to act as ligands that perform non-antigen presentation functions. For example, ligands for natural killer cell receptors have been identified in the cusp region of both classical and non-classical (HLA-E) class I HLA molecules (Boyington et al., 2000; Kaiser et al., 2008). Similarly, in HFE (an empty-grooved HLA class I-like molecule), a transferrin receptor-binding ligand has been mapped to the cusp region (Bennett et al., 2000). Additionally, in M10 (an empty-grooved mouse class I-like MHC molecule), the cusp region has been proposed as a pheromone receptor-binding site (Olson et al., 2005), and the SE, located in the HLA-DR cusp-region, has been recently identified as a calreticulin (CRT)-binding, signal transduction ligand (Ling et al., 2007b).
Thus, the cusp region, whose peculiar tri-dimensional shape has been carefully conserved through MHC evolution independent of antigen presentation, is a hub for signal transduction ligands that interact with a variety of non-MHC receptors and activate important biologic functions. In class II and classical class I HLA molecules, the cusp contains allelic hypervariable regions. As discussed above, antigen presentation per se cannot explain HLA-disease association. Accordingly, rather than assigning the blame exclusively to the antigen presentation function of HLA molecules, the MHC Cusp theory posits that HLA molecules may promote diseases due to their auxiliary allele-specific, yet antigen presentation-independent, biologic effects. Recent findings concerning the functional role of the SE in RA lend support to the theory.
The SE: an HLA-DRB1-Coded Arthritogenic Cusp Ligand
RA is a chronic inflammatory disease, which afflicts millions of individuals worldwide. The disease is multi-systemic, but its best-characterized aspect is chronic joint inflammation and articular bone destruction, mediated by activated osteoclasts (OCs) (Gravallese et al., 2000). The etiology of RA is unclear, although genetic factors, especially the HLA-DRB1 locus (Stastny, 1978), play a major role. Genotypic analyses have revealed that susceptibility to RA is closely associated with a number of HLA-DRB1 alleles that code for SE – a five amino acid sequence motif in residues 70–74 of the HLA-DRβ1 chain (Gregersen et al., 1987). The more common SE-coding alleles (and their coded amino acid sequence motif) include: HLA-DRB1*04:01 (QKRAA); HLA-DRB1*04:04, HLA-DRB1*04:05, HLA-DRB1*04:08, HLA-DRB1*01:01, and HLA-DRB1*01:02 (QRRAA); HLA-DRB1*10:01 (RRRAA). Importantly, SE-coding HLA-DRB1 alleles not only confer disease susceptibility, they are also associated with earlier onset of arthritis and more severe bone damage (Gonzalez-Gay et al., 2002). Another intriguing feature of SE-RA association is that the extent of bone damage correlates positively with the number of SE-coding HLA-DRB1 alleles (Weyand & Goronzy, 1994).
The prevalent hypotheses concerning the mechanism of SE-RA association postulate that the SE motif may permit presentation of arthritogenic antigens (Wucherpfennig & Strominger, 1995), or interfere with T cell repertoire selection with resultant emergence of T cell clones that recognize such antigens (Bhayani & Hedrick, 1991). However, data supporting a direct pathogenic role for antigen-specific immune responses in RA are inconclusive. Additionally, the cause-effect fidelity between SE and RA is not stringent, as several non-RA human diseases and experimental animal models of autoimmunity have been shown to associate with SE-coding alleles as well (discussed by De Almeida & Holoshitz, 2011). Furthermore, as discussed above, antigen presentation-based mechanisms are difficult to reconcile with SE allele-dose effects.
The SE is a 5 amino acid sequence motif in the third allelic hypervariable region, near the tip of the HLA-DR β1 domain cusp. Searching for an allele-based - yet antigen presentation-independent - mechanistic basis for SE-RA association, in recent years my laboratory has examined whether the SE acts as a signal transduction ligand, consistent with its cusp location. The decision to take this road came about rather fortuitously. Studying the cytolytic activity of human γδ T cell clones against EBV-transformed B cell lines, we were surprised to discover that these highly potent cytolytic cells, which can kill indiscriminately all malignant or transformed cells, were selectively unable to kill SE-positive targets. The enigmatic resistance was later found to be due to higher constitutive levels of nitric oxide (NO) in SE-positive targets (Ling et al., 2006). That seminal finding compelled us to explore the role of SE-coding HLA-DRB1 alleles using cDNA transfection, followed by studies with synthetic peptides and recombinant proteins expressing the SE sequence motif (Ling et al., 2007a). In other studies we have identified cell surface CRT – a known innate immune system receptor – as the SE-binding receptor (Ling et al., 2007b), and mapped the SE binding site on that receptor (Ling et al., 2010). SE signaling effects and identification of CRT as the signal transducing receptor have been reviewed in detail elsewhere (de Almeida et al., 2011; Holoshitz et al., 2010; Holoshitz & Ling, 2007).
More recently we demonstrated that engagement of cell surface CRT by the SE ligand activates lineage-dependent functional consequences. For example, in CD8+CD11c+ dendritic cells, the SE ligand inhibits indoleamine 2, 3 deoxygenase, an enzyme known to play an important role in regulatory T (Treg) cell activation. In CD8−CD11c+ dendritic cells, the SE triggers production of IL-6 and IL-23, cytokines known to be involved in activation and expansion of IL-17-producing T (Th17) cells. The end result of these two complementing effects is a potent SE-activated Th17 polarization, both in vitro and in vivo (De Almeida et al., 2010).
Th17 cells are believed to play an important pathogenic role in RA (Shahrara et al., 2008) and can activate OCs (Sato et al., 2006). Given that, and the known association between the SE and erosive disease, we have undertaken to investigate whether the SE has a direct pro-osteoclastogenic effect. The rationale for this study was further strengthened by the fact that the SE is a potent activator of NO and reactive oxygen species (ROS) (Ling et al., 2006; Ling et al., 2007a), signaling molecules which have been previously shown to affect OC activation (Lee et al., 2005; Rahnert et al., 2008). We have discovered that the SE ligand facilitates OC differentiation and functional activation. Importantly, when administered in vivo, the SE ligand enhanced erosive bone damage in collagen-induced arthritis in mice (Holoshitz et al., 2013).
We have recently put the SE ligand hypothesis to a higher level of scrutiny by studying biostable small molecular SE-mimetic ligands. The rationale behind this endeavor was driven by a search for biostable compounds that would more closely resemble the tri-dimensional conformation of the SE in its physiologic HLA-DR context, and would be more effective in vivo compared to the linear 15-mer peptidic SE ligands used in prior studies. A library of such compounds has been recently synthesized using a backbone cyclization method (Naveh et al., 2012). A prototypic SE-mimetic member of that library, containing the SE primary sequence motif QKRAA, was characterized both in vitro and in vivo. This SE-mimetic compound interacted strongly with the SE receptor CRT, potently activated NO and ROS production and markedly facilitated OC differentiation and function in vitro, with potencies 100,000 to 1,000,000-fold higher than those of a linear SE peptidic ligand. When administered in nanogram doses to mice with collagen-induced arthritis, the SE-mimetic ligand enhanced Th17 cell expansion, increased arthritis severity, enhanced OC abundance in synovial tissues and facilitated bone destruction (Fu et al., 2013, in press).
The majority of individuals carrying SE-coding HLA-DRB1 are perfectly healthy, consistent with the consensus in the field that the SE does not cause RA, but rather increases the risk of disease development. The low RA concordance rates among identical twins (Silman et al., 1993) and epidemiologic surveys (Gabriel et al., 1999) together suggest that in addition to genetic factors, environmental influences play critical roles in RA disease onset. In this context, it is worth mentioning that: 1) Anti-citrullinated protein antibodies, a useful diagnostic marker in RA, are closely associated with SE-coding HLA-DRB1 alleles (Klareskog et al., 2008); 2) Biochemical analyses have revealed over-abundance of citrullinated proteins in RA (Yamada et al., 2005); 3) There is strong synergistic effect between SE positivity and cigarette smoking on RA susceptibility (Karlson et al., 2010); 4) Cigarette smoking has been proposed as a potential cause of protein citrullination (Makrygiannakis et al., 2008). Given all that, it is intriguing that we have recently demonstrated that citrullinated CRT, which is over-abundant in RA synovial tissues, displays higher affinity for the SE ligand and transduces more potent SE-activated signaling (Ling et al., 2013). The pathogenic significance of these findings is unclear, but if confirmed in an experimental disease model, these results could provide a mechanistic model of gene-environment interaction in RA.
Thus, using the SE as a test case, our signal transduction data, receptor binding analyses, cell differentiation and activation studies, and in vivo experiments, collectively support the premise of the MHC Cusp theory, by suggesting that in addition to their role in antigen presentation, HLA molecules can contribute to the pathogenesis of autoimmune diseases through allele-coded ligands that activate aberrant signaling events with resultant pathology.
Evolutionary Perspectives
Bearing in mind that teleological arguments are acceptable only when used as a basis to formulate testable hypotheses, in the remaining paragraphs I venture to address the question “why”. In other words, what has been the “purpose” of the substantial energy invested in order to preserve the tri-dimensional structure of the cusp region through evolution, and why has this endeavor produced, paradoxically, detrimental health outcomes?
My proposed answer to the first question is that a protuberant α helical structure may be critically needed to allow effective physical interaction between cusp ligands and their cognate receptors. In this context, it is worth commenting that the modern MHC is likely a descendent of archaic self/non-self-discrimination systems, which, in turn, likely evolved from basic cell-cell contact systems. The theory proposed here argues that the cusp antigen presentation-independent functions may have been preserved through evolution due to their impact on fundamental biologic functions, such as natural killer cell regulation (in classical and non-classical class I HLA molecules), iron metabolism (HFE), mating (M10), or immune auxiliary functions (HLA class II molecules).
Consistent with recent findings concerning the functional role of the SE, it could be argued that a trans-species polymorphic region in class II HLA molecules has been preserved in its cusp-like shape throughout evolution because of its important role in immune auxiliary function, such as T cell polarization. It is also conceivable that at least some cusp region effects may have been evolutionarily conserved thanks to their ability to productively mimic more archaic innate immune stimulants, such as bacterial products. At this juncture it is worth pointing out that the signaling and functional effects of the SE described by us are intriguingly similar to previously reported effects of muramyl dipeptide.
N-acetylmuramic acid L-alanine D-isoglutamine (LD-MDP), a building block of bacterial cell wall, is the minimal structure required for the adjuvanticity of mycobacteria in complete Freund’s adjuvant (Ellouz et al., 1974). Relevant to the focus of this review, MDP demonstrates many functional similarities to the SE ligand (Table 1). For example, similar to the SE, MDP activates production of IL-6 and TNFα (Sanceau et al., 1990; van der Meer et al., 2009), increases NO (Palacios et al., 1992) and ROS (El-Khoury et al., 2011) production, activates OCs (Dewhirst, 1982; Dziak et al., 1982), enhances Th17 differentiation (Manni et al., 2011) and, importantly, facilitates inflammatory arthritis in rodents (Koga et al., 1986; Koga et al., 1980; Kohashi et al., 1986; Zidek & Masek, 1985). Finally, MDP has been reported to bind to the SE receptor, CRT (Chen et al., 2004). Using a cutting-edge biolayer interferometry technique, we have recently confirmed that MDP and CRT do interact physically. Moreover, virtual receptor-ligand docking simulations predicted that the SE ligand and MDP interact with overlapping binding sites on CRT (Figure 1). It is therefore tempting to propose that the SE ligand mimics the adjuvant effects of MDP. Could such mimicry be an underlying mechanisms by which bacterial adjuvants can trigger autoimmunity (Rose, 2010)? Obviously, this question needs to be examined experimentally.
Table 1.
Functional similarities between the SE ligand and MDP
| Reference
|
||
|---|---|---|
| Function
|
SE ligand
|
MDP
|
| Th17 polarization | (De Almeida et al., 2010), (Holoshitz et al., 2013) | (Manni et al., 2011) |
|
| ||
| OC activation | (Holoshitz et al., 2013) | (Dewhirst, 1982),(Dziak et al., 1982) |
|
| ||
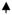 TNFα, IL-6 TNFα, IL-6 |
(De Almeida et al., 2010) | (van der Meer et al., 2009), (Sanceau et al., 1990) |
|
| ||
|
NO |
(Ling et al., 2006) | (Palacios et al., 1992) |
|
| ||
|
ROS |
(Ling et al., 2006) | (El-Khoury et al., 2011) |
|
| ||
| CRT binding | (Ling et al., 2007b), (Ling et al., 2010) | (Chen et al., 2004) |
|
| ||
| Arthritogenicity | (Holoshitz et al., 2013) | (Koga et al., 1986; Koga et al., 1980; Kohashi et al., 1986; Zidek & Masek, 1985) |
Figure 1. Virtual docking of MDP and SE ligands on CRT.
Ligands are shown as colored sticks; CRT surface is in light gray; CRT residues in close proximity to ligands are shown as dark-gray sticks. As can be seen, the two SE ligands, 15-mer linear peptide 65–79*0401 (A) and the SE-mimetic cQKRAA (B), as well as LD-MDP (C), are predicted to occupy overlapping binding sites on CRT, in significant proximity to several key CRT residues.
So, how has it happened that cusp ligands, an evolutionarily conserved fundamental health-promoting mechanism, ended up facilitating disease development? Two mutually non-exclusive answers can be proposed: 1) Cusp-associated diseases are unavoidable outcomes, considering that the hypervariable cusp region represents an MHC mutational “hot spot”. Because most autoimmune diseases appear late in life, they have little impact on genetic fitness; 2) As discussed above, disease development in genetically susceptible individuals is often triggered by environmental exposure. Thus, modern-era environmental pollutants, rather than evolution’s flawed design, should be blamed. Our own data suggest that environmentally induced posttranslational modifications of cusp-binding receptors might lead to higher affinities of receptor-ligand interactions, and potentiate signal transduction. It is conceivable that environmentally-induced posttranslational modifications could occur at the ligand side as well. Consistent with our data, the Cusp theory proposes that such posttranslational modification events in the receptor-ligand junction might increase the intensity of cusp-triggered signal beyond a certain threshold and, in individuals with the right constellation of genetic risk factors, could trigger disease onset.
In closing, the MHC Cusp theory offers a plausible mechanistic basis for HLA-disease association. It should be cautioned, however that alluring as the MHC Cusp road may seem, the long journey to validating this theory has just begun. Only time will tell if taking the road less travelled by will have made all the difference.
Acknowledgments
Dr. Holoshitz is supported by grants from the National Institute of General Medical Sciences and the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Numbers: 5R01GM088560, 5R01AR059085 and 3R01AR059085-03S1.
I thank Ms. Gail Quaderer for administrative assistance.
Footnotes
Disclosure
The author has no known or potential conflicts of interest.
The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health
References
- Ajioka RS, Jorde LB, Gruen JR, Yu P, Dimitrova D, Barrow J, Radisky E, Edwards CQ, Griffen LM, Kushner JP. Haplotype analysis of hemochromatosis: evaluation of different linkage-disequilibrium approaches and evolution of disease chromosomes. Am J Hum Genet. 1997;60(6):1439–1447. doi: 10.1086/515466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MJ, Lebron JA, Bjorkman PJ. Crystal structure of the hereditary haemochromatosis protein HFE complexed with transferrin receptor. Nature. 2000;403(6765):46–53. doi: 10.1038/47417. [DOI] [PubMed] [Google Scholar]
- Bhayani HR, Hedrick SM. The role of polymorphic amino acids of the MHC molecule in the selection of the T cell repertoire. J Immunol. 1991;146(4):1093–1098. [PubMed] [Google Scholar]
- Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. The foreign antigen binding site and T cell recognition regions of class I histocompatibility antigens. Nature. 1987a;329(6139):512–518. doi: 10.1038/329512a0. [DOI] [PubMed] [Google Scholar]
- Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. Structure of the human class I histocompatibility antigen, HLA-A2. Nature. 1987b;329(6139):506–512. doi: 10.1038/329506a0. [DOI] [PubMed] [Google Scholar]
- Boyington JC, Motyka SA, Schuck P, Brooks AG, Sun PD. Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature. 2000;405(6786):537–543. doi: 10.1038/35014520. [DOI] [PubMed] [Google Scholar]
- Brown JH, Jardetzky T, Saper MA, Samraoui B, Bjorkman PJ, Wiley DC. A hypothetical model of the foreign antigen binding site of class II histocompatibility molecules. Nature. 1988;332(6167):845–850. doi: 10.1038/332845a0. [DOI] [PubMed] [Google Scholar]
- Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL, Wiley DC. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature. 1993;364(6432):33–39. doi: 10.1038/364033a0. [DOI] [PubMed] [Google Scholar]
- Cardoso CS, Alves H, Mascarenhas M, Goncalves R, Oliveira P, Rodrigues P, Cruz E, De Sousa M, Porto G. Co-selection of the H63D mutation and the HLA-A29 allele: a new paradigm of linkage disequilibrium? Immunogenetics. 2002;53(12):1002–1008. doi: 10.1007/s00251-001-0414-8. [DOI] [PubMed] [Google Scholar]
- Chen D, Duggan C, Reden TB, Kooragayala LM, Texada DE, Langford MP. Calreticulin is a binding protein for muramyl dipeptide and peptidoglycan in RK13 cells. Biochemistry (Mosc) 2004;43(37):11796–11801. doi: 10.1021/bi0490789. [DOI] [PubMed] [Google Scholar]
- De Almeida DE, Holoshitz J. MHC molecules in health and disease: At the cusp of a paradigm shift. Self Nonself. 2011;2(1):43–48. doi: 10.4161/self.2.1.15757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Almeida DE, Ling S, Holoshitz J. New insights into the functional role of the rheumatoid arthritis shared epitope. FEBS Lett. 2011;585(23):3619–3626. doi: 10.1016/j.febslet.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Almeida DE, Ling S, Pi X, Hartmann-Scruggs AM, Pumpens P, Holoshitz J. Immune dysregulation by the rheumatoid arthritis shared epitope. The Journal of Immunology. 2010;185(3):1927–1934. doi: 10.4049/jimmunol.0904002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewhirst FE. N-acetyl muramyl dipeptide stimulation of bone resorption in tissue culture. Infect Immun. 1982;35(1):133–137. doi: 10.1128/iai.35.1.133-137.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziak R, Rowe DJ, Brown MJ, Hausmann E. Effects of synthetic muramyl dipeptide on bone metabolism in the fetal rat. Arch Oral Biol. 1982;27(10):787–791. doi: 10.1016/0003-9969(82)90031-0. [DOI] [PubMed] [Google Scholar]
- El-Khoury TG, Bahr GM, Echtay KS. Muramyl-dipeptide-induced mitochondrial proton leak in macrophages is associated with upregulation of uncoupling protein 2 and the production of reactive oxygen and reactive nitrogen species. FEBS J. 2011;278(17):3054–3064. doi: 10.1111/j.1742-4658.2011.08226.x. [DOI] [PubMed] [Google Scholar]
- Ellis MC, Hetisimer AH, Ruddy DA, Hansen SL, Kronmal GS, Mcclelland E, Quintana L, Drayna DT, Aldrich MS, Mignot E. HLA class II haplotype and sequence analysis support a role for DQ in narcolepsy. Immunogenetics. 1997;46(5):410–417. doi: 10.1007/s002510050295. [DOI] [PubMed] [Google Scholar]
- Ellouz F, Adam A, Ciorbaru R, Lederer E. Minimal structural requirements for adjuvant activity of bacterial peptidoglycan derivatives. Biochem Biophys Res Commun. 1974;59(4):1317–1325. doi: 10.1016/0006-291x(74)90458-6. [DOI] [PubMed] [Google Scholar]
- Erlich H, Valdes AM, Noble J, Carlson JA, Varney M, Concannon P, Mychaleckyj JC, Todd JA, Bonella P, Fear AL, Lavant E, Louey A, Moonsamy P. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes. 2008;57(4):1084–1092. doi: 10.2337/db07-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Ling S, Liu Y, Yang J, Naveh S, Hannah M, Gilon C, Zhang Y, Holoshitz J. A small shared epitope-mimetic compound potently accelerates osteoclast-mediated bone damage in autoimmune arthritis. J Immunol. 2013 doi: 10.4049/jimmunol.1203231. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel SE, Crowson CS, O’fallon WM. The epidemiology of rheumatoid arthritis in Rochester, Minnesota, 1955–1985. Arthritis Rheum. 1999;42(3):415–420. doi: 10.1002/1529-0131(199904)42:3<415::AID-ANR4>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Gay MA, Garcia-Porrua C, Hajeer AH. Influence of human leukocyte antigen-DRB1 on the susceptibility and severity of rheumatoid arthritis. Semin Arthritis Rheum. 2002;31(6):355–360. doi: 10.1053/sarh.2002.32552. [DOI] [PubMed] [Google Scholar]
- Gravallese EM, Manning C, Tsay A, Naito A, Pan C, Amento E, Goldring SR. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum. 2000;43(2):250–258. doi: 10.1002/1529-0131(200002)43:2<250::AID-ANR3>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. an approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30(11):1205–1213. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- Holoshitz J. The rheumatoid arthritis HLA-DRB1 shared epitope. Curr Opin Rheumatol. 2010;22(3):293–298. doi: 10.1097/BOR.0b013e328336ba63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holoshitz J, De Almeida DE, Ling S. A role for calreticulin in the pathogenesis of rheumatoid arthritis. Ann N Y Acad Sci. 2010;1209(1):91–98. doi: 10.1111/j.1749-6632.2010.05745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holoshitz J, Ling S. Nitric oxide signaling triggered by the rheumatoid arthritis shared epitope: a new paradigm for MHC disease association? Ann N Y Acad Sci. 2007;1110:73–83. doi: 10.1196/annals.1423.009. [DOI] [PubMed] [Google Scholar]
- Holoshitz J, Liu Y, Fu J, Joseph J, Ling S, Colletta A, Sharma P, Begun D, Goldstein S, Taichman R. An HLA-DRB1-coded signal transduction ligand facilitates inflammatory arthritis: a new mechanism of autoimmunity. J Immunol. 2013;190(1):48–57. doi: 10.4049/jimmunol.1202150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jawaheer D, Thomson W, Macgregor AJ, Carthy D, Davidson J, Dyer PA, Silman AJ, Ollier WE. “Homozygosity” for the HLA-DR shared epitope contributes the highest risk for rheumatoid arthritis concordance in identical twins. Arthritis Rheum. 1994;37(5):681–686. doi: 10.1002/art.1780370511. [DOI] [PubMed] [Google Scholar]
- Kaiser BK, Pizarro JC, Kerns J, Strong RK. Structural basis for NKG2A/CD94 recognition of HLA-E. Proc Natl Acad Sci U S A. 2008;105(18):6696–6701. doi: 10.1073/pnas.0802736105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlson EW, Chang SC, Cui J, Chibnik LB, Fraser PA, De Vivo I, Costenbader KH. Gene-environment interaction between HLA-DRB1 shared epitope and heavy cigarette smoking in predicting incident rheumatoid arthritis. Ann Rheum Dis. 2010;69(1):54–60. doi: 10.1136/ard.2008.102962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klareskog L, Ronnelid J, Lundberg K, Padyukov L, Alfredsson L. Immunity to citrullinated proteins in rheumatoid arthritis. Annu Rev Immunol. 2008;26:651–675. doi: 10.1146/annurev.immunol.26.021607.090244. [DOI] [PubMed] [Google Scholar]
- Koga T, Kakimoto K, Hirofuji T, Kotani S, Sumiyoshi A, Saisho K. Muramyl dipeptide induces acute joint inflammation in the mouse. Microbiol Immunol. 1986;30(7):717–723. doi: 10.1111/j.1348-0421.1986.tb02997.x. [DOI] [PubMed] [Google Scholar]
- Koga T, Sakamoto S, Onoue K, Kotani S, Sumiyoshi A. Efficient induction of collagen arthritis by the use of a synthetic muramyl dipeptide. Arthritis Rheum. 1980;23(9):993–997. doi: 10.1002/art.1780230905. [DOI] [PubMed] [Google Scholar]
- Kohashi O, Kohashi Y, Shigematsu N, Ozawa A, Kotani S. Acute and chronic polyarthritis induced by an aqueous form of 6-O-acyl and N-acyl derivatives of N-acetylmuramyl-L-alanyl-D-isoglutamine in euthymic rats and athymic nude rats. Lab Invest. 1986;55(3):337–346. [PubMed] [Google Scholar]
- Lee NK, Choi YG, Baik JY, Han SY, Jeong DW, Bae YS, Kim N, Lee SY. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood. 2005;106(3):852–859. doi: 10.1182/blood-2004-09-3662. [DOI] [PubMed] [Google Scholar]
- Ling S, Cheng A, Pumpens P, Michalak M, Holoshitz J. Identification of the rheumatoid arthritis shared epitope binding site on calreticulin. PLoS ONE. 2010;5(7):e11703. doi: 10.1371/journal.pone.0011703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling S, Cline EN, Haug TS, Fox DA, Holoshitz J. Citrullinated calreticulin potentiates rheumatoid arthritis shared epitope signaling. Arthritis Rheum. 2013;65(3):618–626. doi: 10.1002/art.37814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling S, Lai A, Borschukova O, Pumpens P, Holoshitz J. Activation of nitric oxide signaling by the rheumatoid arthritis shared epitope. Arthritis Rheum. 2006;54(11):3423–3432. doi: 10.1002/art.22178. [DOI] [PubMed] [Google Scholar]
- Ling S, Li Z, Borschukova O, Xiao L, Pumpens P, Holoshitz J. The rheumatoid arthritis shared epitope increases cellular susceptibility to oxidative stress by antagonizing an adenosine-mediated anti-oxidative pathway. Arthritis Res Ther. 2007a;9(1):R5. doi: 10.1186/ar2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling S, Pi X, Holoshitz J. The rheumatoid arthritis shared epitope triggers innate immune signaling via cell surface calreticulin. The Journal of Immunology. 2007b;179(9):6359–6367. doi: 10.4049/jimmunol.179.9.6359. [DOI] [PubMed] [Google Scholar]
- Makrygiannakis D, Hermansson M, Ulfgren A-K, Nicholas AP, Zendman AJW, Eklund A, Grunewald J, Skold CM, Klareskog L, Catrina AI. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann Rheum Dis. 2008;67(10):1488–1492. doi: 10.1136/ard.2007.075192. [DOI] [PubMed] [Google Scholar]
- Manni M, Ding W, Stohl LL, Granstein RD. Muramyl dipeptide induces Th17 polarization through activation of endothelial cells. J Immunol. 2011;186(6):3356–3363. doi: 10.4049/jimmunol.1000847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignot E, Lin X, Arrigoni J, Macaubas C, Olive F, Hallmayer J, Underhill P, Guilleminault C, Dement WC, Grumet FC. DQB1*0602 and DQA1*0102 (DQ1) are better markers than DR2 for narcolepsy in Caucasian and black Americans. Sleep. 1994;17(8 Suppl):S60–67. doi: 10.1093/sleep/17.suppl_8.s60. [DOI] [PubMed] [Google Scholar]
- Naveh S, Tal-Gan Y, Ling S, Hoffman A, Holoshitz J, Gilon C. Developing potent backbone cyclic peptides bearing the shared epitope sequence as rheumatoid arthritis drug-leads. Bioorg Med Chem Lett. 2012;22(1):493–496. doi: 10.1016/j.bmcl.2011.10.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepom GT, Kwok WW. Molecular basis for HLA-DQ associations with IDDM. Diabetes. 1998;47(8):1177–1184. doi: 10.2337/diab.47.8.1177. [DOI] [PubMed] [Google Scholar]
- Nishino S, Okuro M, Kotorii N, Anegawa E, Ishimaru Y, Matsumura M, Kanbayashi T. Hypocretin/orexin and narcolepsy: new basic and clinical insights. Acta Physiol (Oxf) 2010;198(3):209–222. doi: 10.1111/j.1748-1716.2009.02012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldstone MB. Molecular mimicry and immune-mediated diseases. FASEB J. 1998;12(13):1255–1265. doi: 10.1096/fasebj.12.13.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollier WE, Kennedy LJ, Thomson W, Barnes AN, Bell SC, Bennett D, Angles JM, Innes JF, Carter SD. Dog MHC alleles containing the human RA shared epitope confer susceptibility to canine rheumatoid arthritis. Immunogenetics. 2001;53(8):669–673. doi: 10.1007/s002510100372. [DOI] [PubMed] [Google Scholar]
- Olson R, Huey-Tubman KE, Dulac C, Bjorkman PJ. Structure of a pheromone receptor-associated MHC molecule with an open and empty groove. PLoS Biol. 2005;3(8):e257. doi: 10.1371/journal.pbio.0030257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios M, Knowles RG, Moncada S. Enhancers of nonspecific immunity induce nitric oxide synthase: induction does not correlate with toxicity or adjuvancy. Eur J Immunol. 1992;22(9):2303–2307. doi: 10.1002/eji.1830220919. [DOI] [PubMed] [Google Scholar]
- Rahnert J, Fan X, Case N, Murphy TC, Grassi F, Sen B, Rubin J. The role of nitric oxide in the mechanical repression of RANKL in bone stromal cells. Bone. 2008;43(1):48–54. doi: 10.1016/j.bone.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reveille JD. Major histocompatibility genes and ankylosing spondylitis. Best Pract Res Clin Rheumatol. 2006;20(3):601–609. doi: 10.1016/j.berh.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Ridgway WM, Fathman CG. The association of MHC with autoimmune diseases: understanding the pathogenesis of autoimmune diabetes. Clin Immunol Immunopathol. 1998;86(1):3–10. doi: 10.1006/clin.1997.4449. [DOI] [PubMed] [Google Scholar]
- Rose NR. Autoimmunity, infection and adjuvants. Lupus. 2010;19(4):354–358. doi: 10.1177/0961203309360670. [DOI] [PubMed] [Google Scholar]
- Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24:419–466. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- Sabbah E, Savola K, Kulmala P, Reijonen H, Veijola R, Vahasalo P, Karjalainen J, Ilonen J, Akerblom HK, Knip M. Disease-associated autoantibodies and HLA-DQB1 genotypes in children with newly diagnosed insulin-dependent diabetes mellitus (IDDM). The Childhood Diabetes in Finland Study Group. Clin Exp Immunol. 1999;116(1):78–83. doi: 10.1046/j.1365-2249.1999.00863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanceau J, Falcoff R, Beranger F, Carter DB, Wietzerbin J. Secretion of interleukin-6 (IL-6) by human monocytes stimulated by muramyl dipeptide and tumour necrosis factor alpha. Immunology. 1990;69(1):52–56. [PMC free article] [PubMed] [Google Scholar]
- Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y, Cua DJ, Takayanagi H. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203(12):2673–2682. doi: 10.1084/jem.20061775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serjeantson SW, Gao X, Hawkins BR, Higgins DA, Yu YL. Novel HLA-DR2-related haplotypes in Hong Kong Chinese implicate the DQB1*0602 allele in susceptibility to multiple sclerosis. Eur JImmunogenet. 1992;19(1–2):11–19. doi: 10.1111/j.1744-313x.1992.tb00043.x. [DOI] [PubMed] [Google Scholar]
- Setty M, Hormaza L, Guandalini S. Celiac disease: risk assessment, diagnosis, and monitoring. Mol Diagn Ther. 2008;12(5):289–298. doi: 10.1007/BF03256294. [DOI] [PubMed] [Google Scholar]
- Shahrara S, Huang Q, Mandelin AM, 2nd, Pope RM. TH-17 cells in rheumatoid arthritis. Arthritis Res Ther. 2008;10(4):R93. doi: 10.1186/ar2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd CE, Piguet O, Broe GA, Creasey H, Waite LM, Brooks WS, Kril JJ. Histocompatibility antigens, aspirin use and cognitive performance in non-demented elderly subjects. J Neuroimmunol. 2004;148(1–2):178–182. doi: 10.1016/j.jneuroim.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Silman AJ, Macgregor AJ, Thomson W, Holligan S, Carthy D, Farhan A, Ollier WE. Twin concordance rates for rheumatoid arthritis: results from a nationwide study. Br J Rheumatol. 1993;32(10):903–907. doi: 10.1093/rheumatology/32.10.903. [DOI] [PubMed] [Google Scholar]
- Stastny P. HLA-D and Ia antigens in rheumatoid arthritis and systemic lupus erythematosus. Arthritis Rheum. 1978;21(5 Suppl):S139–143. doi: 10.1002/art.1780210921. [DOI] [PubMed] [Google Scholar]
- Tait BD, Drummond BP, Varney MD, Harrison LC. HLA-DRB1*0401 is associated with susceptibility to insulin-dependent diabetes mellitus independently of the DQB1 locus. Eur JImmunogenet. 1995;22(4):289–297. doi: 10.1111/j.1744-313x.1995.tb00245.x. [DOI] [PubMed] [Google Scholar]
- Taneja V, Behrens M, Mangalam A, Griffiths MM, Luthra HS, David CS. New humanized HLA-DR4-transgenic mice that mimic the sex bias of rheumatoid arthritis. Arthritis Rheum. 2007;56(1):69–78. doi: 10.1002/art.22213. [DOI] [PubMed] [Google Scholar]
- Van Der Meer JH, Netea MG, Dinarello CA. Modulation of muramyl dipeptide stimulation of cytokine production by blood components. Clin Exp Immunol. 2009;156(3):428–433. doi: 10.1111/j.1365-2249.2009.03926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyand CM, Goronzy JJ. Disease mechanisms in rheumatoid arthritis: gene dosage effect of HLA-DR haplotypes. J Lab Clin Med. 1994;124(3):335–338. [PubMed] [Google Scholar]
- Wucherpfennig KW, Strominger JL. Selective binding of self peptides to disease-associated major histocompatibility complex (MHC) molecules: a mechanism for MHC-linked susceptibility to human autoimmune diseases. J Exp Med. 1995;181(5):1597–1601. doi: 10.1084/jem.181.5.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada R, Suzuki A, Chang X, Yamamoto K. Citrullinated proteins in rheumatoid arthritis. Front Biosci. 2005;10:54–64. doi: 10.2741/1506. [DOI] [PubMed] [Google Scholar]
- Yin L, Dai S, Clayton G, Gao W, Wang Y, Kappler J, Marrack P. Recognition of self and altered self by T cells in autoimmunity and allergy. Protein Cell. 2013;4(1):8–16. doi: 10.1007/s13238-012-2077-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zidek Z, Masek K. Edemagenic activity of aqueous form of muramyl dipeptide (MDP) in rats. Methods Find Exp Clin Pharmacol. 1985;7(8):419–426. [PubMed] [Google Scholar]
- Zinkernagel RM, Doherty PC. The discovery of MHC restriction. Immunol Today. 1997;18(1):14–17. doi: 10.1016/s0167-5699(97)80008-4. [DOI] [PubMed] [Google Scholar]