

Abstract
Since the discovery of the active metabolite of vitamin D, i.e., 1α,25-dihydroxyvitamin D3, there has been a continuous effort to synthesize analogs able to carry out many of the functions of the native hormone without raising serum calcium concentration. The present report provides a series of previously undescribed analogs wherein this goal is realized. We have prepared 2-methylene-19-nor-1α-hydroxyvitamin D analogs of 1,25-(OH)2D3 that possess only two to four carbons of the side chain without a hydroxyl thereon. Compared to 1,25-(OH)2D3, these analogs are slightly less active in binding to the vitamin D receptor, in causing HL-60 differentiation, and are slightly less active in in vitro transcription assays using the 24-hydroxylase promoter attached to a luciferase reporter gene. Of considerable importance is that these analogs, given to rats at daily doses of up to 70 μg/kg of body weight per day, are either unable or only slightly able to raise serum calcium concentration but are nevertheless able to suppress parathyroid hormone levels in plasma up to 100% and induce 24-hydroxylase mRNA in skin. Because of their ability to act in vivo without raising serum calcium levels, they may be of considerable interest for the systemic treatment of diseases such as psoriasis, cancer, and secondary hyperparathyroidism of renal failure, where a rise in serum calcium is undesirable.
Since the discovery that vitamin D must be metabolized to its biologically active form, 1α,25-dihydroxyvitamin D3 [1,25-(OH)2D3], before it can function, an intense synthetic effort has been placed on preparing analogs of the vitamin D hormone with the hope that these analogs may be selective in their activity and might be useful in treating specific disorders (1). Most notable among new analogs are 19-nor-1,25-dihydroxyvitamin D2 (Zemplar, Abbott) (2), 22-oxa-1α,25-dihydroxyvitamin D2 (3), MC-903 (calcipotriol) (4), 16-ene-23-yne-1α,25-dihydroxyvitamin D3 (5), and 24-difluoro-26,27-dimethyl-16-ene-1α,25-dihydroxyvitamin D3 (6). Calcipotriol has been successfully applied in topical treatment of psoriasis under the trade name of Dovonex (7), whereas the 22-oxa-1α,25-(OH)2D3 has been applied to renal osteodystrophy and psoriasis (3). These two compounds appear to be noncalcemic in their activity, primarily because they are rapidly metabolized and excreted (8, 9). Nevertheless, they have been quite useful in that their metabolic lability renders them considerably safer than the native hormone. Other analogs are also known to be metabolized rapidly (8, 9). The 19-nor-1α,25-dihydroxyvitamin D2 that is successfully marketed for renal osteodystrophy is also less calcemic but not because of rapid metabolism (10). Thus, the search continues for a vitamin D compound that remains in the circulation but is not effective in raising serum calcium, while still retaining noncalcemic activities. This paper reports a series of analogs that are clearly noncalcemic but are nevertheless systemically active.
Materials and Methods
Compounds. Structures of the 1,25-(OH)2D3 analogs are shown in Table 1. 2-Methylene-19-nor-1α-hydroxyhomopregnacalciferol (2MP), 2-methylene-19-nor-20(S)-1α-hydroxy-bishomopregnacalciferol (2MbisP), 2-methylene-19-nor-1α-hydroxypregnacalciferol (2Mpregna), and 1,25-(OH)2D3 were synthesized by Tetrionics (Madison, WI) according to methods devised previously in this laboratory and as modified by Tetrionics (11, 12). The compounds were ≥98% pure and had the following physical characteristics.
Table 1. Summary of in vitro assays of activity.
| VDR binding
|
HL60 differentiation
|
24OHase transcription
|
|||||
|---|---|---|---|---|---|---|---|
| Compound | Chemical structure | Ki, M | Ratio | EC50, M | Ratio | EC50, M | Ratio |
| 1α,25(OH)2D3 | 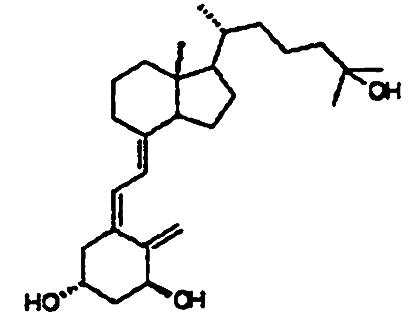 |
7 × 10-11 | 1 | 4 × 10-9 | 1 | 2 × 10-10 | 1 |
| 2MP | 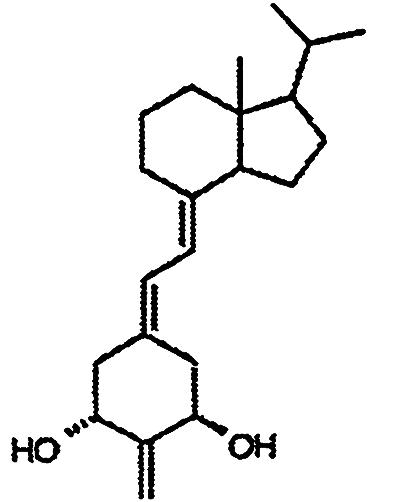 |
3 × 10-10 | 4.3 | 6 × 10-9 | 1.5 | 1 × 10-9 | 5 |
| 2MbisP | 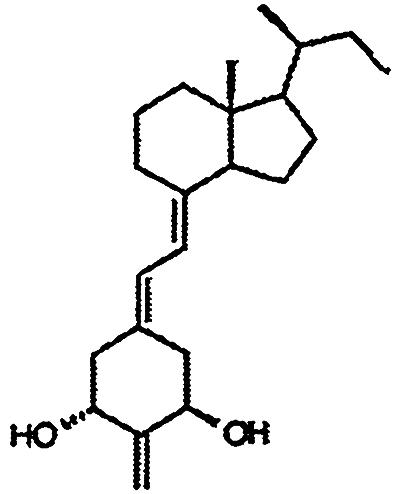 |
6 × 10-10 | 8.6 | 6 × 10-9 | 1.5 | 3 × 10-9 | 15 |
| 2Mpregna | 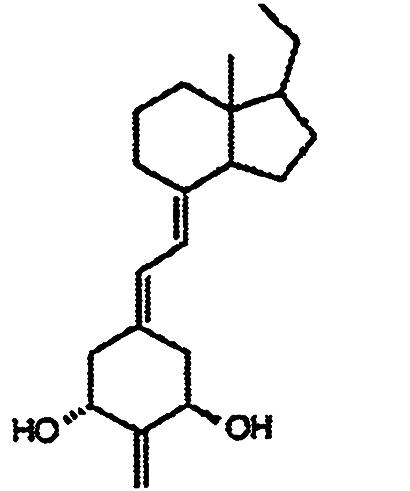 |
3 × 10-10 | 4.3 | 2 × 10-8 | 5 | 3 × 10-9 | 15 |
Two to three separate experiments with two to three replicates in each experiment were performed for all three in vitro assays.
2MP. UV (in ethyl alcohol, EtOH) λmax 244, 252, 262 nm; 1H NMR (CDCl3), 0.547 (3H, s, 18-H3), 0.94 (3H, d, J = 6.6 Hz, 21-H3), 0.86 (3H, d, J = 6.6 Hz, 22-H3), 1.87 (1H, m), 2.00 (2H, m), 2.29 (1H, dd, J = 13.0, 8.3 Hz, 10α-H), 2.34 (1H, dd, J = 13.2, 5.9 Hz, 4β-H), 2.58 (1H, dd, J = 13.2, 3.9 Hz, 4α-H), 2.82 (1H, br d, J = 13 Hz, 9β-H), 2.86 (1H, dd, J = 13.0, 4.6 Hz, 10β-H), 4.49 (2H, m, 1β- and 3α-H), 5.09 and 5.11 (1H and 1H, each s, = CH2), 5.89 and 6.36 (1H and 1H, each d, J = 11.3 Hz, 7- and 6-H); MS m/z (relative intensity) 330 (M+, 100), 287 (M+-C3H7, 22), 269 (M+-C3H7-H2O, 21), 251 (M+-C3H7-2·H2O, 16), 245 (27), 177 (41), 135 (62), 107 (42).
2MbisP. UV (in EtOH) λmax 244, 252, 262 nm; 1H NMR (CDCl3), 0.547 (3H, s, 18-H3), 0.833 (3H, d, J = 6.0 Hz, 21-H3), 0.835 (3H, t, J = 7.3 Hz, 23-H3), 1.87 (1H, m), 2.00 (2H, m), 2.29 (1H, dd, J = 13.2, 8.4 Hz, 10α-H), 2.34 (1H, dd, J = 13.4, 6.0 Hz, 4β-H), 2.58 (1H, dd, J = 13.1, 4.0 Hz, 4α-H), 2.82 (1H, br d, J = 13.3 Hz, 9β-H), 2.86 (1H, dd, J = 13.2, 4.4 Hz, 10β-H), 4.49 (2H, m, 1β- and 3α-H), 5.10 and 5.11 (1H and 1H, each s, = CH2), 5.89 and 6.37 (1H and 1H, each d, J = 11.1 Hz, 7- and 6-H); MS m/z (relative intensity) 344 (M+, 100), 315 (M+-Et, 11), 297 (M+-Et-H2O, 14), 287 (M+-C4H9, 29), 279 (M+-Et-2·H2O, 11), 269 (M+-C4H9-H2O, 25), 259 (45), 251 (M+-C4H9-2·H2O, 21), 191 (39), 135 (70), 107 (55).
2Mpregna. UV (in EtOH) λmax 244, 252, 262 nm; 1H NMR (CDCl3), 0.449 (3H, s, 18-H3), 0.89 (3H, t, J = 7.3 Hz, 20-H3), 1,95 (2H, m), 2.30 (1H, dd, J = 13.6, 8.1 Hz, 10α-H), 2.33 (1H, dd, J = 13.0, 5.8 Hz, 4β-H), 2.58 (1H, dd, J = 13.2, 3.9 Hz, 4α-H), 2.83 (1H, m, 9β-H), 2.86 (1H, m, 10β-H), 4.49 (2H, m, 1β- and 3α-H), 5.09 and 5.11 (1H and 1H, each s, = CH2), 5.89 and 6.36 (1H and 1H, each d, J = 11.2 Hz, 7- and 6-H); MS m/z (relative intensity) 316 (M+, 100), 287 (M+-Et, 16), 269 (M+-Et-H2O, 19), 251 (M+-Et-2·H2O, 5), 231 (76), 163 (56), 135 (61), 107 (88).
Because the chromophores of these three compounds are identical, a molar extinction coefficient of 42,000 at 252 nm was used in the computation of concentration of these compounds from UV absorption data (11). The compounds were dissolved in ethyl alcohol and, with the above molar extinction coefficient, the concentrations were computed for each of them. Aliquots were then taken and added to propylene glycol for i.p. injection.
Animals. Sprague–Dawley male or female rats were obtained from Harlan (Indianapolis), housed in suspended wire cages, and provided the indicated diets and water ad libitum. The diets used were based on diets used by Suda et al. (13) and were highly purified. All animal procedures were approved by the Institutional Review Board of the College of Agricultural and Life Sciences, University of Wisconsin, Madison.
Computation of the Ki of Analog and Vitamin D Receptor. Purified recombinant rat receptor was used as the binding protein (14). Competition for radiolabeled 1,25-(OH)2D3 binding to the recombinant receptor was carried out as described (15). The radiolabeled ligand was added to the receptor with and without increasing amounts of unlabeled analog. After incubation on ice for 16 h, the ligand–receptor complex was separated from free ligand by adsorption to hydroxylapatite as described (15). The hyroxylapatite containing the receptor protein bound to radiolabel was placed in a scintillation vial containing BioSafe-II (Research Products International) and the amount of radioactivity determined in a Packard TriCarb Scinillation Counter (model no. 2300TR). Ki values were calculated by using the Cheng and Prusoff equation (16), with a KD value of 0.2 nM for 1,25-(OH)2[3H]D3 binding to the 1,25-dihydroxyvitamin D3 receptor (VDR) (Table 1).
HL-60 Differentiation. HL-60 cells were seeded at 1 × 105 cells in each plate (60 × 15 mm). Increasing amounts of ligand were added, and differentiation was measured by nitroblue tetrazolinium reduction 4 days after ligand addition as described (17).
Transcriptional Assays. These assays were carried out with osteosarcoma cells 17/2.8 stably transfected with the 24-hydroxylase promoter in front of the luciferase reporter gene (18). The induction of luciferase in these cells after 16 h of exposure to the analog is compared to the active hormone (Table 1).
Intestinal Calcium Transport and Bone Calcium Mobilization. Weanling male rats were fed a 0.47% calcium, 0.3% phosphorus, vitamin D-deficient diet for 1 week, as described (13). They were then switched to a 0.02% calcium-containing diet for 3 weeks and then switched back to the 0.47% calcium, 0.3% phosphorus diet. After 1 week, they were returned to the 0.02% calcium diet, and the experiment was carried out after 1 additional week. The animals were placed into groups of five; the first group served as the vehicle control; the second, third, and fourth groups received increasing doses of the analog, whereas a fifth group received 1,25-(OH)2D3 at the dose level indicated. The same dose was administered every day for 4 days. At the end of this period and 24 h after the last dose, the animals were killed, and serum was taken for determination of calcium; the upper 10 centimeters of duodenum were removed, everted, and used for the measurement of intestinal calcium transport, as described (17). Because these animals were receiving a diet essentially devoid of calcium, rises in serum calcium reflect the calcium coming from bone.
Effect of High Doses of Analogs on Serum Calcium. Normal vitamin D-sufficient rats (adult female Sprague–Dawley) fed a 0.47% calcium, 0.3% phosphorus diet were maintained in the colony, and each day for 7 days, the dose of the indicated analog was provided i.p. in propylene glycol/ethanol (95:5). At various times during the experiment, the animals were weighed, and serum was taken for the determination of calcium.
Parathyroid Hormone (PTH) Suppression. Adult, vitamin D-sufficient, female Sprague–Dawley rats were fed the purified diet containing 0.47% calcium and supplemented with vitamin D (described above). The shortened side chain analogs were administered i.p. for 7 consecutive days and 1,25-(OH)2D3 for 4 consecutive days. Twenty-four hours after the last dose, rats were anesthetized with isofluorane, blood was collected, and the concentration of bioactive serum PTH was measured by using the rat BioActive Intact PTH ELISA kit from Immutopics (San Clemente, CA).
24-Hydroxylase mRNA in Skin. Adult vitamin D-sufficient Sprague–Dawley rats were fed the 0.47% calcium and 0.3% phosphorus purified diet (described above). 2MP was administered i.p. for 7 consecutive days or the native hormone for 4 consecutive days. Twenty-four hours after the last dose, skin from the dorsal side was harvested and immediately frozen in liquid nitrogen. Total RNA was isolated by using the method of Chomczynski and Sacchi (19), and quantitative RT-PCR was performed. Reverse transcription was done by using 10 μg of total RNA and avian myeloblastosis virus reverse transcriptase (Promega). PCR amplification was performed in the presence of SYBR green (FastStart DNA Master SYBR Green I, Roche Applied Science) by using the LightCycler (Roche Applied Science). Reactions for PCR amplification were set up with β-actin primers (5′-TTT GGC ACC ACA CTT TCT AC-3′ and 5′-AGG ATG GCA TGA GGG AGC GC-3′; 95°C denaturation for 15 sec, 63°C annealing for 5 sec, 72°C extension for 12 sec) to control for differences in reverse transcription efficiencies. In addition, because these primers span an intron, these reactions were also used to eliminate any samples containing contaminating genomic DNA. The primers used for quantitation of 24-hydroxylase mRNA were 5′-GCA TGG ATG AGC TGT GCG A-3′ and 5′-AAT GGT GTC CCA AGC CAG C-3′ (95°C denaturation for 15 sec, 55°C annealing for 5 sec, and 72°C extension for 30 sec).
Results
It is indeed quite remarkable that the abbreviated side chain analogs all bind to the VDR with only slightly lower affinity than does the native hormone (Table 1). This is quite surprising inasmuch as these compounds lack a 25-hydroxyl group and a large portion of the side chain, both of which are considered essential for receptor binding (20). These encouraging results led to a study of whether these compounds could induce differentiation of HL-60 cells. Although not quite as effective as 1,25-(OH)2D3, these three compounds are all active in inducing cell differentiation. A further test of the activity of these side chain analogs in transcription assays (Table 1) shows that they are all clearly effective, again indicating that neither the side chain nor the 25-hydroxyl group is required in the genomic mechanism. Taken together, these tests conducted in vitro and in culture indicate that these analogs retain significant activity in VDR binding, in the promotion of cell differentiation, and in the activation of VDR-mediated gene transcription.
A measurement of in vivo activity indicated that the shortened side chain analogs are either unable or less able to support intestinal calcium transport and are inactive in the mobilization of bone calcium, as revealed in vitamin D-deficient rats on a 0.02% calcium diet (Figs. 1 and 2). Doses of 2MbisP as high as 5.4 μg/kg of body weight did not lead to an increase in calcium transport relative to the vehicle-treated group. Although 2MP was clearly less active than 1,25(OH)2D3 in supporting intestinal calcium transport, it showed some slight activity at the highest dose tested in this assay (Fig. 1).
Fig. 1.

Intestinal calcium transport in vitamin D-deficient rats. Values shown are the mean ± standard error. Asterisks denote means statistically different from vehicle control animals (P < 0.05). Statistical analyses were done by performing ANOVA followed by pairwise comparison tests (Tukey's, Scheffé's, and Fisher's least significant difference tests). Those different from vehicle control animals in two of three pairwise comparison tests were deemed statistically significant.
Fig. 2.

Bone calcium mobilization in vitamin D-deficient rats. Values shown are the mean ± standard error. Asterisks denote means statistically different from vehicle control animals (P < 0.05). Statistical comparisons were performed as in Fig. 1.
To emphasize the lack of calcemic activity, Table 2 presents a daily dose study in vitamin D-sufficient female rats. Doses as high as 70 μg/kg per day of the 2MbisP and 2Mpregna failed to increase serum calcium concentration, whereas 2MP at 67 μg/kg per day causes significant hypercalcemia. In other studies, female rats given 2MP at 300 μg/kg per day orally for 30 days had serum calcium values in the normal range (data not shown). Thus, these compounds are very well tolerated even at high doses.
Table 2. Serum calcium levels in adult female rats.
| Compound | Dose level, μg/kg | Serum calcium, mg/dl |
|---|---|---|
| Vehicle | 9.3 ± 0.4 | |
| 1α,25(OH)2D3 | 0.2 | 10.6 ± 0.1* |
| 2MP | 0.8 | 9.5 ± 0.1 |
| 2.3 | 9.8 ± 0.3 | |
| 6.8 | 10.3 ± 0.1* | |
| 67 | 11.2 ± 0.2* | |
| 2MbisP | 0.8 | 9.2 ± 0.1 |
| 2.3 | 9.1 ± 0.1 | |
| 7.0 | 9.1 ± 0.1 | |
| 70 | 9.5 ± 0.1 | |
| 2Mpregna | 0.7 | 9.2 ± 0.1 |
| 2.2 | 8.9 ± 0.1 | |
| 6.5 | 9.0 ± 0.1 | |
| 64 | 9.3 ± 0.1 |
Statistical analysis was done by ANOVA followed by pairwise comparison tests (Tukey's, Scheffe's, and Fisher's least significant difference). Only those values different in two of three post hoc tests were deemed statistically different.
Values statistically different (P < 0.05) from vehicle control values
Of course, the important question is whether these compounds are active at all in vivo. Of great importance is the finding that these three substances are able to suppress circulating PTH levels to virtually zero in the case of 2MP and 2MbisP (Fig. 3). The 2Mpregna, although it causes a 60% suppression, was not quite as effective as the longer side chain analogs (Fig. 3).
Fig. 3.

PTH suppression in adult female rats. Values shown are the mean ± standard error. Asterisks denote means statistically different from vehicle control animals (P < 0.05). Statistical comparisons were performed as in Fig. 1.
To gather further information on systemic activity of the analogs, we studied the induction in vivo of the 24-hydroxylase in keratinocytes by 2MP vs. 1,25-(OH)2D3 (Fig. 4). The results demonstrate that 2MP given i.p. is able to stimulate 24-hydroxylase in vivo in the keratinocyte although not as active as 1,25-(OH)2D3 (data not shown).
Fig. 4.

24-OHase induction in the skin of adult female rats. Values shown are the mean ± standard error. An asterisk denotes means statistically different from vehicle control animals (P < 0.05). Statistical comparisons were performed as in Fig. 1.
These two results suggest that the shortened side chain analogs are systemically active despite the fact that they have no 25-hydroxyl group.
The remarkable lack of calcium elevation by either 2Mpregna or 2MbisP led us to consider whether these compounds even at very high doses can stimulate the genes responsible for raising serum calcium by either an intestinal activity or bone activity. These results show that even as high as 70 μg/kg, the 2-methylene-20(S)-bishomo-1α-hydroxypregnacalciferol is unable to raise serum calcium concentration. Because these doses are well above those found to suppress PTH, it is indeed obvious that this compound is, in fact, selective in its activities. Thus, it is unable to stimulate intestinal calcium transport or bone calcium mobilization, while being able in vivo to suppress PTH in the plasma. The 2Mpregna is also unable to raise serum calcium but is much less effective in suppressing PTH than is the 2MbisP analog. Although 2MP is fully active in suppressing PTH, it is able to raise serum calcium presumably by stimulating intestinal calcium transport.
Discussion
This unique set of analogs of 1,25-(OH)2D3 have three very important features. The first and foremost is that these analogs are able to act even though they have no 25-hydroxyl group and, secondly, have very little side chain. Third, these analogs bind to the receptor, are able to cause cellular differentiation in vitro, and, of considerable importance, they can induce 24-hydroxylase mRNA in vivo in the keratinocyte and suppress secretion of PTH into the blood stream, while having minimal or no ability to activate calcium mobilization. Exactly how the analogs accomplish this array of activities remains to be determined. Very likely they bind to the receptor but must produce different conformational changes in the VDR that either exclude binding a coactivator(s) necessary for calcium mobilization or render the VDR unable by whatever mechanism to stimulate the genes responsible for movement of calcium. On the other hand, both 2MbisP and 2MP are able to induce complete suppression of PTH, while at least 2MP is able to induce the 24-hydroxylase mRNA in the keratinocyte. These properties suggest that these analogs may also be ideal compounds for the treatment of renal osteodystrophy in that (i) they either do not or minimally support intestinal calcium absorption; (ii) they do not support bone calcium mobilization; and (iii) they either do not raise serum calcium levels or at least have little such activity. Further, in unreported results, they appear to lack the ability to support phosphate absorption in the small intestine.
It is of considerable importance that these compounds are able to cause HL-60 differentiation and to suppress growth of those cells. Their application to diseases such as cancer is suggested from these results. Thus, high doses can be used without danger of hypercalcemia, unlike many of the analogs studied for the treatment of cancer to date. Finally, their use in the treatment of autoimmune diseases and psoriasis may also be suggested, because the elimination of the unwanted hypercalcemic activity may allow their safe use at high doses.
The mechanism of selectivity of these shortened side chain noncalcemic analogs remains to be determined. That they bind to the VDR and are able to be transported to keratinocytes and the parathyroid glands suggests that these compounds are selective at the genetic and not the metabolic level. Working out the mechanism whereby this selectivity occurs would be of considerable interest. In the meantime, the possibility that these compounds may be developed for treatment of diseases certainly warrants additional investigation.
Acknowledgments
This work was supported in part by funds from the Wisconsin Alumni Research Foundation.
Abbreviations: 1,25-(OH)2D3, 1,25-dihydroxyvitamin D3; 2Mpregna, 2-methylene-19-nor-1α-hydroxypregnacalciferol; 2MP, 2-methylene-19-nor-1α-hydroxyhomopregnacalciferol; 2MbisP, 2-methylene-19-nor-20(S)-1α-hydroxy-bishomopregnacalciferol; VDR, 1,25-dihydroxyvitamin D3 receptor; PHT, parathyroid hormone.
References
- 1.Jones, G., Strugnell, S. A. & DeLuca, H. F. (1998) Physiol. Rev. 78, 1193-1231. [DOI] [PubMed] [Google Scholar]
- 2.Slatopolsky, E., Finch, J., Ritter, C., Denda, M., Morrissey, J., Brown, A. & DeLuca, H. (1995) Am. J. Kidney Dis. 26, 852-860. [DOI] [PubMed] [Google Scholar]
- 3.Kubodera, N., Sato, K. & Nishii, Y. (1997) in Vitamin D, eds. Feldman, D., Glorieux, F. H. & Pike, J. W. (Academic, New York), Vol. 63, pp. 1071-1086. [Google Scholar]
- 4.Calverley, M. J. (1987) Tetrahedron Lett. 43, 4609-4619. [Google Scholar]
- 5.Uskokovic, M. R., Studzinski, G. P. & Reddy, S. G. (1997) in Vitamin D, eds. Feldman, D., Glorieux, F. H. & Pike, J. W. (Academic, New York), Vol. 62, pp. 1045-1070. [Google Scholar]
- 6.Kensler, T. W., Dolan, P. M., Gange, S. J., Lee, J.-K., Wang, Q. & Posner, G. H. (2000) Carcinogenesis 21, 1341-1345. [PubMed] [Google Scholar]
- 7.Binderup, L., Binderup, E. & Godfredsen, W. O. (1997) in Vitamin D, eds. Feldman, D., Glorieux, F. H. & Pike, J. W. (Academic, New York), Vol. 61, pp. 1027-1043. [Google Scholar]
- 8.Jones, G. (1997) in Vitamin D, eds. Feldman, D., Glorieux, F. H. & Pike, J. W. (Academic, New York), Vol. 58, pp. 973-994. [Google Scholar]
- 9.Brown, A. J. & Slatopolsky, E. (1997) in Vitamin D, eds. Feldman, D., Glorieux, F. H. & Pike, J. W. (Academic, New York), Vol. 59, pp. 995-1009. [Google Scholar]
- 10.Shankar, V. N., Propp, A. E., Schroeder, N. S., Surber, B. W., Makin, H. L. J. & Jones, G. (2001) Arch. Biochem. Biophys. 387, 297-306. [DOI] [PubMed] [Google Scholar]
- 11.Sicinski, R. R., Prahl, J. M., Smith, C. M. & DeLuca, H. F. (1998) J. Med. Chem. 41, 4662-4674. [DOI] [PubMed] [Google Scholar]
- 12.DeLuca, H. F., Gowlugari, S. & Sicinski, R. R. (2003) U.S. patent application P02396.
- 13.Suda, T., DeLuca, H. F. & Tanaka, Y. (1970) J. Nutr. 100, 1049-1052. [DOI] [PubMed] [Google Scholar]
- 14.Vanhooke, J. L., Benning, M. M., Bauer, C. B., Pike, J. W. & DeLuca, H. F. (2004) Biochemistry 43, 4101-4110. [DOI] [PubMed] [Google Scholar]
- 15.Dame, M. C., Pierce, E. A., Prahl, J. M., Hayes, C. E. & DeLuca, H. F. (1986) Biochemistry 25, 4523-4534. [DOI] [PubMed] [Google Scholar]
- 16.Cheng, Y. & Prusoff, W. H. (1973) Biochem. Pharmacol. 22, 3099-3108. [DOI] [PubMed] [Google Scholar]
- 17.Perlman, K., Kutner, A., Prahl, J., Smith, C., Inaba, M., Schnoes, H. K. & DeLuca, H. F. (1990) Biochemistry 29, 190-196. [DOI] [PubMed] [Google Scholar]
- 18.Arbour, N. C., Ross, T. K., Zierold, C., Prahl, J. M. & DeLuca, H. F. (1998) Anal. Biochem. 255, 148-154. [DOI] [PubMed] [Google Scholar]
- 19.Chomczynski, P. & Sacchi, N. (1987) Anal. Biochem. 162, 156-159. [DOI] [PubMed] [Google Scholar]
- 20.Eisman, J. A. & DeLuca, H. F. (1977) Steroids 30, 245-257. [DOI] [PubMed] [Google Scholar]