

Abstract
Prostate cancer (CaP) is the most commonly diagnosed malignancy in males in the Western world with one in six males diagnosed in their lifetime. Current clinical prognostication groupings use pathologic Gleason score, pre-treatment prostatic-specific antigen and Union for International Cancer Control-TNM staging to place patients with localized CaP into low-, intermediate- and high-risk categories. These categories represent an increasing risk of biochemical failure and CaP-specific mortality rates, they also reflect the need for increasing treatment intensity and justification for increased side effects. In this article, we point out that 30–50% of patients will still fail image-guided radiotherapy or surgery despite the judicious use of clinical risk categories owing to interpatient heterogeneity in treatment response. To improve treatment individualization, better predictors of prognosis and radiotherapy treatment response are needed to triage patients to bespoke and intensified CaP treatment protocols. These should include the use of pre-treatment genomic tests based on DNA or RNA indices and/or assays that reflect cancer metabolism, such as hypoxia assays, to define patient-specific CaP progression and aggression. More importantly, it is argued that these novel prognostic assays could be even more useful if combined together to drive forward precision cancer medicine for localized CaP.
BACKGROUND: THE NEED FOR NOVEL BIOLOGICAL END POINTS FOR PROSTATE CANCER PROGNOSIS
Prostate cancer (CaP) is the most commonly diagnosed malignancy in males in the Western world, as >500 000 cases are diagnosed annually and 1 in 34 will die of metastatic disease.1 Treatment options for localized CaP depend on the Union for International Cancer Control-TNM staging [i.e. extent of tumour (local), nodal and distant metastatic cancer burden] of the disease. Using the clinical prognostic variables, local T-category, pre-treatment serum prostate-specific antigen (PSA) and pathologic Gleason score (GS; usually ranging from 5–10) males with localized CaP (e.g. T1–T4N0M0) are placed in low-, intermediate- and high-risk prognostic groups.2,3 These risk groups predict for biochemical failure based on a post-treatment rise in PSA (also referred to as biochemical relapse-free rate) and CaP-specific mortality (PCSM) after local therapies with curative intent.2–4
Active surveillance (AS) is a treatment option for low-risk and probably indolent CaPs, which have PSA values <10 ng ml−1 associated with small volume of GS6 or less in patients' diagnostic biopsies.5,6 Radical prostatectomy or radiotherapy [RT; using either external beam RT (EBRT) or brachytherapy] constitutes the major treatment options for non-indolent intermediate-risk CaP (e.g. T1–T2 lesions, PSA <20 ng ml−1 and GSs 6 or 7; Figure 1). The final choice of treatment will depend on patient preference and other considerations (e.g. operative risk, comorbidities, obstructive urinary symptoms, contraindications to RT etc.).1 However, patients with high-risk or locally advanced disease (e.g. T3–T4 lesions outside the prostate gland and/or GSs ≥8 and/or PSA values >20 ng ml−1) undergo combined modality treatment consisting of either adjuvant or salvage RT following surgery to offset local failure, or undergo combined use of EBRT with androgen deprivation therapy (ADT) to offset the risk of subclinical metastases.1,7,8 In males who develop castrate-resistant and metastatic disease (Figure 1), palliative options include systemic treatment using ADT (luteinizing hormone-releasing hormone agonists/antagonists with secondary hormonal manipulation using enzalutamide or abiraterone), chemotherapy (using docetaxel or cabazitaxel), systemic radionuclides (Radium-223), immunotherapy (Sipuleucel-T) and/or targeted palliative RT (e.g. 8 Gy single dose or 20–30 Gy in daily fractions).9,10 Despite a multitude of treatment options, there are no individualized clinical tests that absolutely tell which patients are unlikely to fail local treatment from those patients who are most likely to fail local treatment within a given clinical risk category. This problem is illustrated by the fact that despite the use of stringent clinical criteria to place patients into clinical prognostic groups, 30–50% of males can still fail precision RT or surgery owing to local resistance and/or systemic spread.1–3 Despite the publication of Phase III dose-escalated EBRT clinical trials in CaP designed to counteract failure due to CaP radioresistance, none of these trials have shown benefit in decreasing PCSM.8 The lack of an effect on survival with EBRT dose escalation can be explained by the fact that in a significant proportion of patients, treatment failure is due to the presence of occult systemic disease rather than local resistance, and that these patients need to be treated with intensification of systemic therapy not EBRT dose intensification, to decrease CaP mortality.1,8 Personalized CaP medicine therefore requires genomic- or biology-based biomarkers, in addition to existing clinical biomarkers, to explain interpatient heterogeneity in outcomes. Furthermore, even if an increased probability of occult metastases can be predicted, even more biomarkers will be required to favour the use of one systemic agent vs another, let alone the scheduling of these agents relative to each other (Figure 1).10
Figure 1.
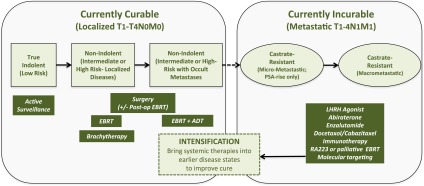
Curative and non-curative states in prostate cancer. Localized prostate cancers (CaPs) can be divided into low-, intermediate- and high-risk (including locally advanced) groups using T-category, pre-treatment prostate-specific antigen (PSA) level and the pathologic Gleason score. These groups have increasing probability of CaP-specific mortality. Low-risk tumours can be aggressively followed using active surveillance. By contrast, intermediate-risk tumours are treated with surgery, external beam radiotherapy (EBRT) or brachytherapy. In cases where a local recurrence occurs after surgery, patients can be treated with post-operative EBRT and convert a local failure into a cure. In high-risk CaP, there is an increased probability for occult systemic metastases, therefore systemic androgen deprivation [androgen-deprivation therapy (ADT)] is used in combination with EBRT. Palliative systemic therapy is the mainstay for patients with castrate-resistant disease in the micrometastatic or macrometastatic stages to increase progression-free survival by months. These therapies include additional ADT (including the use of newer agents, such as abiraterone and enzalutamide), chemotherapy, immunotherapy, systemic radionucleotides (RA233) and use of bespoke molecular-targeted agents. It is argued that an understanding of the genomic and microenvironmental factors that lead to occult metastases could drive intensification protocols using systemic agents in the localized CaP setting to improve the cure rates with radiotherapy and surgery. LHRH, luteinizing hormone-releasing hormone; Post-op, post-operative; RA223, radium-223.
An additional complication to personalized medicine is the knowledge that many low-risk CaPs are indolent and that their overtreatment results in significant morbidity.4,5,11 For example, two-thirds of low-risk CaPs have an indolent course that can be followed without radical treatment when appropriately placed into AS protocols, thereby preventing the side effects and costs of RT or surgery. The corollary is that one-third of these low-risk patients are being inaccurately classified as having indolent cancers and require treatment.5 On an individual basis, there are no assays that can predict with confidence the need for therapy in low-risk CaP.
So, how do we move forward in precision medicine for CaP using precision RT when faced with such clinical conundrums? One approach is to take advantage of technological advances in genomic medicine to determine patient-specific CaP genomics that reflect tumour progression and metastatic disease in addition to novel biology.12,13 State-of-the-art whole-genome sequencing technologies have the capacity for generating a breathtaking amount of genomic data (in excess of 10 billion bases per day) at a fraction of the cost than a decade ago. DNA- and RNA-based prognostic tests to predict CaP recurrence are being actively developed within the industry and academia for clinical use. Finally, there is also a rich history in radiation oncology for characterizing the tumour microenvironment, including assays for subregions of hypoxia within localized CaPs, which have a prognostic impact. This article will now discuss the potential of genomic and hypoxia assays to help attain the goal of implementing precision cancer medicine for patients undergoing curative RT for CaP.
TUMOUR HYPOXIA MEASUREMENTS ARE PROGNOSTIC IN LOCALIZED PROSTATE CANCER
The tumour microenvironment is characterized by subregions of nutrient deprivation, low extracellular pH, high interstitial fluid pressure and hypoxia. Hypoxic areas arise in tumours when oxygen consumption rate exceeds that of supply. The blood vessels within a tumour microenvironment are usually irregularly organized and have abnormal architecture such that tumours will contain regions where the partial oxygen concentration (pO2) is significantly <5 mmHg (i.e. normal tissues range from 10 to 80 mmHg).14 Tumour cells that lie beyond the diffusion distance for oxygen (>100 µm away from blood vessels) can quickly outstrip blood supply and are exposed to chronically low oxygen tensions for hours to days; this is often referred to as “chronic hypoxia”.14,15 Tumour cells remain hypoxic until they die (due to lack of oxygen or nutrients) or are reoxygenated. Hypoxia can also be transiently “acute” or “cycling” due to acute perfusion changes in the tumour vasculature.14,15 Tumours therefore contain a mixture of acute (cycling) and chronic hypoxia subregions with varying biology and varying effects on tumour cell radiosensitivity and genomic stability.14
Intratumoural hypoxia limits the effectiveness of RT and chemotherapy. Cells that are hypoxic or anoxic are usually two to three times more resistant to ionizing radiation when compared with oxic cells unless they are DNA repair deficient (see hypoxia section below).14 Chemotherapy-related tumour cell kill in this scenario is also limited owing to poor drug distribution and decreased tumour cell proliferation that limits the effectiveness of S-phase-specific chemotherapuetics.14 Therapy-resistant cells can adapt to hypoxic regions and result in cycles of selection for aggressive mutator phenotypes with faulty DNA repair and increased genetic instability.15,16 Pre-clinical data have also linked tumour cell hypoxia to increased experimental and spontaneous metastasis.14 Metastasis is a multistep process that involves intravasation through the basement membrane and extracellular matrix into the host vasculature, extravasation through vessel walls and then forming a new nidus with the organ of metastatic spread, and then tumour angiogenesis during secondary tumour growth. Indeed, hypoxia alters metastasis gene expression including that of E-cadherin (cell–cell contact), urokinase-type plasminogen activator receptor (degradation of extracellular matrix proteins), hepatocyte growth factor (cellular motility) and vascular endothelial growth factor (VEGF; angiogenesis and vascular permeability).14 Therefore, pre-clinical data support two general aspects relating to the resistance of hypoxic tumours during RT: increased local tumour cell radioresistance1 and/or an increased capacity for systemic metastases.2
Clinical studies that have attempted to directly measure the level of hypoxia in CaP have used pO2 electrodes, hypoxia imaging [positron emission tomography (PET)] and immunohistochemistry (IHC)17–25 (Table 1). These studies support the concept that hypoxic subregions exist within localized CaPs and are associated with higher rates of biochemical failure following surgery or RT. For example, a prospective clinical trial showed that localized CaPs have uptake of the hypoxia biomarker, pimonidazole, and that this uptake correlated with GS but not vascularity.18 Turaka et al23 showed that decreased prostate-to-muscle oxygen ratio was an important predictor of early biochemical recurrence following brachytherapy secondary to occult metastases at the time of treatment. In the largest clinical study of CaP hypoxia using direct pO2 measurements, Milosevic et al21 showed that hypoxia is associated with both early biochemical relapse and local recurrence in the prostate gland.
Table 1.
Clinical studies of hypoxia assays and prognosis in localized prostate cancer
| Study | N | T-category | Assay | Prognostic value and details |
|---|---|---|---|---|
| Turaka et al23 | 57 | cT1–3 | pO2 probe | Prognostic: lower prostate/muscle pO2 ratio predicted early biochemical failure after brachytherapy |
| Milosevic et al21 | 247 | cT1–2 | pO2 probe | Prognostic: largest study showing that hypoxia predicted early biochemical relapse after radiotherapy and local recurrence |
| Vergis et al24 | 201 (RT); 289 (surgery) | cT1–3 | IHC-VEGF, HIF-1α, OPN | Prognostic: increased expression of VEGF, HIF-1α and, for patients treated with surgery, OPN identified patients at high risk of biochemical failure |
| Carnell et al18 | 43 | cT1–3 | IHC-PIMO | Not tested, but a positive correlation of PIMO +3 binding with Gleason score was demonstrated |
| Boddy et al17 | 149 | cT1–3 | IHC-VEGF, HIF-1α | Not prognostic: there was a significant correlation between HIF-1α and HIF-2α expression, and with AR and VEGF expression. VEGF was also significantly related to the androgen receptor, whereas PHD2 was inversely related to HIF-2α expression. No significant association was shown between HIF-1α or HIF-2α and time to PSA recurrence |
| Green et al20 | 50 | cT3 | IHC | Prognostic: high VEGF expression was associated with lower disease-specific survival |
| Thoms et al22 | 199 (T1–3); 37 (M1) | cT1–T3 | ELISA-OPN | Not prognostic: within localized prostate cancers plasma OPN was not predictive of more aggressive disease or response to radiotherapy or hormone therapy |
| Weber et al25 | 103 | cT1–3 | IHC | Prognostic: high nuclear expression of HIF-1α and low EGFR expression was associated with a good prognosis in patients treated with RT ± ADT |
| Garcia-Parra et al19 | 14 | pT2b–T3a | PET-FAZA + IHC | Not prognostic: negative 18F-FAZA accumulation and CAIX staining in primary prostate cancer despite documented large lesions (up to 4 cm). HIF-1 staining was positive and independent of Gleason score |
ADT, androgen-deprivation therapy; AR, androgen receptor; CAIX, carbonic anhydrase IX; cT, clinical T-category; EGFR, epidermal growth factor receptor; ELISA, enzyme-linked immunosorbent assay; 18F-FAZA, 18F-labelled fluoroazomycin arabinoside; HIF-1, hypoxia-induced factor 1; IHC, immunohistochemistry; OPN, osteopontin; PET, positron emission tomography; PHD, prolyl hydroxylase enzyme; PIMO, pimonidazole; pO2, partial oxygen concentration; PSA, prostate-specific antigen; pT, pathologic T-category; RT, radiotherapy; VEGF, vascular endothelial growth factor.
pO2, measured with pO2 electrode.
Hypoxia leads to an upregulation of the transcription factor, hypoxia inducible factor 1α (HIF-1α), which in turn can increase the expression of downstream proteins such as VEGF, carbonic anhydrase IX (CAIX), glucose transporter 1 (GLUT-1) and osteopontin (OPN).22,26 Vergis et al24 used an IHC-based approach to show that HIF-1α, VEGF and OPN (for surgical patients) and HIF-1α and VEGF (for RT patients) predicted biochemical failure independent of the clinical tumour stage, GS, serum PSA and RT dose. However, a study from the University of Michigan (Ann Arbor, MI) using PET-fluoroazomycin arabinoside analogue (FAZA; as a hypoxia marker) failed to show FAZA uptake or CAIX staining in CaP, suggesting that these biomarkers have little utility in prognostication.19 When taken together, the observation of a low pO2 predicting early failures in the first 2 years following either RT or surgical treatment suggests that hypoxia is associated with a metastatic phenotype.21,23,24 Such hypoxic tumours will require treatment intensification (discussed below) when using EBRT to offset both local radioresistance and systemic metastases.
Despite these data, hypoxia-targeted therapy is still not a standard current cancer treatment. Agents that may be useful in this context might include the use of ADT (given the ability of androgen suppression to improve CaP oxygenation), molecular-targeted agents or hypoxia-targeted systemic agents.15 Importantly, as we will see below, hypoxia can also drive genetic instability by inhibiting DNA repair.27 Understanding these additional consequences of the hypoxic microenvironment on the development of genetic instability may give novel treatment approaches to combat hypoxia-associated resistance.
GENETIC INSTABILITY AND GENOMIC ASSAYS IN LOCALIZED PROSTATE CANCER
Chromosomal instability and aneuploidy are associated with cancer progression and adverse prognosis in CaP.28–32 Patients with tetraploid or aneuploid CaP tumours have increased mortality following radical prostatectomy when compared with patients whose CaP tumours are diploid.30 The specific genomic events that might link to this aspect of aggression are now being understood in the context of abnormal gene copy number loss, gene mutation and abnormal gene expression relating to oncogenes and tumour suppressor genes. Hypothesis-based studies have used array comparative genomic hybridization (using DNA from diagnostic CaP biopsies prior to therapy) to associate specific gene copy number alterations with prognosis following EBRT or radical prostatectomy. Copy number loss of the tumour suppressor genes novel human prostate-specific, androgen-related homeobox gene (NKX3.1) or Phosphatase and tensin homologue (PTEN) or the androgen synthesis genes steroidogenic acute regulatory protein (StAR) and hydroxysteroid (17-beta) dehydrogenase 2 (HSD17B2) are novel and independent genomic prognostic factors (hazard ratio ranges from 2 to 4 for failing local therapy). When associated with copy number gain of the proto-oncogene cMYC,33–35 NKX3.1 loss was also associated with local radioresistance. Furthermore, males who carry germ-line mutations in the breast cancer 2, early onset (BRCA2) tumour suppressor gene develop aggressive CaPs (higher GSs and increased nodal metastases) that have very poor prognosis following surgery or RT.36,37 Surprisingly, the presence or absence of a fusion gene between transmembrane protease serine 2 and the ETS-related gene (TMPRSS2–ERG) fusion (found in >50% of all CaP patients) is not prognostic.38,39
Unbiased genome-wide signatures based on DNA or RNA indices have recently been developed to predict PSA recurrence in the post-operative setting. A set of DNA-based biomarkers (genomic evaluators of metastatic CaP) has improved utility over the sole use of clinical recurrence nomograms (e.g. Kattan nomogram) in the prediction of recurrence following surgery.40 Recent evidence also suggests that miRNAs may also have prognostic value, although only a limited number of studies have correlated genome-wide analysis of miRNA species with differential prognosis.41–43 High levels of miR-96 were prognostic of biochemical recurrence in a series of 155 radical prostatectomies.41 Similarly, others have noted an independent prognostic value (e.g. independent from currently utilized clinical parameters of PSA, T-category and GS) for miR-191, miR-145, miR-100 and miR-122, many of which are arranged in genomic clusters.42,43
Major research efforts have led to the development of RNA-based signatures to better stratify patients between indolent vs aggressive CaPs (Table 2). A 31-gene signature of biochemical recurrence following radical prostatectomy has been reported based on RNA expression of cell cycle progression genes;47 this prognostic assay was also validated in the setting of recurrence following EBRT.49,50 A 22-gene RNA expression panel has been similarly validated across multiple independent cohorts by several groups.45,46,51 Likewise, Wu et al48 have reported a 32-gene RNA expression signature that is prognostic of biochemical recurrence and metastatic disease following radical prostatectomy. Using DNA-based indices, Taylor et al44 have shown independent prognostic utility for six genetic clusters that are prognostic for biochemical recurrence, independent of the GS.
Table 2.
DNA- and RNA-based prognostic signatures for localized prostate cancer
| Signature (DNA or RNA) | Signature development cohort | Outcomes predicted | Validation in separate cohorts (yes/no) | Evaluated in other treatment modality cohorts | References |
|---|---|---|---|---|---|
| DNA-based CNAs for NKX3.1, PTEN, cMYC, StAR | Pre-radiotherapy | BCR | No | Yes: surgery | Zafarana et al35; Locke et al33,34 |
| DNA-based CNA clusters (6 clusters) | Post-surgery recurrence | BCR | No | No | Taylor et al44 |
| RNA 22-gene expression signature | Post-surgery recurrence | M, PCSS, OS | Yes (two cohorts) | No | Cooperberg et al45; Erho et al46 |
| RNA 31-gene expression signature | Post-surgery recurrence post-TURP recurrence | BCR, PCSS | Yes (three cohorts) | Yes (conservatively managed and radiotherapy) | Cuzick et al47 |
| RNA 32-gene expression signature | Post-surgery recurrence | BCR, M | Yes (one cohort) | No | Wu et al48 |
BCR, biochemical recurrence; cMYC, proto-oncogene cMYC; CNA, gene copy number; M, metastases; NKX3.1, novel human prostate-specific, androgen-regulated homeobox gene; OS, overall survival; PCSS, prostate specific-cancer survival; PTEN, phosphatase and tensin homolog; TURP, transurethral resection of the prostate.
Additional DNA epigenetic modifications may drive individualized CaP biology and progression. Using high-throughput genome sequencing and DNA methylation analyses, >147 000 cancer-associated epigenetic alterations were observed in 51 tumour and 53 benign prostate samples; the specific nature of these alterations were dependent on the presence of absence of a TMPRSS2–ERG rearrangement.52 This observation of differential methylation events in fusion-negative tumours (based on enhancer of zeste homologue 2 (EZH2) gene activation) explains CaP carcinogenesis in the 50% of patients who are TMPRSS2–ERG fusion-negative. Systematic overviews of studies for the prognostic role of specific gene methylation (e.g. GSTPi, APC, RAR-β, RASSF1A, PITX2, CCND2, EDNRB and HOX family of genes) in CaP concluded that their prognostic roles are still unknown and require further validation in large clinical cohorts.53,54
Integrating genome-, epigenome-, transcriptome- and proteome-wide data sets to iterate a multimodal genetic test will no doubt improve subgroup prognostication.55,56 Therefore, similar to hypoxia assays, patient-specific indices of genetic instability may be utilized to further define aggressive subsets of CaP for treatment intensification27 (Figure 2). But is there a biological link between genetic instability and hypoxia that leads to adverse prognosis? We will now discuss the potential interplay between hypoxia, DNA repair and genetic instability.
Figure 2.

Combining genomics and hypoxia assays to drive personalized prostate cancer medicine. Genomic signatures (DNA, RNA, epigenetic or miRNA-based) could be combined with hypoxia assays (using imaging such as positron emission tomography–fluoroazomycin arabinoside or intrinsic/extrinsic markers in situ) to triage patients with low probability of systemic metastases to local treatment alone and patients with high probability of metastases to local treatment plus systemic treatment (e.g. combined modality therapy). Systemic treatments could include those shown in Figure 1 that are currently used for metastatic disease, hypoxia-specific cytotoxins or novel agents designed to target abnormal signalling or DNA repair pathways based on susceptibility biomarkers. ADT, androgen-deprivation therapy; EBRT, external beam radiotherapy; PARP1, poly(ADP-ribose) polymerase; TIC, tumour initiating cell.
HYPOXIA AND GENOMIC INSTABILITY IN PROSTATE CANCER: A NOVEL THERAPEUTIC TARGET?
One model of interaction between the CaP tumour microenvironment and CaP genomics is that hypoxic tumour cells have down-regulated DNA repair function (e.g. decreased capacity for the repair of DNA double strand breaks; DSBs) in addition to any genetic instability due to oncogene activation or tumour suppressor gene inactivation.14,27 Such repair-deficient hypoxic tumour cells could adapt to low oxygen levels and acquire an aggressive “mutator” phenotype leading to treatment resistance and metastases.27 For example, there are two major pathways of DSB repair: non-homologous end-joining (NHEJ; active throughout the cell cycle) and homologous recombination (HR; requiring a homologous chromosome available only during S and G2 phases of the cell cycle).57 Hypoxia causes decreased transcription and translation of a series of HR and NHEJ genes, including Rad51, BRCA1, BRCA2 and DNA–protein kinase catalytic subunit (DNA-PKcs). Furthermore, functional studies using isogenic cells have reported hypoxia-induced NHEJ and HR defects.15,16,58–60 As a consequence, despite lower levels of initial DSB formation following ionizing radiation (IR) in hypoxic tumour cells, hypoxia-induced defects in DSBs repair increased the level of unrepaired DSBs and chromosomal aberrations at the first mitosis post-irradiation.27,59 If even a fraction of these hypoxic mutant tumour cells survive subsequent cell division, unstable genetic mutants could undergo clonal selection.
Hypoxia induces activation of common fragile sites throughout the genome (i.e. chromosomal regions prone to breakage) and down-regulation of DSB repair has been implicated as an underlying mechanism to this chromosomal fragility.61 The compromise in DSB resolution in hypoxic cells and the resulting increase in chromosome aberrations is a “perfect storm” towards genomic instability in tumour cells that adapt and continue to proliferate under low oxygen conditions.27 Hypoxia can also lead to a decrease in function of other DNA repair pathways, including that of mismatch repair (MMR), nucleotide excision repair and the Fanconi anaemia (FA) pathway.62–65 The parallel reduction in various DNA damage repair pathways can all potentially contribute to the acquisition of aggressive tumour phenotypes.27
The genetic instability in hypoxic cells would at first seem a complex phenotype to target with standard or molecularly targeted therapies. Any clinical approach would require careful assessment of the tumour microenvironment and genomic status (using assays mentioned in previous sections) to incorporate both hypoxia and genomic assessment as part of a standard of care. However, the repair-defective phenotype might just be the undoing of the hypoxic resistance phenotype, providing an opportunity to specifically target hypoxic tumour cells and improve the therapeutic index.27 Hypoxic tumour cells can be directly targeted using cytotoxic agents that induce DNA damage only under low oxygen; these include the bioreductive drugs, tirapazamine and apaziquone.66,67 A newer drug, TH-302 (a 2-nitroimidazole triggered hypoxia-activated cytotoxin) directly decreases the hypoxic fraction in xenografts of varying histology and is undergoing Phase II–III clinical trials in combination with chemotherapy.68 Additional targeting of hypoxic subregions can be achieved by targeting HIF-1-dependent transcription or targeting the unfolded protein response, which controls gene translation under cellular stress (e.g. targeting the mechanistic target of rapamycin (mTOR) signalling pathway).69–71
One could also target the faulty DNA repair and genetic instability in hypoxic cells using the concept of “contextual” synthetic lethality.72 “Genetic” synthetic lethality is a concept first developed using yeast genetics in which mutations in two genes (e.g. gene A and gene B) result in cell death, while a mutation in only one gene (e.g. either gene A or gene B) results in cell viability.73 This concept has been successfully used to target tumours deficient in HR (e.g. BRCA1- or BRCA2-deficient ovarian cancer, breast cancer and CaPs) by the additional inhibition of the poly (ADP-ribose) polymerase (PARP1) protein.74 PARP1 normally functions in single-strand break and base-excision repair, and its inhibition is synthetically lethal when the HR pathway is also compromised. Similarly, PARP1 inhibition can be toxic to the HR defects associated with hypoxic cells; hence the concept of “contextual” synthetic lethality.72,75 We have shown in principle that PARP1 inhibition can preferentially kill repair-defective hypoxic tumour clonogens, while sparing normal tissues that maintain their DNA repair capability. Other contextual synthetic lethal approaches could be the inhibition of DNA polymerase-β in MMR-deficient hypoxic cells or inhibition of the ataxia telangiectasia mutated (ATM) kinase in hypoxic cells that have defective FA pathway function.8,75,76
Of interest, clinicians may already be combatting genetic instability and hypoxia in CaP with the combined use of ADT and RT.8 This combination has led to improved overall and CaP-specific survival in high-risk and locally advanced CaP.8 The use of neoadjuvant ADT (150 mg per day of bicalutamide) was shown to improve CaP oxygenation (based on pO2 measurements) prior to RT.77 Furthermore, three recent studies have suggested that ADT treatment reduces expression and function of the NHEJ and other DNA repair pathways, supporting the use of DNA-PKcs inhibitors in combination with ADT as a novel treatment for CaP.78–80 Any approach that tries to increase cell kill in hypoxic cells using contextual synthetic lethality or alterations in DNA repair will require assays that can measure the function of NHEJ, HR, MMR and FA proteins in situ to ascertain the fraction of hypoxic cells within a tumour that may be repair deficient. These uses of the predictive biomarkers in addition to pharmacodynamic biomarkers that confirm drug activity in vivo will be required for maximal impact of the use of this targeted approach in combination with RT, surgery or chemotherapy.81
A CAVEAT: THE PROBLEM OF PROSTATE CANCER MULTIFOCALITY AND STEM CELL SUBPOPULATIONS
CaP is unique in that it is a multifocal cancer with clonal subpopulations that have varied histological and molecular abnormalities that could relate to differential outcome. Heterogeneity exists both within and between patients. The vast majority of prostatectomies have more than one malignant focus within a prostate gland, which can be subcategorized by differential genomics based on PTEN, c-MYC and NKX3.1 gene abnormalities, therefore containing CaP foci with differential prognostic information.34,82 It is now well appreciated that tumours with identical GSs may exhibit profound genetic heterogeneity within a single prostate gland.44 It is also unclear whether a focus that is being assayed will potentially fail treatment owing to local radioresistance or because it initially harboured a lethal metastatic clone. Anatomically distinct tumour metastases can be derived from a single progenitor clone;83–85 a concept elegantly proven in renal cancer, whereby single needle biopsies did not predict the genetic heterogeneity within the primary tumour nor distant metastases for an individual patient.86 Studies using circulating tumour cells and circulating cell-free DNA, RNA and miRNA, after improvements in assay specificity and sensitivity, could be useful as a means to detect the most aggressive features of CaP within so-called, “liquid biopsies”, as a function of staging, prognosis and treatment response.87–89
These findings must be also placed into the context of CaP tumour initiating cells (TICs), which constitute that subfraction of cells, which must be sterilized by RT to prevent tumour cell repopulation after treatment (i.e. treatment failure).90,91 Pre-clinical studies suggest that CaP TICs may have increased biological growth under hypoxia and exist as a radioresistant hypoxic niche.92–94 However, rigorous studies are required to delineate the exact TIC markers that will differentiate this subpopulation for specific genomic studies as this relates to individualized prognosis.95,96 Finally, subtumoural heterogeneity in cancer metabolism (e.g. both acute and chronic hypoxia co-exist within a tumour and lead to significant gradients of oxygen consumption) could also confound quantitation and summary statistics for assaying the fraction of tumour hypoxia from one patient to another.14 Intraprostatic heterogeneity must therefore be adequately “sampled” across multiple foci and TIC subpopulations with genomic or hypoxia assays, such that the most aggressive and key features of tumour progression and/or prognosis are not missed.
CONCLUSIONS
A robust understanding of the interplay between hypoxia and genomics in the context of tumour heterogeneity is required to facilitate precision medicine for CaP. We must embrace these complexities if we are to target the most aggressive cases of CaP to improve cure rates (Figure 2). The genome-wide RNA- and DNA-based prognostic signatures developed and validated on post-treatment radical prostatectomy specimens must now be validated using pre-RT biopsies. The enormous reduction in cost and materials required for whole-genome and whole-transcriptome sequencing will further detail the genomics of CaP. Hypoxia-based assays must include functional assessment of DNA repair pathways if the concept of contextual synthetic lethality is to be acted upon within the clinic. Arranging the marriage between these biological end points will highlight key features of aggressive CaP variants that will best explain heterogeneous clinical outcome and provide novel treatments to offset both local and systemic resistance.
ACKNOWLEDGMENTS
This review is dedicated to the late Professor Donal Hollywood.
CONFLICTS OF INTEREST
The views expressed do not necessarily reflect those of the Ontario Ministry of Health and Long Term Care. RGB is a Canadian Cancer Society Research Scientist.
FUNDING
This work was supported by a grant from the Prostate Cancer Canada and Movember Foundation to the CPC-GENE project, the Ontario Institute for Cancer Research, Terry Fox Research Institute to the Canadian Prostate Cancer Biomarker Network, the Durham Motorcycle Ride for Dad fundraiser and the Princess Margaret Cancer Center Foundation. This research was also funded in part by the Ontario Ministry of Health and Long Term Care.
REFERENCES
- 1.Nichol AM, Warde P, Bristow RG. Optimal treatment of intermediate-risk prostate carcinoma with radiotherapy: clinical and translational issues. Cancer 2005; 104: 891–905. doi: 10.1002/cncr.21257 [DOI] [PubMed] [Google Scholar]
- 2.D'Amico AV, Cote K, Loffredo M, Renshaw AA, Chen MH. Pretreatment predictors of time to cancer specific death after prostate specific antigen failure. J Urol 2003; 169: 1320–4. doi: 10.1097/01.ju.0000049200.30192.d1 [DOI] [PubMed] [Google Scholar]
- 3.D'Amico AV, Moul J, Carroll PR, Sun L, Lubeck D, Chen MH. Cancer-specific mortality after surgery or radiation for patients with clinically localized prostate cancer managed during the prostate-specific antigen era. J Clin Oncol 2003; 21: 2163–72. [DOI] [PubMed] [Google Scholar]
- 4.Grimm P, Billiet I, Bostwick D, Dicker AP, Frank S, Immerzeel J, et al. Comparative analysis of prostate-specific antigen free survival outcomes for patients with low, intermediate and high risk prostate cancer treatment by radical therapy. Results from the Prostate Cancer Results Study Group. BJU Int 2012; 110: E431–2. [DOI] [PubMed] [Google Scholar]
- 5.Klotz L. Active surveillance for prostate cancer: overview and update. Curr Treat Options Oncol 2013; 14: 97–108. doi: 10.1007/s11864-012-0221-5 [DOI] [PubMed] [Google Scholar]
- 6.Van der Kwast TH, Roobol MJ. Defining the threshold for significant versus insignificant prostate cancer. Nat Rev Urol 2013; 10: 473–82. doi: 10.1038/nrurol.2013.112 [DOI] [PubMed] [Google Scholar]
- 7.King CR. The timing of salvage radiotherapy after radical prostatectomy: a systematic review. Int J Radiat Oncol Biol Phys 2012; 84: 104–11. doi: 10.1016/j.ijrobp.2011.10.069 [DOI] [PubMed] [Google Scholar]
- 8.Sridharan S, Dal Pra A, Catton C, Bristow RG, Warde P. Locally advanced prostate cancer: current controversies and optimisation opportunities. Clin Oncol (R Coll Radiol) 2013; 25: 499–505. doi: 10.1016/j.clon.2013.04.004 [DOI] [PubMed] [Google Scholar]
- 9.Mukherji D, Eichholz A, De Bono JS. Management of metastatic castration-resistant prostate cancer: recent advances. Drugs 2012; 72: 1011–28. doi: 10.2165/11633360-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 10.Yap TA, Swanton C, de Bono JS. Personalization of prostate cancer prevention and therapy: are clinically qualified biomarkers in the horizon? EPMA J 2012; 3: 3. doi: 10.1007/s13167-011-0138-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kollmeier MA, Zelefsky MJ. How to select the optimal therapy for early-stage prostate cancer. Crit Rev Oncol Hematol 2012; 84(Suppl. 1): e6–15. doi: 10.1016/j.critrevonc.2012.12.002 [DOI] [PubMed] [Google Scholar]
- 12.Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, et al. Punctuated evolution of prostate cancer genomes. Cell 2013; 153: 666–77. doi: 10.1016/j.cell.2013.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kovtun IV, Cheville JC, Murphy SJ, Johnson SH, Zarei S, Kosari F, et al. Lineage relationship of Gleason patterns in Gleason score 7 prostate cancer. Cancer Res 2013; 73: 3275–84. doi: 10.1158/0008-5472.CAN-12-2803 [DOI] [PubMed] [Google Scholar]
- 14.Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer 2008; 8: 180–92. doi: 10.1038/nrc2344 [DOI] [PubMed] [Google Scholar]
- 15.Chan N, Milosevic M, Bristow RG. Tumor hypoxia, DNA repair and prostate cancer progression: new targets and new therapies. Future Oncol 2007; 3: 329–41. doi: 10.2217/14796694.3.3.329 [DOI] [PubMed] [Google Scholar]
- 16.Sprong D, Janssen HL, Vens C, Begg AC. Resistance of hypoxic cells to ionizing radiation is influenced by homologous recombination status. Int J Radiat Oncol Biol Phys 2006; 64: 562–72. doi: 10.1016/j.ijrobp.2005.09.031 [DOI] [PubMed] [Google Scholar]
- 17.Boddy JL, Fox SB, Han C, Campo L, Turley H, Kanga S, et al. The androgen receptor is significantly associated with vascular endothelial growth factor and hypoxia sensing via hypoxia-inducible factors HIF-1a, HIF-2a, and the prolyl hydroxylases in human prostate cancer. Clinical Cancer Res 2005; 11: 7658–63. [DOI] [PubMed] [Google Scholar]
- 18.Carnell DM, Smith RE, Daley FM, Saunders MI, Bentzen SM, Hoskin PJ. An immunohistochemical assessment of hypoxia in prostate carcinoma using pimonidazole: implications for radioresistance. Int J Radiat Oncol Biol Phys 2006; 65: 91–9. [DOI] [PubMed] [Google Scholar]
- 19.Garcia-Parra R, Wood D, Shah RB, Siddiqui J, Hussain H, Park H, et al. Investigation on tumor hypoxia in resectable primary prostate cancer as demonstrated by 18F-FAZA PET/CT utilizing multimodality fusion techniques. Eur J Nucl Med Mol Imaging 2011; 38: 1816–23. doi: 10.1007/s00259-011-1876-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Green MM, Hiley CT, Shanks JH, Bottomley IC, West CM, Cowan RA, et al. Expression of vascular endothelial growth factor (VEGF) in locally invasive prostate cancer is prognostic for radiotherapy outcome. Int J Radiat Oncol Biol Phys 2007; 67: 84–90. [DOI] [PubMed] [Google Scholar]
- 21.Milosevic M, Warde P, Menard C, Chung P, Toi A, Ishkanian A, et al. Tumor hypoxia predicts biochemical failure following radiotherapy for clinically localized prostate cancer. Clinical Cancer Res 2012; 18: 2108–14. [DOI] [PubMed] [Google Scholar]
- 22.Thoms JW, Dal Pra A, Anborgh PH, Christensen E, Fleshner N, Menard C, et al. Plasma osteopontin as a biomarker of prostate cancer aggression: relationship to risk category and treatment response. Br J Cancer 2012; 107: 840–6. doi: 10.1038/bjc.2012.345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turaka A, Buyyounouski MK, Hanlon AL, Horwitz EM, Greenberg RE, Movsas B. Hypoxic prostate/muscle PO2 ratio predicts for outcome in patients with localized prostate cancer: long-term results. Int J Radiat Oncol Biol Phys 2012; 82: e433–9. [DOI] [PubMed] [Google Scholar]
- 24.Vergis R, Corbishley CM, Norman AR, Bartlett J, Jhavar S, Borre M, et al. Intrinsic markers of tumour hypoxia and angiogenesis in localised prostate cancer and outcome of radical treatment: a retrospective analysis of two randomised radiotherapy trials and one surgical cohort study. Lancet Oncol 2008; 9: 342–51. [DOI] [PubMed] [Google Scholar]
- 25.Weber DC, Tille JC, Combescure C, Egger JF, Laouiti M, Hammad K, et al. The prognostic value of expression of HIF1alpha, EGFR and VEGF-A, in localized prostate cancer for intermediate- and high-risk patients treated with radiation therapy with or without androgen deprivation therapy. Radiat Oncol 2012; 7: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell 2012; 148: 399–408. doi: 10.1016/j.cell.2012.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luoto KR, Kumareswaran R, Bristow RG. Tumor hypoxia as a driving force in genetic instability. Genome Integr 2013; 4: 5. doi: 10.1186/2041-9414-4-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michor F, Iwasa Y, Vogelstein B, Lengauer C, Nowak MA. Can chromosomal instability initiate tumorigenesis? Semin Cancer Biol 2005; 15: 43–9. doi: 10.1016/j.semcancer.2004.09.007 [DOI] [PubMed] [Google Scholar]
- 29.Pollack A, Zagars GK, el-Naggar AK, Gauwitz MD, Terry NH. Near-diploidy: a new prognostic factor for clinically localized prostate cancer treated with external beam radiation therapy. Cancer 1994; 73: 1895–903. [DOI] [PubMed] [Google Scholar]
- 30.Pretorius ME, Waehre H, Abeler VM, Davidson B, Vlatkovic L, Lothe RA, et al. Large scale genomic instability as an additive prognostic marker in early prostate cancer. Cell Oncol 2009; 31: 251–9. doi: 10.3233/CLO-2009-0463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ross JS, Figge H, Bui HX, del Rosario AD, Jennings TA, Rifkin MD, et al. Prediction of pathologic stage and postprostatectomy disease recurrence by DNA ploidy analysis of initial needle biopsy specimens of prostate cancer. Cancer 1994; 74: 2811–18. [DOI] [PubMed] [Google Scholar]
- 32.Song J, Cheng WS, Cupps RE, Earle JD, Farrow GM, Lieber MM. Nuclear deoxyribonucleic acid content measured by static cytometry: important prognostic association for patients with clinically localized prostate carcinoma treated by external beam radiotherapy. J Urol 1992; 147: 794–7. [DOI] [PubMed] [Google Scholar]
- 33.Locke JA, Zafarana G, Ishkanian AS, Milosevic M, Thoms J, Have CL, et al. NKX3.1 haploinsufficiency is prognostic for prostate cancer relapse following surgery or image-guided radiotherapy. Clinical Cancer Res 2012; 18: 308–16. [DOI] [PubMed] [Google Scholar]
- 34.Locke JA, Zafarana G, Malloff CA, Lam WL, Sykes J, Pintilie M, et al. Allelic loss of the loci containing the androgen synthesis gene, StAR, is prognostic for relapse in intermediate-risk prostate cancer. Prostate 2012; 72: 1295–305. doi: 10.1002/pros.22478 [DOI] [PubMed] [Google Scholar]
- 35.Zafarana G, Ishkanian AS, Malloff CA, Locke JA, Sykes J, Thoms J, et al. Copy number alterations of c-MYC and PTEN are prognostic factors for relapse after prostate cancer radiotherapy. Cancer 2012; 118: 4053–62. doi: 10.1002/cncr.26729 [DOI] [PubMed] [Google Scholar]
- 36.Castro E, Goh C, Olmos D, Saunders E, Leongamornlert D, Tymrakiewicz M, et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol 2013; 31: 1748–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Narod SA, Neuhausen S, Vichodez G, Armel S, Lynch HT, Ghadirian P, et al. Rapid progression of prostate cancer in men with a BRCA2 mutation. Br J Cancer 2008; 99: 371–4. doi: 10.1038/sj.bjc.6604453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dal Pra A, Lalonde E, Sykes J, Warde F, Ishkanian A, Meng A, et al. TMPRSS2-ERG status is not prognostic following prostate cancer radiotherapy: implications for fusion status and DSB repair. Clin Cancer Res 2013; 19: 5202–9. [DOI] [PubMed] [Google Scholar]
- 39.Minner S, Enodien M, Sirma H, Luebke AM, Krohn A, Mayer PS, et al. ERG status is unrelated to PSA recurrence in radically operated prostate cancer in the absence of antihormonal therapy. Clin Cancer Res 2011; 17: 5878–88. [DOI] [PubMed] [Google Scholar]
- 40.Paris PL, Weinberg V, Albo G, Roy R, Burke C, Simko J, et al. A group of genome-based biomarkers that add to a Kattan nomogram for predicting progression in men with high-risk prostate cancer. Clin Cancer Res 2010; 16: 195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leite KR, Tomiyama A, Reis ST, Sousa-Canavez JM, Sanudo A, Dall'Oglio MF, et al. MicroRNA-100 expression is independently related to biochemical recurrence of prostate cancer. J Urol 2011; 185: 1118–22. doi: 10.1016/j.juro.2010.10.035 [DOI] [PubMed] [Google Scholar]
- 42.Martens-Uzunova ES, Jalava SE, Dits NF, van Leenders GJ, Moller S, Trapman J, et al. Diagnostic and prognostic signatures from the small non-coding RNA transcriptome in prostate cancer. Oncogene 2012; 31: 978–91. doi: 10.1038/onc.2011.304 [DOI] [PubMed] [Google Scholar]
- 43.Spahn M, Kneitz S, Scholz CJ, Stenger N, Rudiger T, Strobel P, et al. Expression of microRNA-221 is progressively reduced in aggressive prostate cancer and metastasis and predicts clinical recurrence. Int J Cancer 2010; 127: 394–403. [DOI] [PubMed] [Google Scholar]
- 44.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010; 18: 11–22. doi: 10.1016/j.ccr.2010.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cooperberg MR, Simko JP, Cowan JE, Reid JE, Djalilvand A, Bhatnagar S, et al. Validation of a cell-cycle progression gene panel to improve risk stratification in a contemporary prostatectomy cohort. J Clin Oncol 2013; 31: 1428–34. doi: 10.1200/JCO.2012.46.4396 [DOI] [PubMed] [Google Scholar]
- 46.Erho N, Crisan A, Vergara IA, Mitra AP, Ghadessi M, Buerki C, et al. Discovery and validation of a prostate cancer genomic classifier that predicts early metastasis following radical prostatectomy. PLoS One 2013; 8: e66855. doi: 10.1371/journal.pone.0066855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cuzick J, Swanson GP, Fisher G, Brothman AR, Berney DM, Reid JE, et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: a retrospective study. Lancet Oncol 2011; 12: 245–55. doi: 10.1016/S1470-2045(10)70295-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu CL, Schroeder BE, Ma XJ, Cutie CJ, Wu S, Salunga R, et al. Development and validation of a 32-gene prognostic index for prostate cancer progression. Proc Natl Acad Sci U S A 2013; 110: 6121–6. doi: 10.1073/pnas.1215870110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cuzick J, Berney DM, Fisher G, Mesher D, Moller H, Reid JE, et al. Prognostic value of a cell cycle progression signature for prostate cancer death in a conservatively managed needle biopsy cohort. Br J Cancer 2012; 106: 1095–9. doi: 10.1038/bjc.2012.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Freedland SJ, Gerber L, Reid J, Welbourn W, Tikishvili E, Park J, et al. Prognostic utility of cell cycle progression score in men with prostate cancer after primary external beam radiation therapy. Int J Radiat Oncol Biol Phys 2013; 86: 848–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karnes RJ, Bergstralh EJ, Davicioni E, Ghadessi M, Buerki C, Mitra AP, et al. Validation of a genomic classifier that predicts metastasis following radical prostatectomy in an at risk patient population. J Urol 2013; 190: 2047–53. doi: 10.1016/j.juro.2013.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Borno ST, Fischer A, Kerick M, Falth M, Laible M, Brase JC, et al. Genome-wide DNA methylation events in TMPRSS2-ERG fusion-negative prostate cancers implicate an EZH2-dependent mechanism with miR-26a hypermethylation. Cancer Discov 2012; 2: 1024–35. doi: 10.1158/2159-8290.CD-12-0041 [DOI] [PubMed] [Google Scholar]
- 53.Chao C, Chi M, Preciado M, Black MH. Methylation markers for prostate cancer prognosis: a systematic review. Cancer Causes Control 2013; 24: 1615–41. doi: 10.1007/s10552-013-0249-2 [DOI] [PubMed] [Google Scholar]
- 54.Kron K, Trudel D, Pethe V, Briollais L, Fleshner N, van der Kwast T, et al. Altered DNA methylation landscapes of polycomb-repressed loci are associated with prostate cancer progression and ERG oncogene expression in prostate cancer. Clinical Cancer Res 2013; 19: 3450–61. [DOI] [PubMed] [Google Scholar]
- 55.Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov 2011; 1: 487–95. doi: 10.1158/2159-8290.CD-11-0130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature 2007; 446: 153–8. doi: 10.1038/nature05610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Helleday T, Lo J, van Gent DC, Engelward BP. DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair (Amst) 2007; 6: 923–35. doi: 10.1016/j.dnarep.2007.02.006 [DOI] [PubMed] [Google Scholar]
- 58.Chan N, Koritzinsky M, Zhao H, Bindra R, Glazer PM, Powell S, et al. Chronic hypoxia decreases synthesis of homologous recombination proteins to offset chemoresistance and radioresistance. Cancer Res 2008; 68: 605–14. doi: 10.1158/0008-5472.CAN-07-5472 [DOI] [PubMed] [Google Scholar]
- 59.Kumareswaran R, Ludkovski O, Meng A, Sykes J, Pintilie M, Bristow RG. Chronic hypoxia compromises repair of DNA double-strand breaks to drive genetic instability. J Cell Sci 2012; 125: 189–99. doi: 10.1242/jcs.092262 [DOI] [PubMed] [Google Scholar]
- 60.Meng AX, Jalali F, Cuddihy A, Chan N, Bindra RS, Glazer PM, et al. Hypoxia down-regulates DNA double strand break repair gene expression in prostate cancer cells. Radiother Oncol 2005; 76: 168–76. doi: 10.1016/j.radonc.2005.06.025 [DOI] [PubMed] [Google Scholar]
- 61.Coquelle A, Toledo F, Stern S, Bieth A, Debatisse M. A new role for hypoxia in tumor progression: induction of fragile site triggering genomic rearrangements and formation of complex DMs and HSRs. Mol Cell 1998; 2: 259–65. [DOI] [PubMed] [Google Scholar]
- 62.Kuhnert VM, Kachnic LA, Li L, Purschke M, Gheorghiu L, Lee R, et al. FANCD2-deficient human fibroblasts are hypersensitive to ionising radiation at oxygen concentrations of 0% and 3% but not under normoxic conditions. Int J Radiat Biol 2009; 85: 523–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mihaylova VT, Bindra RS, Yuan J, Campisi D, Narayanan L, Jensen R, et al. Decreased expression of the DNA mismatch repair gene Mlh1 under hypoxic stress in mammalian cells. Mol Cell Biol 2003; 23: 3265–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nakamura H, Tanimoto K, Hiyama K, Yunokawa M, Kawamoto T, Kato Y, et al. Human mismatch repair gene, MLH1, is transcriptionally repressed by the hypoxia-inducible transcription factors, DEC1 and DEC2. Oncogene 2008; 27: 4200–9. doi: 10.1038/onc.2008.58 [DOI] [PubMed] [Google Scholar]
- 65.Rezvani HR, Mahfouf W, Ali N, Chemin C, Ged C, Kim AL, et al. Hypoxia-inducible factor-1alpha regulates the expression of nucleotide excision repair proteins in keratinocytes. Nucleic Acids Res 2010; 38: 797–809. doi: 10.1093/nar/gkp1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ahn GO, Brown M. Targeting tumors with hypoxia-activated cytotoxins. Front Biosci 2007; 12: 3483–501. [DOI] [PubMed] [Google Scholar]
- 67.Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer 2011; 11: 393–410. doi: 10.1038/nrc3064 [DOI] [PubMed] [Google Scholar]
- 68.Meng F, Evans JW, Bhupathi D, Banica M, Lan L, Lorente G, et al. Molecular and cellular pharmacology of the hypoxia-activated prodrug TH-302. Mol Cancer Ther 2012; 11: 740–51. doi: 10.1158/1535-7163.MCT-11-0634 [DOI] [PubMed] [Google Scholar]
- 69.Choudhury A, Zhao H, Jalali F, Al Rashid S, Ran J, Supiot S, et al. Targeting homologous recombination using imatinib results in enhanced tumor cell chemosensitivity and radiosensitivity. Mol Cancer Ther 2009; 8: 203–13. doi: 10.1158/1535-7163.MCT-08-0959 [DOI] [PubMed] [Google Scholar]
- 70.Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 2008; 8: 851–64. doi: 10.1038/nrc2501 [DOI] [PubMed] [Google Scholar]
- 71.Zhao H, Luoto KR, Meng AX, Bristow RG. The receptor tyrosine kinase inhibitor amuvatinib (MP470) sensitizes tumor cells to radio- and chemo-therapies in part by inhibiting homologous recombination. Radiother Oncol 2011; 101: 59–65. [DOI] [PubMed] [Google Scholar]
- 72.Chan N, Pires IM, Bencokova Z, Coackley C, Luoto KR, Bhogal N, et al. Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer Res 2010; 70: 8045–54. doi: 10.1158/0008-5472.CAN-10-2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 2005; 5: 689–98. doi: 10.1038/nrc1691 [DOI] [PubMed] [Google Scholar]
- 74.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009; 361: 123–34. doi: 10.1056/NEJMoa0900212 [DOI] [PubMed] [Google Scholar]
- 75.Chan N, Bristow RG. “Contextual” synthetic lethality and/or loss of heterozygosity: tumor hypoxia and modification of DNA repair. Clin Cancer Res 2010; 16: 4553–60. doi: 10.1158/1078-0432.CCR-10-0527 [DOI] [PubMed] [Google Scholar]
- 76.Martin SA, McCabe N, Mullarkey M, Cummins R, Burgess DJ, Nakabeppu Y, et al. DNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1. Cancer Cell 2010; 17: 235–48. doi: 10.1016/j.ccr.2009.12.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Milosevic M, Chung P, Parker C, Bristow R, Toi A, Panzarella T, et al. Androgen withdrawal in patients reduces prostate cancer hypoxia: implications for disease progression and radiation response. Cancer Res 2007; 67: 6022–5. [DOI] [PubMed] [Google Scholar]
- 78.Al-Ubaidi FL, Schultz N, Loseva O, Egevad L, Granfors T, Helleday T. Castration therapy results in decreased Ku70 levels in prostate cancer. Clin Cancer Res 2013; 19: 1547–56. [DOI] [PubMed] [Google Scholar]
- 79.Goodwin JF, Schiewer MJ, Dean JL, Schrecengost RS, de Leeuw R, Han S, et al. A Hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov 2013; 3: 1254–71. doi: 10.1158/2159-8290.CD-13-0108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov 2013; 3: 1245–53. doi: 10.1158/2159-8290.CD-13-0172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chalmers AJ, Lakshman M, Chan N, Bristow RG. Poly(ADP-ribose) polymerase inhibition as a model for synthetic lethality in developing radiation oncology targets. Semin Radiat Oncol 2010; 20: 274–81. doi: 10.1016/j.semradonc.2010.06.001 [DOI] [PubMed] [Google Scholar]
- 82.Cooper CS, Foster CS. Concepts of epigenetics in prostate cancer development. Br J Cancer 2009; 100: 240–5. doi: 10.1038/sj.bjc.6604771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aryee MJ, Liu W, Engelmann JC, Nuhn P, Gurel M, Haffner MC, et al. DNA methylation alterations exhibit intraindividual stability and interindividual heterogeneity in prostate cancer metastases. Sci Transl Med 2013; 5: 169ra10. doi: 10.1126/scitranslmed.3005211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu W, Laitinen S, Khan S, Vihinen M, Kowalski J, Yu G, et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat Med 2009; 15: 559–65. doi: 10.1038/nm.1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mehra R, Tomlins SA, Yu J, Cao X, Wang L, Menon A, et al. Characterization of TMPRSS2-ETS gene aberrations in androgen-independent metastatic prostate cancer. Cancer Res 2008; 68: 3584–90. doi: 10.1158/0008-5472.CAN-07-6154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012; 366: 883–92. doi: 10.1056/NEJMoa1113205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sita-Lumsden A, Fletcher CE, Dart DA, Brooke GN, Waxman J, Bevan CL. Circulating nucleic acids as biomarkers of prostate cancer. Biomark Med 2013; 7: 867–77. doi: 10.2217/bmm.13.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Harouaka R, Kang Z, Zheng SY, Cao L. Circulating tumor cells: advances in isolation and analysis, and challenges for clinical applications. Pharmacol Ther 2014; 141: 209–21. doi: 10.1016/j.pharmthera.2013.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Danila DC, Fleisher M, Scher HI. Circulating tumor cells as biomarkers in prostate cancer. Clin Cancer Res 2011; 17: 3903–12. doi: 10.1158/1078-0432.CCR-10-2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Baumann M, Krause M, Thames H, Trott K, Zips D. Cancer stem cells and radiotherapy. Int J Radiat Biol 2009; 85: 391–402. doi: 10.1080/09553000902836404 [DOI] [PubMed] [Google Scholar]
- 91.Woodward WA, Bristow RG. Radiosensitivity of cancer-initiating cells and normal stem cells (or what the Heisenberg uncertainly principle has to do with biology). Semi Radiat Oncol 2009; 19: 87–95. doi: 10.1016/j.semradonc.2008.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ma Y, Liang D, Liu J, Axcrona K, Kvalheim G, Stokke T, et al. Prostate cancer cell lines under hypoxia exhibit greater stem-like properties. PLoS One 2011; 6: e29170. doi: 10.1371/journal.pone.0029170 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 93.Anderson KM, Guinan P, Rubenstein M. The effect of normoxia and hypoxia on a prostate (PC-3) CD44/CD41 cell side fraction. Anticancer Res 2011; 31: 487–94. [PubMed] [Google Scholar]
- 94.Frame FM, Pellacani D, Collins AT, Simms MS, Mann VM, Jones G, et al. HDAC inhibitor confers radiosensitivity to prostate stem-like cells. Br J Cancer 2013; 109: 3023–33. doi: 10.1038/bjc.2013.691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sharpe B, Beresford M, Bowen R, Mitchard J, Chalmers AD. Searching for prostate cancer stem cells: markers and methods. Stem Cell Rev 2013; 9: 721–30. doi: 10.1007/s12015-013-9453-4 [DOI] [PubMed] [Google Scholar]
- 96.Chen X, Rycaj K, Liu X, Tang DG. New insights into prostate cancer stem cells. Cell Cycle 2013; 12: 579–86. doi: 10.4161/cc.23721 [DOI] [PMC free article] [PubMed] [Google Scholar]