

Abstract
Proinflammatory CD4+CD28null T cells are frequently found in the circulation of RA patients, but are less common in the rheumatic joint. In the present study we sought to identify functional differences between CD4+CD28null T cells from blood and synovial fluid in comparison to conventional CD28 expressing CD4+ T cells. 44 RA patients, displaying a distinct CD4+CD28null T cell population in blood, were recruited for this study and the methylation status of the IFNG locus was examined in isolated T cell subsets, and intracellular cytokine production (IFN-γ, TNF, IL-17) and chemokine receptor expression (CXCR3, CCR6 and CCR7) were assessed by flow cytometry on T cells from the two compartments. Circulating CD4+CD28null T cells were significantly more hypomethylated in the CNS-1 region of the IFNG locus than conventional CD4+CD28+ T cells and produced higher levels of both IFN-γ and TNF after TCR crosslinking. CD4+CD28null T cells from the site of inflammation expressed significantly more CXCR3 and CCR6 compared to their counterparts in blood. While IL-17A production could hardly be detected in CD4+CD28null cells from the blood, a significant production was observed in CD4+CD28null T cells from synovial fluid. CD4+CD28null T cells were not only found to differ from conventional CD4+CD28+ T cells in the circulation, but we could also demonstrate that synovial CD4+CD28null T cells showed additional effector functions (IL-17 co-production) as compared to the same subset in peripheral blood, suggesting an active role for these cells in the perpetuation of inflammation in the subset of patients having a CD28null population.
Introduction
CD4+CD28null cells are highly differentiated effector memory CD4+ T cells that have downregulated the costimulatory molecule CD28, due to loss of an CD28-specific initiator complex [1, 2]. CD28 is progressively lost after replicative senescence [3] (continuous viral or autoantigen stimulation) or under proinflammatory conditions, e.g. by TNF [4]. CD4+CD28null cells differ from conventional CD4+ T cells with respect to shortened telomeres [5], reduced TCR diversity [6, 7], by displaying cytotoxic capacity [8, 9], expression of NK cell receptors [10] and resistance to apoptosis [11]. While being independent of classical co-stimulation, CD4+CD28null T cells are not anergic, but respond rapidly to stimulation [12]. Because of their proinflammatory features (cytokine production and cytotoxicity) it is likely that they contribute to disease progression of several inflammatory disorders. Increased frequencies of CD4+CD28null cells can be found in the peripheral circulation of various immune disorders, such as Rheumatoid Arthritis (RA) [12], Multiple Sclerosis [13, 14] and Inflammatory Bowel Disease [15, 16]. CD4+CD28null T cells can be found in about one third of RA patients. The frequency in the circulation varies, but can be up to 50% of all CD4+ T cells [17]. Notably, despite their presence in the circulation at significant frequencies, CD4+CD28null cells are infrequent in the joints of RA patients (synovial fluid and synovial membrane) [17]; the nature behind this observation has remained largely unexplored.
Since the distribution of these cells differs between the circulation and the target site of disease we wanted to examine if the CD4+CD28null cells that are present in the joint have different features than the CD28null population seen in peripheral blood. For this purpose we examined the ability of CD4+CD28null T cells from blood and synovial fluid of RA patients to produce cytokines by studying both the methylation status of the IFNG locus, since CD4+CD28null T cells are well known to make this cytokine, and their cytokine secretion capacity by comparing IFN-γ, TNF and IL-17 production, i.e. cytokines implicated in RA pathogenesis. Furthermore since it is not known why only some of these cells migrate to the joint we characterized the cells with regard to chemokine receptor expression and compared CD4+CD28null T cells to conventional CD4+CD28+ T cells.
We found that cells from the joints of RA patients differ to those found in the periphery with regard to methylation status, cytokine production and chemokine receptor expression.
Materials and Methods
Patients and samples
All together 44 RA patients were enrolled in the study. The diagnosis was determined by rheumatologists according to the American College of Rheumatology 1987 revised criteria for the classification of RA [18]. All patients had at least 5% CD28null cells in their circulation. PB and SF samples from 14 patients were used for methylation status. 23 RA patients were included in cytokine analysis and 12 in the chemokine receptor expression. Some of the patients were included in several experiments. The mean age was 57 years (range 18–86); 75% were female. 3 of the patients had a disease duration of less than a year, 7 patients less than 5 years and 34 patients had a disease duration over 5 up to 44 years. All patients attended either the Rheumatology Clinic at the Karolinska University Hospital or were participants in the Benaroya Research Institute (BRI) Rheumatic Disease Registry. Peripheral blood and synovial fluid samples (captured in heparin tubes) were prepared by ficoll (Ficoll-Paque Plus, GE Healthcare, Uppsala, Sweden) separation and cryopreserved until use. Informed consent was obtained from all subjects under protocols approved by the institutional review board at BRI or the Karolinska Hospital.
T cell isolation
T cells were isolated by flow cytometry on a MoFlo (Beckman Coulter) by sorting for CD3+CD4+ and either CD28+ or CD28−. The purity of isolated subsets was on average 98%.
All surface staining was carried out in PBS supplemented with 1% human sera and 0.05 % sodium azide using anti-CD3 PC5 (Beckman Coulter) or anti-CD3 FITC, anti-CD4 FITC or anti-CD4 PC5 or anti-CD4 APCCY7, CD45RO PC5 and anti-CD28 PE (all BD) antibody conjugates.
DNA methylation status of the IFNG 5′CNS1 enhancer
Genomic DNA was isolated and bisulphite treated as previously described [19]. The 5′CNS1 region of the IFNG gene was amplified by PCR with (fwd: TGG TTA GAA GGT ATA AAA GAA AAG G, rev: ATC ACA TCC TAA CAC TCA CAA C) Primers designed for the IFNG locus (Cybergene, Stockholm, Sweden). For the Cobra method these primers were conjugated to 6-FAM.
Cobra PCR products were digested with MaeII in excess (New England Biolabs). Following a phenol chloroform extraction, digested products were loaded onto a 310 Genetic Analyzer (Applied Biosystems, Foster City, CA) and fragment analysis was performed using Gene scan v3.7 application software (Applied Biosystems).
For Ms-Snupe the PCR products were treated with exonuclease I and calf intestinal phosphatase (New England Biolabs, Ontario, Canada). Methylation-sensitive single nucleotide primer extension was performed with the following primer: GAT TTA TAT TGA GTG GAA GAA ATT TA using SNaPshot Multiplex kit (Applied Biosystems) according to the manufacturer’s instructions. Products were loaded onto a 310 Genetic Analyzer (Applied Biosystems), and fragment analysis was performed using Gene Scan version 3.7 application software (Applied Biosystems).
Intracellular cytokine staining
Cells were cultured for 6h in complete media (RPMI, HEPES, L-Glutamine, Penicillin, Streptomycin) containing 5% human serum and stimulated with plate-bound α-CD3 monoclonal antibody (plate coated with 2.5 μg/ml, clone OKT-3) and incubated at 37°C, 5% CO2. In order to prevent produced cytokines from being secreted, 10 μg/ml Brefeldin A (Sigma-Aldrich, Steinheim, Germany) was added to the cultures 4h prior to harvesting. Extracellular and intracellular cytokine staining was performed using Cytofix/Cytoperm Kit (BD) according to the manufacturer’s instructions. Antibodies: Anti-CD28 PE or anti-CD28 APC, anti-CD14 APC Cy7, anti-CD4 PeCy7 or anti-CD4 PerCP, anti-IFN-γ FITC (all BD), anti-CD3 PB (BD or Biolegend), anti-TNF PerCP Cy5.5 (Biolegend), anti-IL17 Alexa 647 (Biolegend) or anti-IL17A PE (eBioscience). LIVE/DEAD Aqua Dead Cell Stain (Invitrogen) was used to exclude dead cells. The PBMCs were run on a Beckman Coulter CyAn. Analyses were performed with FlowJo software, version 8.1.0 or higher (Treestar Inc.).
Chemokine receptor expression analysis
Cells were stained with the following antibody-conjugates:
Anti-CD14 FITC or anti-CD14 APC, anti-CD19 FITC or anti-CD19 APC, anti-CD197 PE or anti-CD197 Alexa Fluor700, anti-CD196 PerCpCy5.5, anti-CD4 V500, anti-CD28 APC or anti-CD28 PE Cy7 or anti-CD28 V450, anti-CD45RO APC Cy7 or anti-CD45RO eFluor650NC, anti-CD183 PECy5 (all BD) and anti-CD183 BV421or anti-CD183 Pacific Blue. Annexin V FITC or Annexin V APC was used to exclude dead/dying cells.
Cells were run on a LSRII (BD) or Gallios (Beckman Coulter) flow cytometer and analysed with FlowJo software, version 9.3.3 or higher.
Statistical analysis
A nonparametric Mann-Whitney test was used to detect differences in methylation status, cytokine secretion and chemokine receptor expression between the subsets. No correction for multiple comparisons was done. P values less than 0.05 were considered statistically significant. All statistical analyses were performed using Prism 5.0 (GraphPad Software, La Jolla, CA, USA).
Results
Methylation status of the IFNG locus
We examined a key regulatory element in the enhancer of the IFNG locus in conventional CD4+CD28+ and CD4+CD28null T cells from both blood and synovial fluid obtained from RA patients. In peripheral blood cells we observed a highly hypomethylated IFNG locus amongst the CD4+CD28null T cells, with a significant difference in comparison to conventional CD4+CD28+ T cells (Figure 1). In contrast, we could not detect a difference between CD4+CD28+ and CD4+CD28null T cells derived from the inflamed joints of RA patients, as they both showed a similar degree of methylation at the IFNG locus (Figure 1). When we compared conventional CD4+CD28+ T cells derived from the blood with those derived from synovial fluid we could detect a significant difference. There was no significant difference between CD4+CD28null T cells from the joint compared to those from peripheral blood (Figure 1).
Figure 1. Methylation status of the IFNG promoter locus.

PBMC (black bars) or SFMC (grey bars) were sorted for CD4+CD28+ or CD4+CD28− (i.e. CD4+CD28null) and the methylation status of the promoter region of IFN-γ was assessed. Displayed is the percentage of methylation, reported as mean+SD. CD4+CD28+ PB (n=9), CD4+CD28+ SF (n=8), CD4+CD28− PB (n=7) and CD4+CD28− SF (n=6). A Mann-Whitney test was used to determine differences between subsets. P values less than 0.05 were considered statistically significant. A P value of 0.01 to 0.05 is indicated with * and a P value of 0.001 to 0.01 with **.
Cytokine production
To further elucidate the different effector functions and also to validate the IFNG expression we studied the cytokine production of CD4+CD28+ and CD4+CD28null cells from blood and synovial fluid. After polyclonal stimulation with α-CD3, T cells show profound cytokine production as exemplified in Figure 2A–C.
Figure 2. Intracellular cytokine staining from CD4+CD28+ and CD4+CD28− T cells for IFN-γ, TNF and IL-17A.

PBMC (black dots) or SFMC (grey dots) were stimulated in vitro with 2.5μg/ml α-CD3 (OKT-3) for 6h and intracellular cytokine staining was performed for IFN-γ, TNF and IL-17A. Cells were gated for CD14− and CD3+ (A–F). Representative FACS staining from PBMC for IFN-γ (A), TNF (B) and IL-17A (C). Cytokine production from PBMC (n=21) and SFMC (n=3) for IFN-γ (D), TNF production from PBMC (n=16) (E) and IL-17A production from PBMC (n=21) and SFMC (n=3) (F). Results displayed as mean+SD. A Mann-Whitney test was used to determine differences between subsets. P values less than 0.05 were considered statistically significant. A P value of 0.001 to 0.01 is indicated with ** and a P value lower than 0.001 with ***.
CD4+CD28null T cells from the periphery produced significantly higher amounts of Th1 cytokines, namely IFN-γ and TNF (Figure 2D and E) than conventional CD4+CD28+ cells, while in synovial fluid there was no detectable difference in IFN-γ production between conventional and CD4+CD28null cells (Figure 2D).
CD4+CD28null T cells from the periphery did not produce IL-17, whereas there was significantly more production from conventional CD4+CD28+ T cells (Figure 2F). However, in synovial fluid, CD4+CD28null T cells were capable of IL-17 production and produced significantly more than CD4+CD28null T cells from blood (Figure 2F).
Chemokine receptor expression
The expression of specific chemokine receptors characterizes different subsets of T cells and drives their localization to specific tissues [20]. We therefore compared the expression of chemokine receptors CXCR3, CCR6 and CCR7 on the surface of memory CD4+CD28+ and CD4+CD28null T cells from blood and synovial fluid of RA patients.
CD4+CD28null T cells from synovial fluid expressed significantly more CXCR3 than their counterparts from blood (Figure 3A). While CD4+CD28null and conventional T cells from synovial fluid had comparable expression for CCR6 and CCR7, CD4+CD28null cells from blood expressed significantly less CCR6 and CCR7 then conventional cells (Figure 3B and C). There was no difference between blood and synovial fluid with respect to the rare expression of CCR7 in the CD28null subsets (Figure 3C).
Figure 3. Chemokine receptor expression.
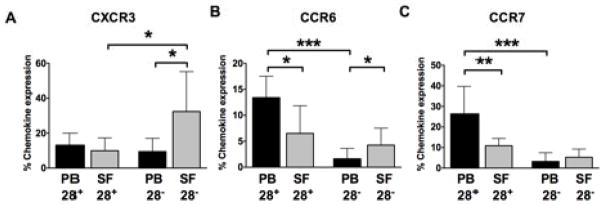
PBMC (black bars) and SFMC (grey bars) were gated on CD14−CD4+, subdivided into 2 populations: CD4+CD28+CD45RO+ and CD4+CD28−, and analysed for the expression of CXCR3 (A), CCR6 (B) and CCR7 (C). All values are reported as mean+SD. A Mann-Whitney test was used to determine differences between subsets. P values less than 0.05 were considered statistically significant. A P value of 0.01 to 0.05 is indicated with *, 0.001 to 0.01 with ** and a P value lower than 0.001 with ***.
To distinguish Th17 cells (CCR6+ single positive) from Th1 cells that co-produce IL-17 and IFN-γ (CXCR3+CCR6+) we analysed to what extent CCR6+ cells also co-expressed CXCR3. In both blood and synovial fluid the majority of CD4+CD28+ T cells that expressed CCR6 were CXCR3−, whereas in the CD28null subset a substantial proportion of the CCR6+ cells were also CXCR3+ (Data not shown).
Discussion
In this study we sought to identify functional differences in CD4+CD28null T cells in the context of rheumatoid arthritis by comparison with on the one hand conventional CD4+CD28+ T cells and on the other hand we compared the CD28null subset derived from blood versus synovial fluid.
CD4+CD28null T cells are known to readily produce IFN-γ upon re-activation, but it is still a matter of debate whether these cells originate form the Th1 subset or not. Th1 cell differentiation induces hypomethylation of the IFNG promoter and its upstream CNS1 enhancer region, and this hypomethylation during commitment to Th1 precedes IFNG expression. [21]. A key hypomethylated CpG site within the IFNG 5′CNS enhancer of Th1 cells has been described [19, 22]. This region binds important transcription factors for Th1 cell development such as T-bet and NF-AT [23, 24].
Dysregulated silencing of genes has been implicated as the cause of an unleashed transcriptional program, which can lead to breakdown of tolerance [25]. We have previously investigated the methylation status of the global CD4+ T cell compartment from RA patients. In paired samples from blood and synovial fluid significant hypomethylation of the IFNG locus in CD4+ T cells from the joints was observed, in contrast to blood [22]. This is in line with our results for conventional CD4+CD28+ T cells, where we also detected a high level of hypomethylation in cells from the joint. In contrast, CD4+CD28null T cells from peripheral blood showed the highest level of hypomethylation and also displayed the highest production of IFN-γ. The high extent of hypomethylation indeed indicates that the cells are more accessible for transcription factors and more likely to have higher IFN-γ production. In synovial fluid the IFNG promoter was hypomethylated in both CD4+CD28+ and CD4+CD28null T cells and both subsets consequently produced high levels of IFN-γ, demonstrating the regulating link between methylation status and cytokine production.
CD4+CD28null T cells have been previously proposed to be of a Th1 subtype [26, 27]. Our results on the epigenetic profiling of the IFNG locus confirm this notion. However, recently a new transcription factor has been put forward in the context of terminally differentiated T cells [28]. In the absence of the helper T cell master regulator ThPOK, MHC class II restricted CD4+ T cells developed cytotoxic properties. It will be interesting to see if this will have implications also for CD4+ CD28null T cells.
CD4+CD28null T cells from blood and the joints are readily triggered to produce cytokines, e.g. IFN-γ (and TNF) [17, 29], but so far no IL-17 production has been demonstrated by CD4+CD28null T cells. Yamada et al. investigated PBMC from RA patients, but could not detect any CD4+IL17+CD57+ (a differentiation marker overlapping with CD28null cells [30]) after stimulation with PMA and ionomycin [26]. However, they found scarce IL17+ producing cells in the joints, but it was not further reported whether these were conventional CD4+CD28+ T cells or CD4+CD28null cells. In our hands CD4+CD28null T cells from the blood showed hardly any IL-17 production, whereas cells from the site of inflammation displayed capacity to produce IL-17. Indeed, T cells making IL-17 have been observed by other groups in synovial cells of RA patients [31, 32] and high levels of IL-17 were reported in synovial fluid of RA patients [31, 33], but without addressing the CD28 phenotype.
Chemokines play critical roles in regulating immune responses and inflammation and have been implicated to play a role in RA [34], by recruiting various types of leukocytes to the inflamed joints [35].
Whereas CXCR3 is used as marker for Th1 cells, CCR6 is expressed on the Th17 subset. Cells expressing both CXCR3 and CCR6 also belong to the Th1 subset, and have the capacity to produce IL-17 in addition to IFN-γ [36]. CCR7 is expressed on naïve T cells and central memory cells that home to lymph nodes. For the rheumatic joint this marker is of interest since lymphocyte aggregates are often seen in the inflamed synovial tissue and in severe cases even form ectopic lymph node structures; and T cells that express CCR7 are involved in lymphocyte migration to these structures [37, 38]. Clearly CD4+CD28null T cells are not central memory T cells and are not likely to contribute to these events since they virtually lack CCR7 expression. This is in line with previous results that demonstrated a lack of CCR7 expression on this T cell subset [30].
We observed high production of IFN-γ from CD4+CD28null T cells from the joint and also increased expression of CXCR3 and CCR6 on CD4+CD28null T cells from the joints compared to those from the periphery. Both receptors CXCR3 and CCR6 have been implicated to facilitate migration of T lymphocytes to inflamed tissues [36, 39, 40]. In the setting of juvenile arthritis Wedderburn et al. have reported enrichment of IFN-γ producing T cells that express high levels of CXCR3 and CCR5 in the joints of JIA patients [41] and furthermore increased expression of CXCR3 has been reported on CD4+ and CD8+ subpopulations of T cells from the synovial fluid in ankylosing spondylitis patients [42].
CCR6 expression on CD4+CD28null T cells was primarily seen in combination with CXCR3 expression, both in the joint and in the periphery, suggesting that the CXCR3+ CD4+CD28null T cells have gained CCR6 co-expression at the site of inflammation. The IL-17 producing cells we found in the joint are therefore likely to be positive for both CXCR3 and CCR6, but this was not investigated in our study.
TNF can downregulate the expression of CD28 on T cells [4]. Compared to in circulation, the level of TNF is higher in synovial fluid and this might induce local differentiation of CD28null cells in the synovial joint. Still, although TNF levels are elevated in the joint, there are significantly fewer CD4+CD28null T cells [17]. We therefore propose that these cells migrate from blood to the joint, but this has yet to be proven.
Conclusions
The joints of rheumatoid arthritis patients are sites of chronic and/or recurrent inflammation, with the time of joint effusion coinciding with local disease activity. It is therefore not entirely surprising that CD4+CD28null T cells from the joint show different properties than their counterparts in the periphery. We utilized several complementary approaches, namely epigenetic analysis, intracellular cytokine stainings and chemokine receptor expression, to show that peripheral CD4+CD28null T cells can be functionally and characteristically distinguished from conventional T cells. We also could show for the first time that joint-derived CD4+CD28null T cells can produce IL-17. The proinflammatory phenotype of the CD4+CD28null T cells suggests an active role for these cells in the perpetuation of inflammation in the subset of RA patients that have an extended CD28null population.
Acknowledgments
The authors thank the staff and patients at the Rheumatology Clinic of Karolinska University Hospital, especially Eva Jemseby, Gull-Britt Almgren and Julia Boström for organizing the sampling, storage, and administration of biomaterial, and Annika van Vollenhoven for excellent cell sorting. We would also like to thank the Virginia Mason Clinic’s Rheumatology group, and the Translational Research Program at the Benaroya Research Institute in Seattle. Work performed at KI was supported by grants from the Margaretha af Ugglas Foundation, the Swedish Association against Rheumatism, the Swedish Medical Association, the King Gustaf V 80 year Foundation, the Swedish Research Council, the EU FP7 project Masterswitch (HEALTH-F2-2008-223404) and the IMI JU funded project BTCure 115142-2. Work performed at BRI was supported by grants from the NIH including NIAID 5U19 AI050864, NIMS 5 R01 AR037296, and NIAID UO1 AI101981.
Footnotes
Author statement
JP carried out and analysed the methylation study, intracellular cytokine study and chemokine receptor expression study, participated in its design and drafted the manuscript. SJ participated in methylation study and intracellular cytokine study. OS participated in cytokine study. LL participated in methylation study. MR participated in chemokine receptor expression study. OW helped design the methylation study. JB contributed to chemokine receptor expression study. RvV was responsible for patient inclusion at KI and assisted in drafting the manuscript. VM conceived the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Declaration of commercial interest
No competing interests.
References
- 1.Vallejo AN, Brandes JC, Weyand CM, Goronzy JJ. Modulation of CD28 expression: distinct regulatory pathways during activation and replicative senescence. Journal of immunology. 1999 Jun 1;162:6572–9. [PubMed] [Google Scholar]
- 2.Vallejo AN, Bryl E, Klarskov K, Naylor S, Weyand CM, Goronzy JJ. Molecular basis for the loss of CD28 expression in senescent T cells. J Biol Chem. 2002 Dec 6;277:46940–9. doi: 10.1074/jbc.M207352200. [DOI] [PubMed] [Google Scholar]
- 3.Broux B, Markovic-Plese S, Stinissen P, Hellings N. Pathogenic features of CD4+CD28- T cells in immune disorders. Trends Mol Med. 2012 Aug;18:446–53. doi: 10.1016/j.molmed.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 4.Bryl E, Vallejo AN, Weyand CM, Goronzy JJ. Down-regulation of CD28 expression by TNF-alpha. Journal of immunology. 2001 Sep 15;167:3231–8. doi: 10.4049/jimmunol.167.6.3231. [DOI] [PubMed] [Google Scholar]
- 5.Schonland SO, Lopez C, Widmann T, et al. Premature telomeric loss in rheumatoid arthritis is genetically determined and involves both myeloid and lymphoid cell lineages. Proc Natl Acad Sci U S A. 2003 Nov 11;100:13471–6. doi: 10.1073/pnas.2233561100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmidt D, Martens PB, Weyand CM, Goronzy JJ. The repertoire of CD4+ CD28- T cells in rheumatoid arthritis. Mol Med. 1996 Sep;2:608–18. [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner U, Pierer M, Kaltenhauser S, et al. Clonally expanded CD4+CD28null T cells in rheumatoid arthritis use distinct combinations of T cell receptor BV and BJ elements. Eur J Immunol. 2003 Jan;33:79–84. doi: 10.1002/immu.200390010. [DOI] [PubMed] [Google Scholar]
- 8.Nakajima T, Schulte S, Warrington KJ, et al. T-cell-mediated lysis of endothelial cells in acute coronary syndromes. Circulation. 2002 Feb 5;105:570–5. doi: 10.1161/hc0502.103348. [DOI] [PubMed] [Google Scholar]
- 9.van Leeuwen EM, Remmerswaal EB, Vossen MT, et al. Emergence of a CD4+CD28- granzyme B+, cytomegalovirus-specific T cell subset after recovery of primary cytomegalovirus infection. Journal of immunology. 2004 Aug 1;173:1834–41. doi: 10.4049/jimmunol.173.3.1834. [DOI] [PubMed] [Google Scholar]
- 10.Fasth AE, Bjorkstrom NK, Anthoni M, Malmberg KJ, Malmstrom V. Activating NK-cell receptors co-stimulate CD4(+)CD28(-) T cells in patients with rheumatoid arthritis. Eur J Immunol. 2010 Feb;40:378–87. doi: 10.1002/eji.200939399. [DOI] [PubMed] [Google Scholar]
- 11.Vallejo AN, Schirmer M, Weyand CM, Goronzy JJ. Clonality and longevity of CD4+CD28null T cells are associated with defects in apoptotic pathways. Journal of immunology. 2000 Dec 1;165:6301–7. doi: 10.4049/jimmunol.165.11.6301. [DOI] [PubMed] [Google Scholar]
- 12.Schmidt D, Goronzy JJ, Weyand CM. CD4+ CD7- CD28- T cells are expanded in rheumatoid arthritis and are characterized by autoreactivity. The Journal of clinical investigation. 1996 May 1;97:2027–37. doi: 10.1172/JCI118638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scholz C, Patton KT, Anderson DE, Freeman GJ, Hafler DA. Expansion of autoreactive T cells in multiple sclerosis is independent of exogenous B7 costimulation. Journal of immunology. 1998 Feb 1;160:1532–8. [PubMed] [Google Scholar]
- 14.Lovett-Racke AE, Trotter JL, Lauber J, Perrin PJ, June CH, Racke MK. Decreased dependence of myelin basic protein-reactive T cells on CD28-mediated costimulation in multiple sclerosis patients. A marker of activated/memory T cells. The Journal of clinical investigation. 1998 Feb 15;101:725–30. doi: 10.1172/JCI1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allez M, Tieng V, Nakazawa A, et al. CD4+NKG2D+ T cells in Crohn’s disease mediate inflammatory and cytotoxic responses through MICA interactions. Gastroenterology. 2007 Jun;132:2346–58. doi: 10.1053/j.gastro.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 16.Yokoyama Y, Fukunaga K, Ikeuchi H, et al. The CD4+CD28null and the regulatory CD4+CD25High T-cell phenotypes in patients with ulcerative colitis during active and quiescent disease, and following colectomy. Cytokine. 2011 Nov;56:466–70. doi: 10.1016/j.cyto.2011.06.021. [DOI] [PubMed] [Google Scholar]
- 17.Fasth AE, Snir O, Johansson AA, et al. Skewed distribution of proinflammatory CD4+CD28null T cells in rheumatoid arthritis. Arthritis research & therapy. 2007;9:R87. doi: 10.1186/ar2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnett FC, Edworthy SM, Bloch DA, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988 Mar;31:315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 19.Janson PC, Marits P, Thorn M, Ohlsson R, Winqvist O. CpG methylation of the IFNG gene as a mechanism to induce immunosuppression [correction of immunosupression] in tumor-infiltrating lymphocytes. Journal of immunology. 2008 Aug 15;181:2878–86. doi: 10.4049/jimmunol.181.4.2878. [DOI] [PubMed] [Google Scholar]
- 20.Schaerli P, Moser B. Chemokines: control of primary and memory T-cell traffic. Immunol Res. 2005;31:57–74. doi: 10.1385/IR:31:1:57. [DOI] [PubMed] [Google Scholar]
- 21.Wilson CB, Makar KW, Shnyreva M, Fitzpatrick DR. DNA methylation and the expanding epigenetics of T cell lineage commitment. Semin Immunol. 2005 Apr;17:105–19. doi: 10.1016/j.smim.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 22.Janson PC, Linton LB, Bergman EA, et al. Profiling of CD4+ T cells with epigenetic immune lineage analysis. Journal of immunology. 2011 Jan 1;186:92–102. doi: 10.4049/jimmunol.1000960. [DOI] [PubMed] [Google Scholar]
- 23.Lee DU, Avni O, Chen L, Rao A. A distal enhancer in the interferon-gamma (IFN-gamma) locus revealed by genome sequence comparison. J Biol Chem. 2004 Feb 6;279:4802–10. doi: 10.1074/jbc.M307904200. [DOI] [PubMed] [Google Scholar]
- 24.Shnyreva M, Weaver WM, Blanchette M, et al. Evolutionarily conserved sequence elements that positively regulate IFN-gamma expression in T cells. Proc Natl Acad Sci U S A. 2004 Aug 24;101:12622–7. doi: 10.1073/pnas.0400849101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strickland FM, Richardson BC. Epigenetics in human autoimmunity. Epigenetics in autoimmunity - DNA methylation in systemic lupus erythematosus and beyond. Autoimmunity. 2008 May;41:278–86. doi: 10.1080/08916930802024616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamada H, Nakashima Y, Okazaki K, et al. Th1 but not Th17 cells predominate in the joints of patients with rheumatoid arthritis. Ann Rheum Dis. 2008 Sep;67:1299–304. doi: 10.1136/ard.2007.080341. [DOI] [PubMed] [Google Scholar]
- 27.Brugaletta S, Biasucci LM, Pinnelli M, et al. Novel anti-inflammatory effect of statins: reduction of CD4+CD28null T lymphocyte frequency in patients with unstable angina. Heart. 2006 Feb;92:249–50. doi: 10.1136/hrt.2004.052282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mucida D, Husain MM, Muroi S, et al. Transcriptional reprogramming of mature CD4(+) helper T cells generates distinct MHC class II-restricted cytotoxic T lymphocytes. Nat Immunol. 2013 Mar;14:281–9. doi: 10.1038/ni.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park W, Weyand CM, Schmidt D, Goronzy JJ. Co-stimulatory pathways controlling activation and peripheral tolerance of human CD4+CD28- T cells. Eur J Immunol. 1997 May;27:1082–90. doi: 10.1002/eji.1830270507. [DOI] [PubMed] [Google Scholar]
- 30.Fasth AE, Cao D, van Vollenhoven R, Trollmo C, Malmstrom V. CD28nullCD4+ T cells--characterization of an effector memory T-cell population in patients with rheumatoid arthritis. Scandinavian journal of immunology. 2004 Jul-Aug;60:199–208. doi: 10.1111/j.0300-9475.2004.01464.x. [DOI] [PubMed] [Google Scholar]
- 31.Chabaud M, Durand JM, Buchs N, et al. Human interleukin-17: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis and rheumatism. 1999 May;42:963–70. doi: 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 32.Aarvak T, Chabaud M, Kallberg E, Miossec P, Natvig JB. Change in the Th1/Th2 phenotype of memory T-cell clones from rheumatoid arthritis synovium. Scandinavian journal of immunology. 1999 Jul;50:1–9. doi: 10.1046/j.1365-3083.1999.00581.x. [DOI] [PubMed] [Google Scholar]
- 33.Kotake S, Udagawa N, Takahashi N, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. The Journal of clinical investigation. 1999 May;103:1345–52. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007 Jun;7:429–42. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- 35.Szekanecz Z, Vegvari A, Szabo Z, Koch AE. Chemokines and chemokine receptors in arthritis. Front Biosci (Schol Ed) 2010;2:153–67. doi: 10.2741/s53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Acosta-Rodriguez EV, Rivino L, Geginat J, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007 Jun;8:639–46. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 37.Wengner AM, Hopken UE, Petrow PK, et al. CXCR5- and CCR7-dependent lymphoid neogenesis in a murine model of chronic antigen-induced arthritis. Arthritis and rheumatism. 2007 Oct;56:3271–83. doi: 10.1002/art.22939. [DOI] [PubMed] [Google Scholar]
- 38.Cantaert T, Kolln J, Timmer T, et al. B lymphocyte autoimmunity in rheumatoid synovitis is independent of ectopic lymphoid neogenesis. Journal of immunology. 2008 Jul 1;181:785–94. doi: 10.4049/jimmunol.181.1.785. [DOI] [PubMed] [Google Scholar]
- 39.Syrbe U, Siveke J, Hamann A. Th1/Th2 subsets: distinct differences in homing and chemokine receptor expression? Springer Semin Immunopathol. 1999;21:263–85. doi: 10.1007/BF00812257. [DOI] [PubMed] [Google Scholar]
- 40.Nistala K, Moncrieffe H, Newton KR, Varsani H, Hunter P, Wedderburn LR. Interleukin-17-producing T cells are enriched in the joints of children with arthritis, but have a reciprocal relationship to regulatory T cell numbers. Arthritis and rheumatism. 2008 Mar;58:875–87. doi: 10.1002/art.23291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wedderburn LR, Robinson N, Patel A, Varsani H, Woo P. Selective recruitment of polarized T cells expressing CCR5 and CXCR3 to the inflamed joints of children with juvenile idiopathic arthritis. Arthritis and rheumatism. 2000 Apr;43:765–74. doi: 10.1002/1529-0131(200004)43:4<765::AID-ANR7>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 42.Duftner C, Dejaco C, Kullich W, et al. Preferential type 1 chemokine receptors and cytokine production of CD28- T cells in ankylosing spondylitis. Ann Rheum Dis. 2006 May;65:647–53. doi: 10.1136/ard.2005.042085. [DOI] [PMC free article] [PubMed] [Google Scholar]