

Abstract
Cyclin E1 regulates the initiation of S phase in cellular division. However, in many cancers cyclin E1 is aberrantly overexpressed and this molecular phenotype correlates with increased tumor aggressiveness and poor patient survival. The molecular cause(s) of cyclin E1 abnormalities in cancers is poorly understood. Here, we show cyclin E1 overexpression in cancer is promoted by dysregulation of the protein phosphatase PP2A-B55β. PP2A-B55β targets the N- and C-terminal phosphodegrons of cyclin E1 for dephosphorylation, thus protecting it from degradation mediated by the SCFFbxw7 ubiquitin ligase. Augmented B55β expression stabilizes cyclin E1 and promotes its overexpression in cancer-derived cell lines and breast tumors. Conversely, B55β ablation enforces the degradation of cyclin E1 and inhibits cancer cell proliferation in vitro and tumor formation in vivo. Therefore, PP2A-B55β promotes cyclin E1 overexpression by antagonizing its degradation and its inhibition could represent a therapeutic mechanism for abrogating cyclin E1 function in cancers.
Keywords: B55β, PPP2R2B, Cyclin E, SCFFbxw7, Fbw7
INTRODUCTION
Cyclin E1, an activator of cyclin-dependent kinase 2 (Cdk2), is an important regulator of the G1/S phase transition of the cell division cycle. Cyclin E1 initiates an S phase program by promoting histone biosynthesis, centrosome duplication, and licensing of DNA origins of replication, among other functions (1). Cyclin E1 protein level is tightly regulated during cellular division through coordinated transcriptional and proteolytic controls, which ensure its expression is restricted to the G1/S phase transition. CCNE1, which encodes cyclin E1, is expressed in late G1 phase stimulated by E2F and c-Myc activities through Cdk2-dependent positive feedback loops (1). Once S phase is initiated, cyclin E1 is targeted for ubiquitin-dependent proteolysis by the E3 ubiquitin ligase SCFFbxw7 (2, 3). Ubiquitylation of cyclin E1 is triggered by phosphorylation of residues Thr395, Ser399, and to a lesser extent Thr77, mediated by autophosphorylation and GSK3 kinase, creating N- and C-terminal phosphodegrons (also termed CPDs) that promote its recognition by SCFFbxw7 (4).
A wealth of clinical studies have shown that cyclin E1 is aberrantly overexpressed in many human cancers and this molecular phenotype often correlates with increased tumor aggressiveness and poor patient survival. For example, ~30% of breast cancers overexpress cyclin E1 and this molecular abnormality has been shown to be a stronger prognostic marker of poor patient outcome than important clinicopathological factors such as tumor grade/stage, hormone receptor status, or lymph node metastasis (5). Cyclin E1 overexpression is also a major molecular determinant of basal-like triple negative breast cancers (TNBCs; ER−, PR−, Her2−), a highly aggressive breast cancer sub-type that is common in BRCA1 mutation carriers, and is an independent predictor of poor patient outcome (6). Cyclin E1 overexpression has also been associated with increased resistance to the anti-Her2 therapy trastuzumab in breast cancers (7). At the molecular level, cyclin E1 overexpression alters gene expression programs that regulate cellular division, differentiation, survival, and senescence (1). It has also been shown to promote genetic instability by causing defects in chromosome segregation (8–10), centrosome hyperamplification (11), interference with the pre-replication complex assembly (12), and formation of unstable DNA replication intermediates (13, 14).
Despite the importance of cyclin E1 in cancer the molecular mechanism(s) that promote its dysregulation are poorly understood. In breast cancers, genetic amplifications of CCNE1 occur in only ~5% of cases (15). Furthermore, mutations of FBXW7, which encodes the substrate recognition component of SCFFbxw7, are exceedingly rare (<1%; 16). Therefore, other molecular mechanisms must be responsible for cyclin E1 overexpression in these cancers, which allow for its hyper-accumulation in the presence of functional degradation machinery.
MATERIALS and METHODS
Patient Specimens and Cell Lines
Breast cancer specimens were obtained from patients treated at the Department of Obstetrics and Gynecology of the Innsbruck Medical University in Austria using Institutional Review Board approved procedures. Clinicopathological information for tumor specimens is provided in Table S1. Cell lines were purchased from the American Tissue Culture Collection (ATCC) and grown in the recommended culture mediums. Detailed information regarding tumor analysis, cell line treatments, and plasmid and siRNAs transfections is provided in Supplemental Material and Methods.
Ubiquitylation Assays
In vivo ubiquitylation assays were performed by co-transfecting HEK293T cells with the indicated expression plasmids and siRNAs and 48 hrs later treating cells with 10 µM MG132 for 4 hrs prior to harvesting. Cells were lysed using denaturing conditions (1% SDS) as described (17). The Thr77Ala/Thr395Ala mutant of cyclin E1 was created using the Quikchange Site-Directed Mutagenesis Kit (Strategene). In vitro ubiquitylation reactions were performed as described previously (18). Human recombinant SCFFbxw7 was generated by multi-infecting Sf9 insect cells with the relevant expressing baculoviruses. Recombinant cyclin E1-Cdk2 was purchased from EMD Millipore.
Cell Fractionation
Cells were suspended in buffer containing 10 mM Tris (pH 7.4), 5 mM MgCl2, 10 mM KCl, 300 mM sucrose, 0.1 mM EDTA, 0.5 mM DTT, with added phosphatase and protease inhibitors and incubated on ice for 10 min. NP-40 was then added to a final concentration of 0.125%, the cells vortexed for 10 sec and the cytoplasmic fraction clarified by centrifugation @ 1000 rpm in a microfuge. The nuclear pellet was then washed 4× in buffer containing 10 mM Hepes (pH 7.4), 50 mM NaCl, 25% glycerol, and 0.1 mM EDTA, with the last wash spun @ 6000 rpm in a microfuge. The nuclear fraction was then isolated by incubation in buffer containing 20 mM Tris (pH 7.4), 5 mM MgCl2, 300 mM KCl, 0.2 mM EDTA, 0.5 mM DTT, and 25% glycerol, with phosphatase and protease inhibitors for 15 min, and the lysate clarified by centrifugation @ 13,000 rpm for 15 min.
Phosphatase and Kinase Assays
For phosphatase assays, human recombinant cyclin E1-Cdk2 was incubated with PP2A core enzyme (EMD Millipore) in phosphatase reaction buffer (12.5 mM Tris (pH 7.0), 25 µM CaCl2) for 30 min at 30°C. Okadaic acid (EMD Millipore) and λPPase (New England Biolabs) were added to the indicated reactions. For PP2A-B55β phosphatase assays, PP2A-B55β complexes were assembled in HEK293T cells, immunopurified, and reactions performed as described above using recombinant cyclin E1-Cdk2 as substrate. Anti-Flag antibodies were purchased from Sigma-Aldrich. Kinase assays were performed by incubating human recombinant pRb with cyclin E1 immunoprecipitated from cell extracts (250 µg) with antibody HE172. Reactions were performed at 30°C for 30min in reaction buffer containing 20mM Tris (pH 7.5), 7.5 mM MgCl2, and γ-32p-ATP.
Mouse Tumor Studies
MDA-MB-231-luc-D3H2LN cells were transduced with control or B55β shRNA expressing lentiviruses, combined with Matrigel (BD Biosciences), and 2×106 cells injected into the mammary fat pads of 4–6 week old female beige nude mice. Twelve mice were injected for each group. Tumor growth and metastasis were monitored using a Xenogen IVIS 200 Imaging System.
Statistical Analysis
Linear and nonlinear regression analyses were used to evaluate potential correlations between expression of PP2A β-subunits, Fbxw7α, and CCNE1 and cyclin E1 protein levels in breast tumors and differences in cell and tumor growth rates, respectively. A p-value <0.05 was considered statistically significant. All calculations were performed using GraphPad Prizm 5 software (GraphPad Software Inc.).
RESULTS
PP2A dephosphorylates cyclin E1 and protects it from ubiquitylation by SCFFbxw7
Since cyclin E1 degradation is triggered by Cdk2 and GSK3 phosphorylations, we explored whether cyclin E1 overexpression in cancers could possibly be caused by dysregulation of a protein phosphatase that counteracted these activities. Cyclin E1 and components of protein phosphatase 2A (PP2A) were previously identified in immunocomplexes of DNA polymerase α-primase (Pol-Prim) (19). Interestingly, PP2A had been previously shown to regulate the phosphorylation status and degradation of c-Myc, another SCFFbxw7 substrate (20, 21). PP2A is a Ser/Thr phosphatase which plays important roles in cell division control and cancer. We confirmed that both expressed and endogenous cyclin E1 interacted with the Aα (structural) and Cα (catalytic) subunits of PP2A by immunoprecitiation (IP)-Western blot analysis (Figs. 1A and B). Incubation of recombinant cyclin E1-Cdk2 complexes with recombinant PP2A core enzyme showed that PP2A could dephosphorylate both the N- and C-terminal phosphodegrons of cyclin E1 (Fig. 1C). In support of this in vitro result, knockdown of Cα subunit expression in HeLa cells by siRNA was found to induce significant increases in phosphorylation of both Thr77 and Thr395 of cyclin E1 (Fig. 1D). These data suggested that PP2A might function to protect cyclin E1 from ubiquitylation by counteracting Cdk2-and GSK3-mediated phosphorylations. To test this, we co-expressed cyclin E1, Cdk2, Fbxw7α, and HA-ubiquitin in HEK293T cells and then knocked down Cα expression by siRNA. IP-Western blot analysis revealed that Cα knockdown significantly increased ubiquitylation of cyclin E1 in vivo (Fig. 1E). However, no increase in ubiquitylation was observed in similar experiments that used a mutant version of cyclin E1 that had both its phosphodegrons mutated (Thr77Ala/Thr395Ala), confirming the effect was mediated through cyclin E1 phosphorylation (Fig. 1E). Furthermore, addition of PP2A core enzyme to in vitro ubiquitylation reactions of cyclin E1 by SCFFbxw7 was found to decrease the ubiquitylation efficiency (Fig. 1F). Collectively, these results indicated that PP2A counteracted cyclin E1 phosphorylations, thus protecting it from ubiquitylation by SCFFbxw7.
Figure 1. PP2A dephosphorylates cyclin E1 and antagonizes its ubiquitylation by SCFFbxw7.
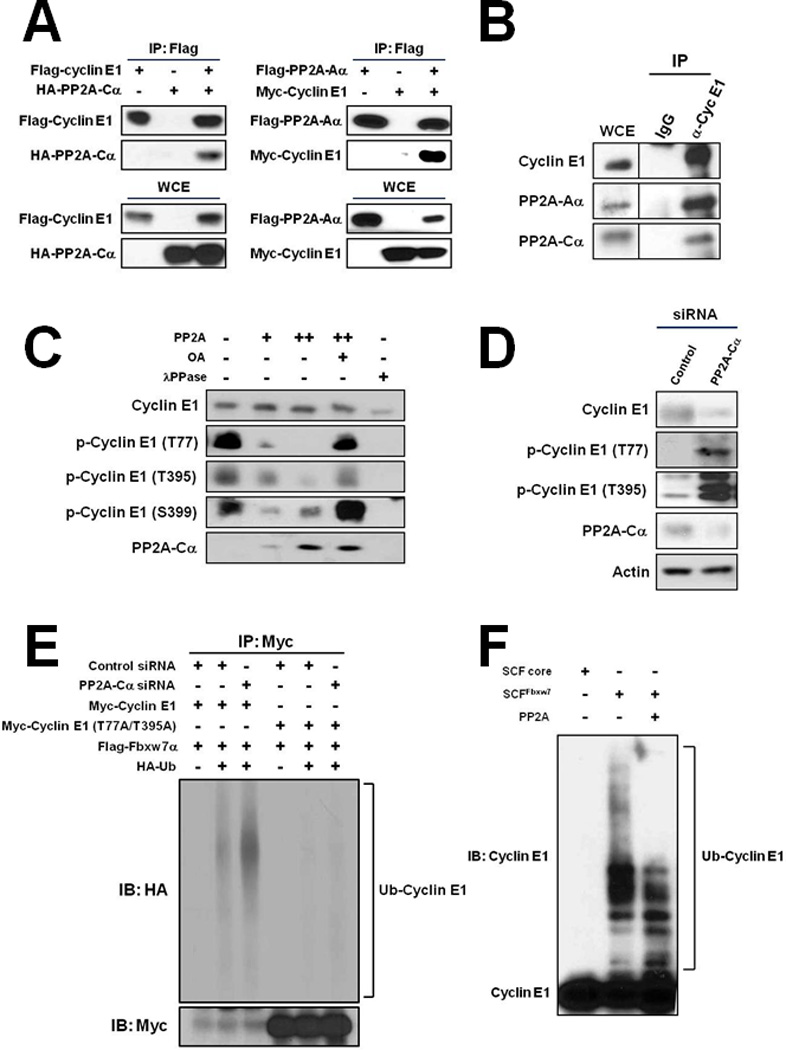
A, Cyclin E1 co-IPs with PP2A subunits Cα (left) and Aα (right) expressed in HEK293T cells. WCE, whole cell extract. B, Endogenous cyclin E1 co-IPs with endogenous Aα and Cα from HeLa cells. C, Increased phosphorylation of the N- and C-terminal phosphodegrons (p-Thr77, p-Thr395) of cyclin E1 in HeLa cells transfected with Cα siRNA. Actin is shown as a loading control. MG132 (10 µM) was added to the cells 1 hr prior to harvesting to stabilize phosphorylated forms of cyclin E1. D, PP2A dephosphorylates cyclin E1 phosphodegrons in vitro. Immunoblot of p-Thr77, p-Thr395, and p-Thr399 of recombinant cyclin E1-Cdk2 incubated with recombinant PP2A core enzyme (A/C subunits). PP2A inhibitor okadaic acid (OA) and non-specific phosphatase λPPase are shown as controls. E, PP2A protects cyclin E1 from ubiquitylation in vivo. HEK293T cells were transfected with the indicated expression plasmids and siRNAs, Myc-cyclin E1 or mutant Myc-cyclin E1 (Thr77Ala/Thr395Ala) immunoprecipitated, and ubiquitylated forms detected by immunoblotting with anti-HA antibodies. Exposure below demonstrates IP efficiency of Myc-cyclin E1 and Myc-cyclin E1 (Thr77Ala/Thr395Ala). F, PP2A reduces E1 ubiquitylation by SCFFbxw7α in vitro. Recombinant cyclin E1-Cdk2 complexes were incubated with recombinant SCFFbxw7α. PP2A core enzyme was added to the indicated reaction. SCF core is SCFFbxw7α without substrate recognition component Fbxw7α.
PP2A-B55β regulates cyclin E1 phosphorylation
PP2A exists as a dimeric core composed of A and C subunits bound to a regulatory (B) subunit, which dictates sub-cellular localization and substrate specificity (22). Humans express 2-A subunits, 2-C subunits, and 16-B subunits, allowing for a diverse array of enzyme compositions, each with presumably a unique set of substrates. Ectopic co-expression experiments revealed that cyclin E1 co-precipitated with several subunits of the B55 and B56 β-subunit families in vivo (Fig. 2A). Knockdown of expression of these β-subunits in HeLa cells showed that B55β and B56δ, and a lesser extent B56β, reduced cyclin E1 protein levels comparable to Cα knockdown (Fig. 2B), as would be expected if their loss of function de-protected cyclin E1 from ubiquitin-dependent proteolysis. Several lines of evidence led us to exclude both B56β and B56δ as potential regulators of cyclin E1. Firstly, we could not verify an in vivo interaction between these β-subunits and endogenous cyclin E1 protein (Fig. S1). Secondly, qPCR analysis failed to detect any correlation between B56β or B56δ expression and cyclin E1 protein levels in breast cancer specimens (described below). Thirdly, the expression of both B56β and B56δ during the cell division cycle were not consistent with a potential role in cyclin E1 degradation, with both peaking in S phase when cyclin E1 degradation is maximal (see Fig. 3B). In addition, mass spectrometry analysis of B55β identified several β-subunits as potential interacting proteins, suggesting the observed interactions between B56β or B56δ and cyclin E1 could be indirect (data not shown). We therefore focused on B55β, encoded by PPP2R2B, and its potential regulation of cyclin E1. Knockdown of B55β expression using 3 different siRNAs confirmed a reductive effect on cyclin E1 protein level, even though CCNE1 expression and cell cycle kinetics were not adversely affected (Figs. S2 & S3). B55β was found to co-precipitate with endogenous cyclin E1 protein (Fig. 2C). Cell fractionation experiments also showed that B55β localized to both the cytoplasm and nucleus in HeLa cells, mirroring cyclin E1 (Fig. 2D). This result was contrary to previous reports which found B55β is localized to the cytoplasm (23, 24). In addition, treatment of cells with proteasome inhibitor MG132 or chemical inhibitors of Cdk2 and GSK3 kinases was found to diminish the reduction in cyclin E1 caused by B55β knockdown, confirming that the effect of B55β on cyclin E1 was both proteasome- and phosphorylation-dependent (Figs. 2E & 2F). Moreover, PP2A-B55β complexes assembled in HEK293T cells were found to efficiently dephosphorylate both the N- and C-terminal phosphodegrons of recombinant cyclin E1 in vitro (Fig. 2G). Collectively, these data demonstrated that PP2A-B55β selectively targets the N- and C-terminal phosphodegrons of cyclin E1 for dephosphorylation.
Figure 2. PP2A-B55β regulates cyclin E1 phosphorylation.
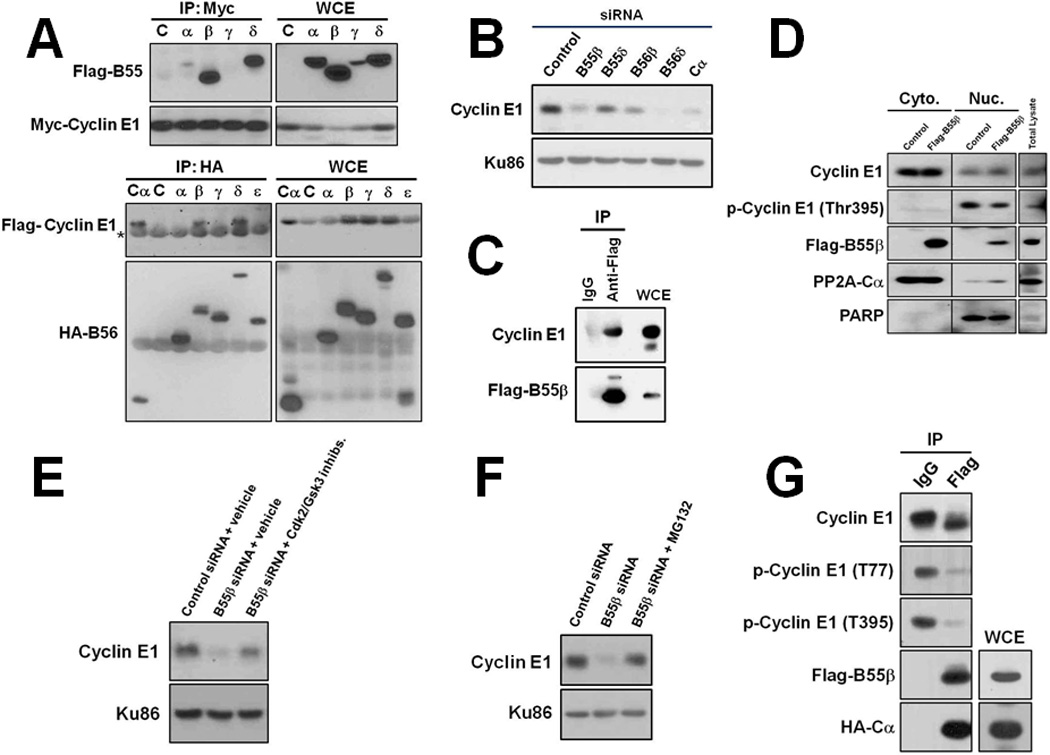
A, Cyclin E1 co-IPs with B-regulatory subunits B55β, B55δ, B56β, and B56δ in HEK293T cells. Asterisk indicates IgG heavy chain. B, Knockdown of B55β, B56β and B56δ expression in MDA-MB-231 cells decreases cyclin E1 protein levels. Ku86 is shown as a loading control. C, Endogenous cyclin E1 co-IPs with Flag-B55β expressed in HeLa cells. D, B55β is localized to the cytoplasm and nucleus in cells. HeLa cells were transfected with a plasmid that expresses Flag-B55β and cytoplasmic and nuclear fractions isolated. Exclusive nuclear protein PARP is shown as verification of fractionation efficiency. E, B55β knockdown-induced decrease of cyclin E1 in MDA-MB-231 cells is proteasome-dependent. B55β expression was knocked-down for 48 hrs and proteasome inhibitor Mg132 added 4 hrs prior to harvesting cells. F, B55β knockdown-induced decrease of cyclin E1 is phosphorylation-dependent. Cells were treated with Cdk2 and GSK3 inhibitors for 4 hrs prior to harvesting. G, PP2A-B55β dephosphorylates cyclin E1 in vitro. PP2A-B55β complexes were assembled in HEK293T cells and used in in vitro phosphatase reactions with recombinant cyclin E1-Cdk2 as substrate.
Figure 3. B55β regulates cyclin E1 stability and determines its level in cell lines and cell division cycles.
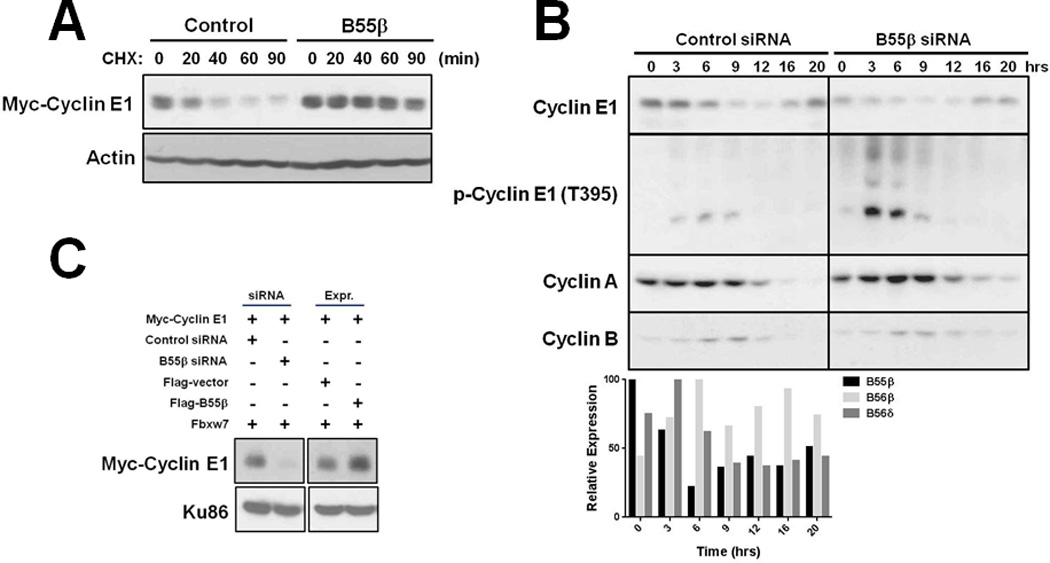
A, B55β stabilizes cyclin E1. HEK293T cells were co-transfected with plasmids that express Myc-cyclin E1, Cdk2, Flag-Fbxw7α, and Flag-B55β or empty vector, protein synthesis halted by cyclohexamide (CHX) treatment, and Myc-cyclin E1 stability analyzed by immunoblotting. B, B55β is required for proper accumulation of cyclin E1 at the G1/S transition. HeLa cells were transfected with control or B55β siRNA, the cell cycle blocked at the G1/S boundary, and then released from block and cell cycle proteins analyzed by immunoblotting. qPCR data for expression of B55β, B56β and B56δ is shown below. C, B55β determines cyclin E1 levels in MDA-MB-231 breast cancer cells.
PP2A-B55β regulates cyclin E1 stability and determines its level in cell division cycles and cancer-derived cell lines
We next evaluated the potential importance of PP2A-B55β in determining cyclin E1 levels in cellular division and cancer. Cyclohexamide (CHX) half-life determination experiments showed that B55β expression significantly increased the stability of cyclin E1 expressed in HEK293T cells (20 min vs. >90 min) (Fig. 3A), consistent with its role in protecting cyclin E1 from ubiquitin-dependent proteolysis by SCFFbxw7. To determine whether B55β could play a role in the regulation of cyclin E1 in cell division cycles, we synchronized HeLa cells transfected with control or B55β siRNAs at the G1/S boundary and then analyzed cyclin E1 levels by Western blotting following release into the cell division cycle. Knockdown of B55β expression was found to reduce the accumulation of cyclin E1 at the G1/S boundary and this was accompanied by increased Thr395 phosphorylation, whereas cyclin A and B1 levels were unaffected (Fig. 3B). In addition, qPCR analysis revealed B55β expression mirrored cyclin E1 protein; peaking at the G1/S boundary and decreasing as cells progressed into S phase (Fig. 3B). To examine the potential influence of B55β on cyclin E1 levels in cancer cells, we transfected MDA-MB-231 TNBC cells with plasmids that express Myc-cyclin E1 and all components required for its degradation (e.g. Fbxw7, Cdk2), and then modulated B55β expression by siRNA or ectopic expression. Whereas B55β knockdown significantly reduced cyclin E1 protein level, its ectopic expression induced cyclin E1 hyperaccumulation (Fig. 3C). These results suggested that cyclin E1 levels in cancer cells were largely determined by PP2A-B55β. To test this hypothesis, we quantified the levels of cyclin E1 protein and B55β and CCNE1 expression in breast cancer specimens by Western blotting and qPCR, respectively. Remarkably, cyclin E1 protein level was found to correlate more strongly with B55β expression than CCNE1 expression (R2=0.41 vs. 0.14) (Fig. 4A), and this correlation was enhanced in breast cancers that expressed CCNE1 at ≥50% above the mean (R2=0.70 vs. 0.10) (Fig. 4B). Since cyclin E1 degradation is regulated by SCFFbxw7, we also analyzed its potential influence on determining cyclin E1 levels in breast cancers. qPCR analysis failed to uncover a significant correlation between cyclin E1 protein level and Fbxw7α expression in these specimens (Fig. S4). Furthermore, our previous analysis showed that these same breast cancer specimens do not contain mutations of the FBXW7 gene, a potential mechanism of SCFFbxw7 inactivation in cancers (16). Of note, we also found no correlations between cyclin E1 protein levels and expression of B56β or B56δ (Fig. S4). Therefore, these data demonstrated that the level of cyclin E1 in cancer cells is largely determined by B55β.
Figure 4. B55β expression correlates with cyclin E1 levels in breast cancers and its knockdown inhibits breast cancer cell proliferation.
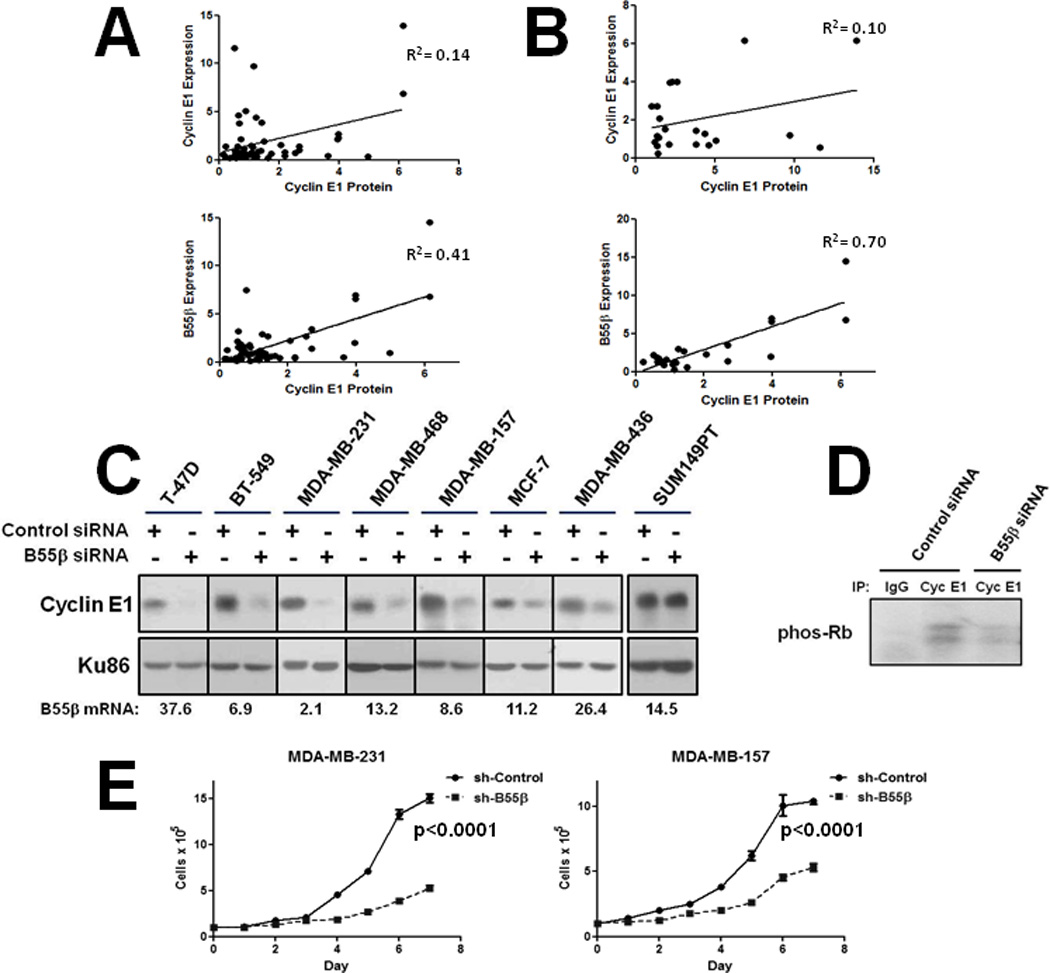
A & B, B55β expression correlates with cyclin E1 protein level in breast cancers. B55β and CCNE1 expression were quantified by qPCR and cyclin E1 protein by Western blot analysis. A, All tumors. B, Breast cancer specimens that express CCNE1 at ≥50% above the mean. C, B55β expression knockdown reduces cyclin E1 levels in a panel of breast cancer cell lines with functional SCFFbxw7. qPCR data showing B55β knockdown efficiency is shown below. Cell line SUM149PT contains defective FBXW7 alleles. D, B55β knockdown reduces cyclin E1-associated kinase activity in breast cancer cells. Cyclin E1-Cdk2 was immunoprecipitated from extracts and used in in vitro kinase assays with human recombinant pRb as substrate. E, B55β knockdown inhibits breast cancer cell proliferation. TNBC cell lines MDA-MB-231 and MDA-MB-157 were transduced with control or B55β shRNA-expressing lentiviruses and cells counted. Experiments were performed in triplicate.
PP2A-B55β inhibition reduces breast cancer cell growth in vitro and tumor formation in vivo
We next tested the potential therapeutic application of PP2A-B55β inhibition as a means of decreasing cyclin E1 levels in breast cancer cells. Knockdown of B55β expression in a panel of breast cancer cell lines representing both luminal hormone receptor positive (ER+, PR+; MCF-7, T47-D) and basal-like TNBCs (ER−, PR−, Her2−; BT-549, MDA-MB-231, MDA-MB-468, MDA-MB-157, MDA-MB-436, SUM149PT) was found to decrease cyclin E1 protein levels in all cell lines except SUM149PT (Fig. 4C), which has been shown to harbor FBXW7 gene mutations (25). Of note, B55β knockdown even reduced cyclin E1 levels in MDA-MB-157 cells, which contain a CCNE1 gene amplification and overexpress cyclin E1 at 64-fold the level of normal breast epithelial cells (26). B55β expression knockdown also resulted in reduced cyclin E1-Cdk2 associated kinase activity (Fig. 4D). To evaluate the effects of B55β expression knockdown on breast cancer cell growth, we tranduced two basal-like TNBC cell lines (MDA-MB-231 and MDA-MB-157) with B55β shRNA-expressing or control lentiviruses. B55β knockdown was found to significantly reduce cell proliferation long-term (Fig. 4E). Moreover, MDA-MB-231-luc cells transduced with B55β shRNA-expressing lentiviruses displayed a significantly reduced growth rate and ability to form tumors when implanted into the mammary fat pads of nude mice compared to control cells (Figs. 5A and B). The B55β-shRNA expressing tumors also failed to metastasize to bronchial lymph nodes (0/12 vs. 2/12 for control tumors) (Fig. 5C), the preferred metastatic site in this model system. These results demonstrated that B55β inhibition can efficiently reduce cyclin E1 levels in breast cancer cells, and limits their growth and tumorigenicity.
Figure 5. B55β knockdown inhibits breast cancer formation in nude mice and model of PP2A-B55β regulation of cyclin E1 in cellular division and cancer.
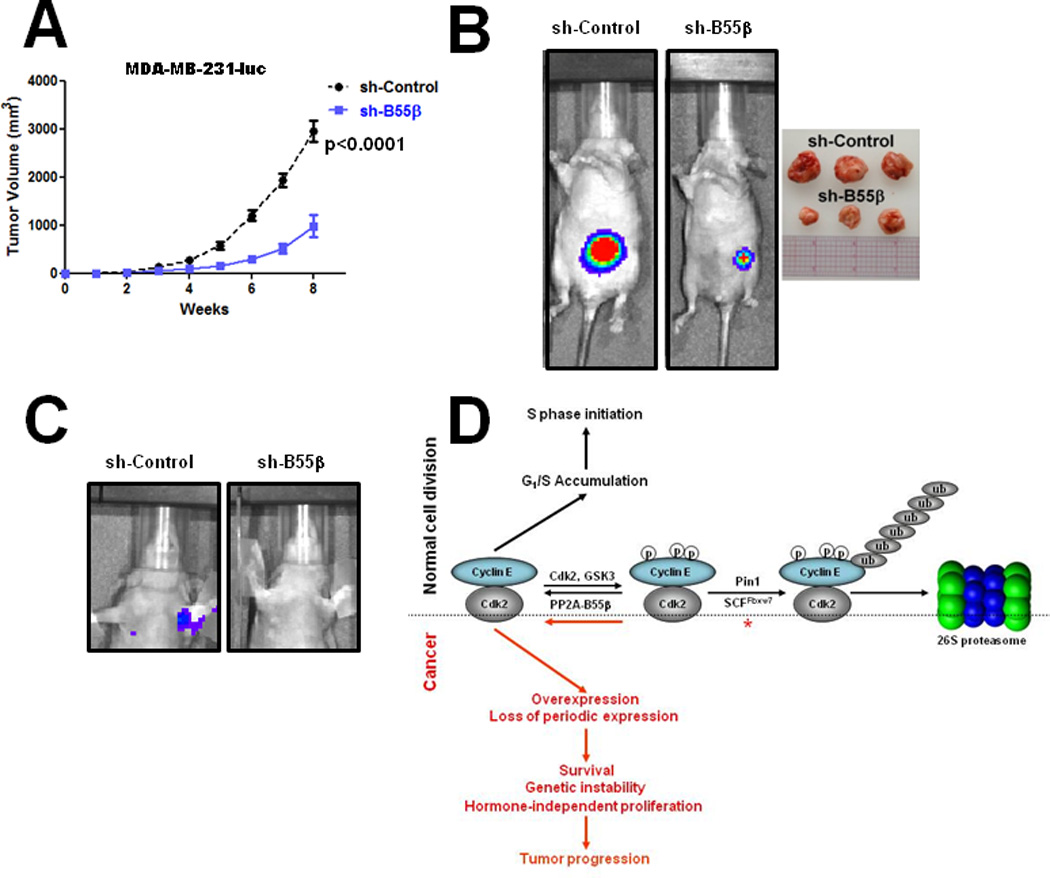
A, Growth curves of control and B55β shRNA-expressing breast cancers. MDA-MB-231-luc cells were transduced with control or B55β shRNA-expressing lentiviruses and then implanted into the mammary fat pads of nude mice. Twelve mice were analyzed for each cell line. B, Representative luminescent images of tumors 6 weeks post-implantation. Examples of excised tumors at 8 weeks post-implantation are shown on right. C, B55β knockdown inhibits metastasis progression. Luminescent images of mice 8 weeks post-implantation showing bronchial metastases of a control cell tumor but not B55β shRNA-expressing tumor. D, Model of B55β regulation of cyclin E1 in cellular division and cancer. Details explained in text.
DISCUSSION
Although cyclin E1 overexpression is an important prognostic marker in breast and other cancers, the molecular cause(s) of these alterations was poorly understood. Our data shows that the protein phosphatase PP2A-B55β regulates cyclin E1 levels in cellular division cycles and contributes to its dysregulation in cancers (Fig. 5D). During G1 phase PP2A-B55β antagonizes phosphorylations of cyclin E1 mediated by autophosphorylation and GSK3 kinase, allowing for cyclin E1 to accumulate and initiate an S phase program. After S phase is initiated, B55β levels decline which de-protects cyclin E1 from phosphorylation thus triggers its recognition and ubiquitylation by SCFFbxw7. In support of a possible involvement of PP2A in cyclin E1 regulation, a recent report found that in yeast PP2ACdc55 controls the stability of G1 cyclin Cln2 through regulation of its phosphorylation state (27). In cancers, this regulation can be aberrant and augmented B55β expression functions to antagonize cyclin E1 degradation, leading to its hyperaccumulation especially in cells that overexpress CCNE1. The overexpressed cyclin E1 then promotes cancer cell survival, hormone-independent growth, genetic instability, and progression to advanced disease.
The ability of augmented B55β to promote cyclin E1 overexpression suggests that it could function as an oncoprotein in human tumorigenesis. Indeed, a search of the ONCOMINE database (28) revealed B55β is overexpressed in several cancer types including breast, ovary, brain, and liver (Fig. S5). However, a previous report showed that PPP2R2B is often transcriptionally repressed by promoter hypermethylation in colorectal cancers, suggesting it functions as a tumor suppressor in these cancers (29). Contrary to this study though, we found that knockdown of B55β expression in colorectal cancer cell lines HT29 and HCT116, which contain hypermethylated PPP2R2B alleles, significantly reduced their proliferation and cyclin E1 levels (Fig. S6). A possible explanation for this discrepancy could be that reduced B55β expression imparts tumorigenic properties on colorectal cancer cells that are independent of cyclin E1, though B55β expression is indispensible for proliferation.
PP2A-B55β ablation reduced cyclin E1 protein levels in breast cancer cells which contain functional SCFFbxw7, suggesting its inhibition could represent a rational therapeutic strategy for inactivating cyclin E1 function in cancers. The oncogenic potential of cyclin E1 has been shown to be mediated through both Cdk2 dependent and independent functions, the latter of which includes promotion of G0-S phase transition and oncogene mediated transformation (30). Therefore, PP2A-B55β inhibition could represent a more effective therapeutic mechanism for abrogating cyclin E1 functions in cancers than drugs that target Cdk2 kinase activity, since both Cdk2 dependent and independent functions would be affected. PP2A-B55β directed therapies might be particularly effective for treatment of highly aggressive basal-like TNBCs, which typically overexpress cyclin E1, lack targeted and effective therapies, and whose growth and survival has been shown to depend on these abnormalities (31). They might also be effective as a prophylactic treatment for carriers of BRCA1 mutations, who exhibit an increased risk of developing basal-like TNBCs in their lifetime.
Supplementary Material
ACKNOWLEDGEMENTS
We thank D. Perrotti for plasmid pHM6-PP2A-Cα and B. Wadzinski for B55 family expression plasmids. We thank B. Clurman for anti-p-cyclin E1 (S399) antibody. We thank S. Reed for anti-cyclin E1 antibodies HE12 and HE172 and human recombinant pRb. We also thank Stefan Grotegut for help with Oncomine database searches.
Financial support: Department of Defense Breast Cancer Research Program
GRANT SUPPORT
This work was supported by a grant from the DOD Breast Cancer Research Program (W81XWH-11-1-0176) to C.S.
Footnotes
Conflicts of interest: None
REFERENCES
- 1.Siu KT, Rosner M, Minella AC. An integrated view of cyclin E function and regulation. Cell Cycle. 2012;1:57–64. doi: 10.4161/cc.11.1.18775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 3.Tan Y, Sangfelt O, Spruck C. The Fbxw7/hCdc4 tumor suppressor in human cancer. Cancer Lett. 2008;271:1–12. doi: 10.1016/j.canlet.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 4.Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP. Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell. 2007;26:131–143. doi: 10.1016/j.molcel.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 5.Keyomarsi K, Tucker SL, Buchholz TA, Callister M, Ding Y, Hortobagyi GN, et al. Cyclin E and survival in patients with breast cancer. N Engl J Med. 2002;347:1566–1575. doi: 10.1056/NEJMoa021153. [DOI] [PubMed] [Google Scholar]
- 6.Foulkes WD, Brunet JS, Stefansson IM, Straume O, Chappuis PO, Begin LR, et al. The prognostic implications of the basal-like (cyclin E(high)/p27(low)/p53+/glomeruloid-microvascular-proliferation+) phenotype of BRCA1-related breast cancer. Cancer Res. 2004;64:830–835. doi: 10.1158/0008-5472.can-03-2970. [DOI] [PubMed] [Google Scholar]
- 7.Scaltriti M, Eichhorn PJ, Cortes J, Prudkin L, Aura C, Jimenez J, et al. Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in Her2+ breast cancer patients. Proc Natl Acad Sci USA. 2011;108:3761–3766. doi: 10.1073/pnas.1014835108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spruck CH, Won KA, Reed SI. Deregulated cyclin E induces chromosome instability. Nature. 1999;401:297–300. doi: 10.1038/45836. [DOI] [PubMed] [Google Scholar]
- 9.Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, et al. Inactivation of hCDC4 can cause chromosome instability. Nature. 2004;428:77–81. doi: 10.1038/nature02313. [DOI] [PubMed] [Google Scholar]
- 10.Keck JM, Summers MK, Tedesco D, Ekholm-Reed S, Chuang LC, Jackson PK, et al. Cyclin E overexpression impairs progression through mitosis by inhibiting APC(Cdh1) J Cell Biol. 2007;178:371–385. doi: 10.1083/jcb.200703202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saavedra HI, Maiti B, Timmers C, Altura R, Tokuyama Y, Fukasawa K, et al. Inactivation of E2F3 results in centrosome amplification. Cancer Cell. 2003;3:333–346. doi: 10.1016/s1535-6108(03)00083-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ekholm-Reed S, Mendez J, Tedesco D, Zetterberg A, Stillman B, Reed SI. Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J Cell Biol. 2004;165:789–800. doi: 10.1083/jcb.200404092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 14.Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–446. doi: 10.1016/j.cell.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Natrajan R, Mackay A, Wilkerson PM, Lambros MB, Wetterskog D, Arnedos M, et al. Functional characterization of the 19q12 amplicon in grade III breast cancers. Breast Cancer Res. 2012;20:R53. doi: 10.1186/bcr3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007;67:9006–9012. doi: 10.1158/0008-5472.CAN-07-1320. [DOI] [PubMed] [Google Scholar]
- 17.Spruck C, Strohmaier H, Watson M, Smith AP, Ryan A, Krek TW, et al. A CDK-independent function of mammalian Cks1: targeting of SCF(Skp2) to the CDK inhibitor p27Kip1. Mol Cell. 2001;7:639–650. doi: 10.1016/s1097-2765(01)00210-6. [DOI] [PubMed] [Google Scholar]
- 18.Klotz K, Cepeda D, Tan Y, Sun D, Sangfelt O, Spruck C. SCF(Fbxw7/hCdc4) targets cyclin E2 for ubiquitin-dependent proteolysis. Exp Cell Res. 2008;315:1832–1839. doi: 10.1016/j.yexcr.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 19.Dehde S, Rohaly G, Schub O, Nasheuer HP, Bohn W, Chemnitz J, et al. Two Immunologically distinct human DNA polymerase alpha-primase subpopulations are involved in cellular DNA replication. Mol Cell Biol. 2001;21:2581–2593. doi: 10.1128/MCB.21.7.2581-2593.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004;6:308–318. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- 21.Arnold HK, Sears RC. Protein phosphatase 2A regulatory subunit B56alpha associates with c-myc and negatively regulates c-Myc accumulation. Mol Cell Biol. 2006;26:2832–2844. doi: 10.1128/MCB.26.7.2832-2844.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Virshup DM, Shenolikar S. From promiscuity to precision: protein phosphatases get a makeover. Mol Cell. 2009;33:537–545. doi: 10.1016/j.molcel.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 23.Dagda RK, Barwacz CA, Cribbs JT, Strack S. Unfolding-resistant translocase targeting: a novel mechanism for outer mitochondrial membrane localization exemplified by the Bbeta2 regulatory subunit of protein phosphatase 2A. J Biol Chem. 2005;280:27375–27382. doi: 10.1074/jbc.M503693200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang YC, Lee CM, Lee LC, Tung LC, Hsieh-Li HM, Lee-Chen GJ, et al. Mitochondrial dysfunction and oxidative stress contribute to the pathogenesis of spinocerebellar ataxia type 12 (SCA12) J Biol Chem. 2011;285:21742–21754. doi: 10.1074/jbc.M110.160697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature. 2001;413:316–322. doi: 10.1038/35095076. [DOI] [PubMed] [Google Scholar]
- 26.Keyomarsi K, Pardee AB. Redundant cyclin overexpression and gene amplification in breast cancer cells. Proc Natl Acad Sci USA. 1993;90:1112–1116. doi: 10.1073/pnas.90.3.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCourt P, Gallo-Ebert C, Gonghong Y, Jiang Y, Nickels JT., Jr PP2A(Cdc55) regulates G1 cyclin stability. Cell Cycle. 2013;12:1201–1210. doi: 10.4161/cc.24231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–180. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan J, Lee PL, Li Z, Jiang X, Lim YC, Hooi SC, et al. B55β-associated PP2A complex controls PDK1-directed Myc signaling and modulates rapamycin sensitivity in colorectal cancer. Cancer Cell. 2010;18:459–471. doi: 10.1016/j.ccr.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 30.Geng Y, Yu Q, Sicinska E, Das M, Schneider JE, Bhattacharya S, et al. Cyclin E ablation in the mouse. Cell. 2003;114:431–443. doi: 10.1016/s0092-8674(03)00645-7. [DOI] [PubMed] [Google Scholar]
- 31.Liang Y, Gao H, Lin SY, Goss JA, Brunicardi FC, Li K. siRNA-based targeting of cyclin E overexpression inhibits breast cancer cell growth and suppresses tumor development in breast cancer mouse models. PLoS ONE. 2010;5:e12860. doi: 10.1371/journal.pone.0012860. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.