

Abstract
Idiopathic pulmonary arterial hypertension (iPAH) is associated with high morbidity and mortality. We evaluated whether luminal delivery of the human prostacyclin synthase (hPGIS) cDNA with adeno-associated virus (AAV) vectors could attenuate PAH. AAV serotype 5 (AAV5) and AAV9 vectors containing the hPGIS cDNA under the control of a cytomegalovirus-enhanced chicken β-actin (CB) promoter or vehicle (saline) were instilled into lungs of rats. Two days later, rats were injected with monocrotaline (MCT, 60 mg/kg) or saline. Biochemical, hemodynamic, and morphologic assessments were performed when the rats developed symptoms (3–4 weeks) or at 6 weeks. Luminal (airway) administration of AAV5 and AAV9CBhPGIS vectors (MCT-AAV5 and MCT-AAV9 rats) significantly increased plasma levels of 6-keto-PGF1α as compared with MCT-controls, and closely resembled levels measured in rats not treated with MCT (saline–saline). Right ventricular (RV)/left ventricular (LV)+septum (S) ratios and RV systolic pressure (RVSP) were greater in MCT-control rats than in saline–saline rats, whereas the ratios and RVSP in MCT-AAV5CBhPGIS and MCT-AAV9CBhPGIS rats were similar to saline–saline rats. Thickening of the muscular media of small pulmonary arteries of MCT-control rats was detected in histological sections, whereas the thickness of the muscular media in MCT-AAV5CBhPGIS and MCT-AAV9CBhPGIS rats was similar to saline–saline controls. In experiments with different promoters, a trend toward increased levels of PGF1α expression was detected in lung homogenates, but not plasma, of MCT-treated rats transduced with an AAV9-hPGIS vector containing a CB promoter. This correlated with significant reductions in the RV/LV+S ratio and RVSP in MCT-AAV9CBhPGIS rats that resembled levels in saline–saline rats. No changes in levels of PGF1α, RV/LV+S, or RVSP were detected in rats transduced with AAV9-hPGIS vectors containing a modified CB promoter (CB7) or a distal epithelial cell-specific promoter (CC10). Thus, AAV9CBhPGIS vectors prevented development of MCT-induced PAH and associated pulmonary vascular remodeling.
Introduction
Idiopathic pulmonary arterial hypertension (iPAH) is a potentially fatal disease of the pulmonary vasculature characterized by pulmonary artery endothelial cell, smooth muscle cell, and adventitial cell proliferation leading to vascular luminal obliteration, increased pulmonary vascular pressures and resistance, and right-sided heart failure with resultant high morbidity and mortality (McLaughlin et al., 2002). Current therapies, including prostacyclin and its analogs, endothelial receptor antagonists, and phophodiesterase-5 inhibitors, have led to improvements in dyspnea, exercise tolerance, and survival (Rubin et al., 2002; Galie and Branzi, 2005; Galie et al., 2005; McLaughlin et al., 2005; Sitbon et al., 2005; Mclaughlin, 2013). However, far too many affected individuals still die prematurely.
Although familial disease has been linked to mutations in BMPR2, Alk1, endoglin, and SMAD9 (Deng et al., 2000; International PPH Consortium et al., 2000; Shintani et al., 2009), the etiology of the majority of cases of PAH is complex with multiple pathophysiological mechanisms. A variety of molecular approaches have been considered for therapy of PAH, including cell-based approaches and direct vector delivery to the lung or vasculature (Campbell et al., 1999, 2001; Nagaya et al., 2000; Kido et al., 2005; McMurtry et al., 2005; Zhao et al., 2005). Prostacyclin synthase (PGIS), the rate -limiting enzyme for prostacyclin production, is an attractive therapeutic transgene for molecular approaches, as prostacyclin infusions have been reasonably well tolerated clinically and improve exercise tolerance and survival in patients with PAH. Attenuation of chronic hypoxic PAH by overexpression of the rat PGIS cDNA under the control of the epithelial-specific human surfactant protein C (hSPC) promoter established the feasibility of paracrine delivery of prostacyclin to the pulmonary vasculature (Geraci et al., 1999). Intratracheal and direct hepatic injections of the hemagglutinating virus of Japan (HVJ)-PGIS liposomes have confirmed this concept (Ono et al., 2004). Delivery of adeno-associated virus serotype 1 (AAV1)- and AAV2-hPGIS vectors by intramuscular injection has been shown to attenuate monocrotaline (MCT)-induced and chronic hypoxic PAH in rodents (Ito et al., 2007; Kawakami et al., 2007; Kataoka et al., 2013). Recently, attenuation of established MCT-induced PAH by aerosolized airway luminal delivery of AAV1-SERCA2 with a Penn Century Device has been reported (Hadri et al., 2013).
In this study, we evaluated whether noninvasive luminal airway delivery of AAV5- and AAV9-hPGIS serotype vectors could attenuate MCT-induced PAH. We also tested the ability of constitutive and tissue-specific promoters to mediate efficient gene transfer and attenuate MCT-induced PAH.
Materials and Methods
Vector production
Several vector constructs were generated for this project (Fig. 1A). The hPGIS cDNA was originally cloned from human mRNA by Dr. Wanda O'Neal (University of North Carolina at Chapel Hill, NC) and cloned into an AAV2 plasmid vector backbone (R. Jude Samulski, University of North Carolina at Chapel Hill, NC). This transgene was also cloned from human mRNA into the AAV backbone by Dr. Julie Johnston (Penn Vector Core, Philadelphia, PA). Three different promoters provided by the Penn Vector Core were used for our studies: the cytomegalovirus (CMV)-enhanced chicken β-actin (CB) promoter (Fisher et al., 1996; Auricchio et al., 2002), a modified CB (CB7) promoter (Penn Vector Core), and the CC10 promoter (Stripp et al., 1992; Auricchio et al., 2002). The CB7 promoter contains 282 bp of the core CB promoter as compared with 260 bp within the CB promoter, a second CMV enhancer element, and a large hybrid intron with a CB splice donor and rabbit β-globin splice acceptor. The AAV5 and AAV9 capsids were also provided by the Penn Vector Core. For this study, vectors produced with the AAV5 and AAV9 capsids contain an AAV2 vector genome and are referred to henceforth as AAV5 and AAV9 vectors.
FIG. 1.
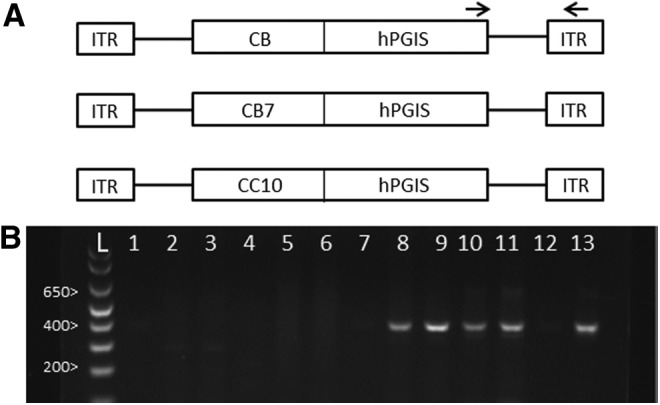
(A) Schematic of AAV vectors. CB, cytomegalovirus-enhanced chicken β-actin promoter; CB7, modified CB promoter; CC10, clara cell secretory protein promoter. (B) PCR analysis of AAV-CBhPGIS vector DNA in rat lungs 4–6 weeks after AAV-CBhPGIS administration. Lanes 1 and 2, saline–saline; lanes 3 and 4, MCT-control; lanes 5–7, MCT-AAV-CBhPLAP; lanes 8–11, MCT-AAV-CBhPGIS; lane 12, negative control (water); lane 13, positive control (pAAVCBhPGIS plasmid); lane L, 1 kb ladder. Arrows in (A) depict the location of PCR primers selected for (B). AAV, adeno-associated virus; MCT, monocrotaline; PCR, polymerase chain reaction.
The AAV9CC10hPGIS, AAV5CB7hPGIS, and initial lot of AAV9CBhPGIS vectors were produced locally and titered by dot blot assay as previously described (Grieger et al., 2006). Clinical-grade AAV9CBhPGIS and AAV9CB7hPGIS for experiments were obtained from the Penn Vector Core under the aegis of the NHLBI's Gene Therapy Resource Program.
Vector administration
All animal experiments and procedures were approved by the Institutional Animal Care and Use Committees of the University of Arkansas for Medical Sciences and the Central Arkansas Veterans Healthcare System. Rats (∼150 g) were purchased from Charles River Laboratories. Experiments were conducted in two different strains of rats. The Fisher 344 rat strain was employed in our initial studies of AAV5 versus AAV9 serotypes (Figs. 2 and 3). However, subsequent studies with different promoters were conducted in Sprague Dawley rats because of the inability to obtain adequate numbers of age- and size-matched Fisher rats from vendors (Fig. 4). Rats were anesthetized with ketamine (80 mg/kg) and xylazine (15 mg/kg) by IP injection. AAV5 or AAV9hPGIS vector, irrelevant AAV9-human placental alkaline phosphatase (hPLAP) or AAV9-empty transgene vector (200 μl; 2×1011 vector genomes), or vehicle (saline, 200 μl) was instilled into the lungs of rats by orotracheal instillation (Johnson et al., 2003). Forty-eight hours later, rats were injected with MCT (60 mg/kg IP) or an equal volume of saline. Rats were monitored daily until they developed symptoms of pulmonary hypertension (typically 3–4 weeks) such as weight loss and tachypnea, or until 6 weeks after vector administration. Hemodynamic, morphologic, and biochemical assessments were performed at this time.
FIG. 2.

Biochemical and functional assessment of AAV-mediated hPGIS expression in MCT-treated rats. (A) PGF1α in plasma of MCT-treated rats (n>8, p=0.018). (B) PGF1α in lung, liver, and heart tissue 4–6 weeks after AAV5 or AAV9-CBhPGIS administration to MCT-treated rats (n>9). (C) RV/LV+S ratios 4–6 weeks after AAV5 or AAV9-CBhPGIS administration to MCT-treated rats (n≥9, p≤0.001). (D) RVSP measurements 4–6 weeks after delivery of AAV5 or AAV9-CBhPGIS to MCT-treated rats (n≥6, p=0.008). †Significantly different from MCT-control. hPGIS, human prostacyclin synthase.
FIG. 3.
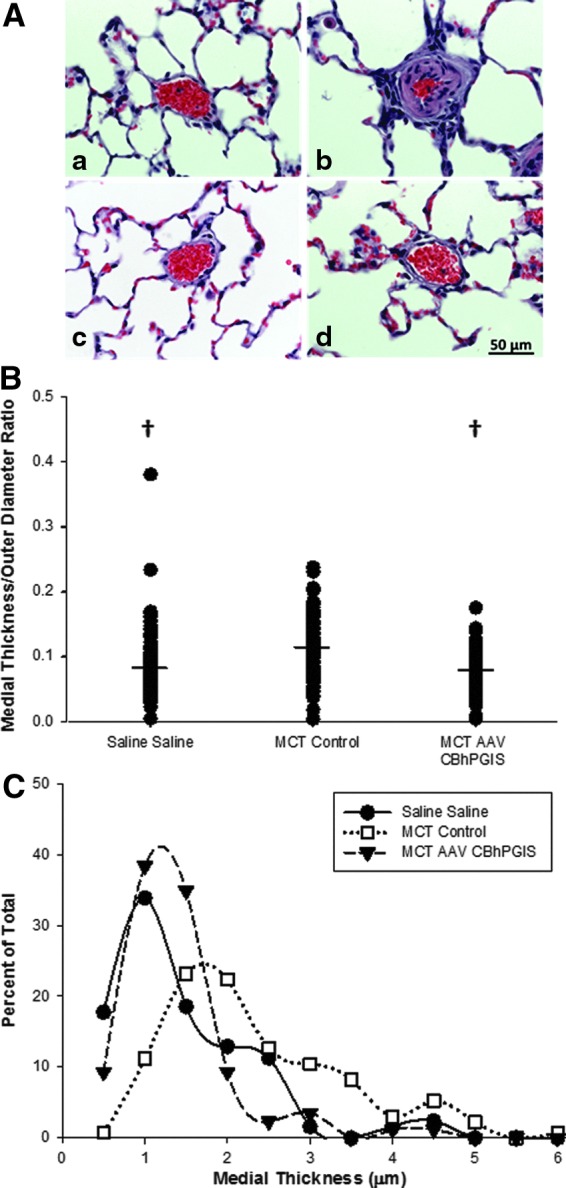
Effect of AAV-hPGIS on pulmonary vascular remodeling. (A) Hematoxylin and eosin stains of (a) saline–saline control, (b) MCT-control, (c) MCT-AAV5-hPGIS, and (d) MCT-AAV9-hPGIS sections of rat lung. (B) Ratios of vascular medial (smooth muscle) thickness to outer diameter (total vessel wall thickness) of small pulmonary arteries (n=4 [AAV5=3, AAV9=1], p≤0.001). †Significantly different from MCT-control. (C) Distribution of vascular medial thickness in small pulmonary arteries of AAV and control rats.
FIG. 4.

Biochemical and functional assessment of AAV-hPGIS vectors containing different promoters. (A) PGF1α in plasma 4–6 weeks after delivery of AAV9-hPGIS vectors with CB (n=12), CB7 (n=23), or CC10 (n=8) promoters to lungs of MCT-treated rats (p=0.012). (B) PGF1α in lung homogenates 4–6 weeks after AAV9-hPGIS administration to MCT-treated rats [n is same as for (A), p≤0.001]. (C) RV/LV+S ratios 4–6 weeks after administration of AAV9-hPGIS vectors to MCT-treated rats [n is same as for (A), p≤0.001]. (D) RVSP measurements in MCT-treated rats after delivery of AAV9-hPGIS vectors containing CB (n=9), CB7 (n=17), or CC10 (n=8) promoters (p≤0.001). A smaller n for RVSP measurements reflects the inability to obtain hemodynamic data from some MCT-treated rats. †Significantly different from MCT–saline control. RV/LV+S, right ventricle/(left ventricle+septum).
Polymerase chain reaction detection of AAVCBhPGIS vector DNA
DNA was extracted from formalin-fixed paraffin wax-embedded lungs and/or from frozen lung samples taken from MCT-treated rats 4–6 weeks after AAV-CBhPGIS administration using the RecoverAll total RNA Isolation kit (Ambion). Polymerase chain reaction (PCR) was performed on 300 ng of the isolated DNA using primers that spanned the hPGIS cDNA and the 3′ AAV inverted terminal repeat (forward primer, AGG TAC GGC TTC GGT CTG AT; reverse primer, AGT GGC CAA CTC CAT CAC TAG G) to amplify a 400 bp product. A negative control (water) and positive control (pAAV-CBhPGIS) were included in the PCR process. The PCR products were electrophoresed on a 1% agarose gel stained with ethidium bromide and visualized using UV light.
Measurement of hPGIS expression
To assess expression of the PGIS transgene, blood was collected by cardiac puncture at the time of euthanasia via CO2 asphyxiation and centrifuged at 514×g for 5 min, and the supernatant collected. Levels of 6-keto PGF1α, the nonenzymatic hydrolysis product of PGI2, were measured by enzyme immunoassay kit (EIA Assay Design, Inc.) in the supernatant (plasma). For measurement of 6-keto PGF1α in the tissues, organs were harvested and homogenized.
Measurement of right ventricular mass and right ventricular pressures
The right internal jugular vein of anesthetized rats was surgically exposed and cannulated with a 2F pressure transducer catheter (Millar Instruments) interfaced to a control unit (PC-2000) and a data acquisition system (Power Lab 8/30; AD Instruments). The catheter was advanced into the right ventricle and the pressures recorded. Right ventricular systolic pressure (RVSP) was measured in the vast majority, but not all rats, as either some MCT-treated rats had severe symptoms that prohibited collection of hemodynamic data or the measurements were technically inadequate for interpretation. After euthanasia, the hearts of all experimental animals were removed and blotted, and the RV was dissected free of the left ventricle+septum (LV+S) and each portion was weighed.
Histology
After euthanasia, the chest was opened and trachea exposed by sharp dissection. The trachea was cannulated and the lungs were inflation-fixed at 20 cm H2O with buffered paraformaldehyde (4%). Lungs were removed and subsequently immersed in the paraformaldehyde solution. Specimens were then embedded in paraffin, sectioned (5 μm) at the hilum, and stained with hematoxylin and eosin. Parallel sections were stained with an Elastic von Giessen stain for measurement of vascular media and outer-diameter wall thickness. Images were captured by visualization under a Zeiss Axioimager light microscope interfaced to a computer. Morphometric analysis was performed to assess vessel diameter.
Statistics
Data are presented as the mean±SE. The data were analyzed with the application of the one-way ANOVA method. A p-value <0.05 was considered statistically significant. Corrections for multiple comparisons were performed using Dunn's method.
Results
Vector delivery
We performed PCR analysis of vector DNA in rat lung 4–6 weeks after administration of AAVCBhPGIS. The forward primer was selected from the hPGIS cDNA and the reverse primer was selected from the ITR to generate a 400 bp PCR product specific for AAV-hPGIS vector DNA. As shown in Fig. 1B, vector DNA was detected in the lungs of rats 4 weeks postdelivery of AAV9CBhPGIS vectors (lanes 8–11), but not in the lungs of rats that received luminal administration of an AAV9CBhPLAP vector (lanes 5–7) or lungs of rats that received vehicle (saline, lanes 1–4). These data confirmed the specificity of the PCR for the AAVhPGIS vectors and established delivery of the AAVCBhPGIS vector genome to the lung.
Expression of hPGIS
Intraluminal administration of AAV5 and AAV9CBhPGIS vectors increased serum PGF1α levels by ∼3-fold and ∼7-fold, respectively, as compared with MCT-control rats, which closely resembled levels measured in rats not treated with MCT (Fig. 2A). A trend toward an increase in the levels of PGF1α was detected in the lung and heart homogenates of MCT-AAV9CBhPGIS rats as compared with MCT-control or saline–saline rats (Fig. 2B), but did not reach significance. No differences in PGF1α levels were detected in liver homogenates from rats.
To assess the effects of hPGIS overexpression mediated by AAV5 and AAV9CBhPGIS vectors on function, we measured RV/LV+S ratios and RVSP. The RV/LV+S weight ratios were greater in MCT-control rats (0.306±0.013) than in saline–saline control rats (0.228±0.005), consistent with the development of PAH induced by MCT. The ratios in MCT rats transduced with AAV5 and AAV9CBhPGIS vectors (0.232±0.005 and 0.235±0.008, respectively) were also different from MCT-control ratios, but similar to the ratios in saline–saline controls, consistent with prevention of MCT-induced cardiac remodeling (Fig. 2C). Similarly, the RVSP was nearly twofold greater in MCT-control rats (36.0±3.3 mm Hg) than in saline–saline control rats (23.6±1.8 mm Hg), whereas RVSP in MCT-AAV5 and MCT-AAV9CBhPGIS rats (22.4±3.6 and 25.5±0.9 mm Hg, respectively) was similar to measurements in saline–saline rats (Fig. 2D). Thus, AAV5 and AAV9CBhPGIS vectors attenuated the development of MCT-induced PAH.
Prevention of pulmonary vascular remodeling by AAV-mediated hPGIS expression
The effects of AAV-mediated expression of hPGIS on pulmonary vascular remodeling were measured (Fig. 3) in paraffin sections of rat lung. MCT-control rats exhibited a markedly thickened muscular media in the small pulmonary arteries, consistent with pulmonary hypertension, as compared with small pulmonary arteries in saline–saline rats (Fig. 3A). In contrast, the small pulmonary arteries of the MCT-AAV5CBhPGIS and MCT-AAV9CBhPGIS were similar to saline–saline control rats, consistent with prevention of MCT-induced PH.
Morphometric analysis of Elastic von Giessen–stained sections confirmed the thickening of pulmonary artery smooth muscle media in MCT-control rats as compared with MCT rats transduced with AAVCBhPGIS vectors (Fig. 3B). The diameters of the muscular medial thickness and the outer wall of small pulmonary arteries were measured, and the ratio of the media to total wall thickness was calculated. The ratios of medial thickness to outer wall diameter for the AAV vector-transduced groups were compared with the control groups (Fig. 3B). MCT-control rats had a greater ratio of medial thickness to outer vessel wall diameter than saline–saline control rats, whereas MCT rats transduced with AAV5 or AAV9CBhPGIS vectors had ratios that were not different from saline–saline controls. The distribution of medial wall thickness in PAs <50 μm in size is depicted in Fig. 3C. MCT-control rats had an increased medial thickness of small pulmonary arteries as compared with the saline–saline control group or the MCT-AAV9CBhPGIS rats (n=4 [3 AAV5 and 1 AAV9 specimen]). These data are consistent with the attenuation of pulmonary vascular remodeling in rats transduced with AAV5 or AAV9CBhPGIS vectors.
Promoter effects on hPGIS expression
Because of promising preliminary results with AAV9 vectors containing the CB7 promoter by others in vivo (Julie Johnston, PhD, Penn Vector Core, Philadelphia, PA, personal communication), we began a series of experiments with clinical-grade AAV-CB7hPGIS vectors in the MCT rat model. Disappointing results led us to perform experiments with the clinical-grade AAV9CBhPGIS vectors and a locally produced vector containing the CC10 epithelial-specific promoter, AAV9CC10hPGIS. The summary data for these experiments are presented in Fig. 4. No significant increases in PGF1α levels in in plasma (Fig. 4A) or lung homogenates (Fig. 4B) were detected in MCT-AAV9CBhPGIS-transduced rats as the responses were highly variable. However, significant reductions in RV mass (RV/LV+S ratios) and RVSP as compared with MCT-control rats (Fig. 4C and D) were detected, consistent with attenuation of MCT-induced PAH. These hemodynamic findings were similar to the measurements in naïve rats (saline–saline) that did not receive vector or MCT. The attenuation in RV mass and pressures was restricted to rats treated with AAV9-hPGIS vectors containing the CB promoter and could not be reproduced with AAV5CBhPGIS vectors in these latter experiments (data not shown). No significant changes in levels of PGF1α, RV/LV+S, or RVSP were detected in MCT rats transduced with AAV9-hPGIS vectors containing the CB7 or CC10 promoters as compared with MCT-controls. Surprisingly, PGF1α was increased in rats treated with an AAV9CBhPLAP vector (p=0.012), possibly reflecting some cross reactivity of the transgene, but no evidence of functional changes in RV mass or RVSP was detected. Thus, AAV9CBhPGIS vectors transduced MCT-treated rats and attenuated PAH, whereas AAV5-hPGIS vectors with CB7 or CC10 promoters did not.
Discussion
This study demonstrated that expression of hPGIS mediated by a single luminal (airway) instillation of AAV9-hPGIS vector containing a CB promoter prevented the development of MCT-induced PAH and associated pulmonary vascular remodeling. An AAV5-hPGIS vector with the same CB promoter also prevented the development of MCT-induced PAH. Measurements of the nonenzymatic hydrolysis product of prostacyclin, PGF1α, were highly variable, limiting the ability of this measure to confirm hPGIS expression statistically, although some trends may be present. This is not an unexpected result as the assay for detection of 6-keto-PGF1α cannot differentiate between the human and rat molecules.
One of the aims of the study was to determine which of the two selected AAV serotypes would be the best for gene delivery. A variety of serotypes have been considered for gene transfer to the lung, including AAV serotypes 1, 2, 5, 6, and 9 (Zabner et al., 2000; Auricchio et al., 2002; Limberis and Wilson, 2006; Limberis et al., 2009). Previous studies of AAV-hPGIS vectors in chronic hypoxic mice or MCT-treated rats relied extensively on the AAV1 and AAV2 serotype vectors delivered by intramuscular injection (Ito et al., 2007; Kawakami et al., 2007; Kataoka et al., 2013). While AAV1 has been used for lung gene transfer, its utility in muscle gene transfer has been recognized in many fields of clinical gene transfer (Song et al., 1998; Chao et al., 2000; Flotte et al., 2011). Noninvasive aerosol lung delivery of AAV2 vectors has been tested in human clinical trials of cystic fibrosis, but showed limited clinical efficacy (Moss et al., 2004). Subsequently, AAV5, AAV6, and AAV9 pseudotyped vectors were developed as promising vectors for improved airway and distal lung delivery (Halbert et al., 2001; Auricchio et al., 2002; Limberis et al., 2009). In preliminary studies, AAV5 vectors were superior to AAV1 vectors for mediating efficient hPLAP reporter gene transfer to the lungs of mice after luminal airway delivery (data not shown). In contrast, Flotte et al. (2010) reported greater efficiency of AAV1 vectors as compared with AAV5 vectors in a chimpanzee in vivo airway model that was predictive of findings in cultures of polarized human airway epithelia. Our study focused on delivery of AAV5 and AAV9 vectors to distal lung, with prior studies showing that AAV5 transduces both airway and alveolar epithelial cells, whereas AAV9 vectors primarily transduce alveolar (type II) epithelial cells (Auricchio et al., 2002; Limberis and Wilson, 2006; Limberis et al., 2009). Published data also suggested that AAV9 vectors could efficiently transfer a variety of transgenes under the control of various promoters after luminal airway instillation (Auricchio et al., 2002; Limberis et al., 2009). The possibility that the AAV9 capsid vector with AAV2 genome backbone could be readministered, even in the presence of circulating neutralizing antibodies, as early as 1 month after initial exposure, made it a promising gene transfer vector candidate for use in humans (Auricchio et al., 2002; Limberis and Wilson, 2006; Limberis et al., 2009). Accordingly, we selected AAV5 and AAV9 vectors for the current study.
One of the hallmarks of the prior in vivo gene transfer studies with AAV-hPGIS vectors was the consistent selection of the CB or CAG promoters to drive transcription of the hPGIS cDNA (Ito et al., 2007; Kawakami et al., 2007; Kataoka et al., 2013). We tested the CB promoter against a modified version of the CB promoter known as CB7 (provided by the Penn Vector Core) and against the epithelial-specific promoter, CC10 (Stripp et al., 1992; Auricchio et al., 2002). The results of our study supported the selection of the constitutive CB promoter chosen by others, as AAV9-hPGIS vectors containing the CC10 and CB7 promoters did not attenuate MCT-induced PAH. These results were unexpected as the CC10 promoters had previously been shown to efficiently mediate gene transfer to murine lungs (Auricchio et al., 2002). Because data from a personal communication (Julie Johnston, PhD) suggested that the CB7 promoter was a highly effective promoter, we devoted significant resources to testing this vector in vivo, but found that this promoter failed to mediate efficient transgene expression in rats. We were also unable to reproduce functional expression of AAV5CBhPGIS in Sprague Dawley rats (data not shown). We do not know whether the failure of these promoters and AAV serotype 5 to mediate gene expression in Sprague Dawley rats was a species-specific phenomenon or whether unforeseen epigenetic factors played a role.
A limitation of our study was the MCT-induced PAH prevention model in which rats received noninvasive lung delivery of vector 2 days before MCT administration. Prior studies of intramuscular injection of AAV1 and AAV2-hPGIS vectors also used a prevention model approach (Ito et al., 2007; Kawakami et al., 2007; Kataoka et al., 2013). In contrast, Hadri et al. (2013) administered an AAV1-SERCA2 vector by luminal airway aerosol administration with a Penn Century Device in both prevention and established MCT-PAH models with similar results. Unlike our study, Hadri and associates performed their studies in older male Sprague Dawley rats (350 g), whereas our studies were performed in younger females and males (150 g). Moreover, the severity of PAH was greater in the Hadri study with an RVSP of 50–60 mm Hg as compared with an RVSP of ∼40 mm Hg in our study, with equivalent doses of MCT delivered by intraperitoneal injection. This difference in RVSP between the two studies may have reflected gender differences in MCT-induced PAH severity between male and female rats (Bal et al., 2013). However, our findings that the RV\LV+S ratios and RVSP measured in MCT-injected male rats did not differ from the measurements in female rats (data not shown) fail to support the gender difference argument. Rather, our data likely demonstrated strain differences as the RV/LV+S ratios and RVSP in MCT-injected Fisher 344 rats (Fig. 2C and D) were lower than the same measurements in Sprague Dawley rats (Fig. 4C and D), which are consistent with data from Hadri et al. (2013) (Pan et al., 1993). Despite these strain differences, the data presented from our study compare favorably to the data from the intramuscular injection of AAV1 and AAV2-hPGIS vectors and luminal airway aerosol administration of AAV-SERCA2 vectors in a prevention of MCT-induced PAH model (Ito et al., 2007; Kawakami et al., 2007; Hadri et al., 2013; Kataoka et al., 2013).
In conclusion, noninvasive luminal lung delivery of AAV5- and AAV9CBhPGIS gene transfer vectors significantly attenuated the severity of MCT-induced PAH and prevented cardiac and pulmonary vascular remodeling. Although the constitutive CB promoter was best for in vivo delivery to rat lung, we do not know if a CB promoter would be ideal for human studies. Overall, these data demonstrate a potential role for noninvasive luminal airway delivery of AAV-hPGIS vectors for paracrine delivery of prostacyclin to the pulmonary vasculature in human clinical trials of PAH.
Acknowledgments
The authors thank Mr. Gabor Molnar and Dr. Ruth Everett for their assistance in developing the techniques for this article.
The use of the facilities in the University of Arkansas for Medical Sciences Digital and Confocal Microscopy Laboratory supported by National Institutes of Health (NIH) Grant 2 P20 RR 16460 (PI: Larry Cornett, INBRE, Partnerships for Biomedical Research in Arkansas) is acknowledged.
The study was supported by a VA Merit Grant (L.G.J.) from the U.S. Department of Veterans Affairs and a grant from the Arkansas Biosciences Institute (L.G.J.). The NIH NHLBI Gene Therapy Resource Program provided vector through the aegis of the Penn Vector Core in support of this project.
Disclaimer
The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of National Center for Toxicological Research or NIH.
Author Disclosure Statement
No competing financial interests exist.
References
- Auricchio A., O'connor E., Weiner D., et al. (2002). Noninvasive gene transfer to the lung for systemic delivery of therapeutic proteins. J. Clin. Invest. 110, 499–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal E., Ilgin S., Atli O., et al. (2013). The effects of gender difference on monocrotaline-induced pulmonary hypertension in rats. Hum. Exp. Toxicol. 32, 766–774 [DOI] [PubMed] [Google Scholar]
- Campbell A.I., Kuliszewski M.A., and Stewart D.J. (1999). Cell-based gene transfer to the pulmonary vasculature: endothelial nitric oxide synthase overexpression inhibits monocrotaline-induced pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 21, 567–575 [DOI] [PubMed] [Google Scholar]
- Campbell A.I., Zhao Y., Sandhu R., and Stewart D.J. (2001). Cell-based gene transfer of vascular endothelial growth factor attenuates monocrotaline-induced pulmonary hypertension. Circulation 104, 2242–2248 [DOI] [PubMed] [Google Scholar]
- Chao H., Liu Y., Rabinowitz J., et al. (2000). Several log increase in therapeutic transgene delivery by distinct adeno-associated viral serotype vectors. Mol. Ther. 2, 619–623 [DOI] [PubMed] [Google Scholar]
- Deng Z., Morse J.H., Slager S.L., et al. (2000). Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am. J. Hum. Genet. 67, 737–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher K.J., Gao G.P., Weitzman M.D., et al. (1996). Transduction with recombinant adeno-associated virus for gene therapy is limited by leading-strand synthesis. J. Virol. 70, 520–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flotte T.R., Fischer A.C., Goetzmann J., et al. (2010). Dual reporter comparative indexing of rAAV pseudotyped vectors in chimpanzee airway. Mol. Ther. 18, 594–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flotte T.R., Trapnell B.C., Humphries M., et al. (2011). Phase 2 clinical trial of a recombinant adeno-associated viral vector expressing alpha1-antitrypsin: interim results. Hum. Gene Ther. 22, 1239–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galie N., and Branzi A. (2005). Pulmonary arterial hypertension: therapeutic algorithm. Ital. Heart J. 6, 856–860 [PubMed] [Google Scholar]
- Galie N., Badesch D., Oudiz R., et al. (2005). Ambrisentan therapy for pulmonary arterial hypertension. J. Am. Coll. Cardiol. 46, 529–535 [DOI] [PubMed] [Google Scholar]
- Geraci M.W., Gao B., Shepherd D.C., et al. (1999). Pulmonary prostacyclin synthase overexpression in transgenic mice protects against development of hypoxic pulmonary hypertension. J. Clin. Invest. 103, 1509–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieger J.C., Choi V.W., and Samulski R.J. (2006). Production and characterization of adeno-associated viral vectors. Nat. Protoc. 1, 1412–1428 [DOI] [PubMed] [Google Scholar]
- Hadri L., Kratlian R.G., Benard L., et al. (2013). Therapeutic efficacy of AAV1.SERCA2a in monocrotaline-induced pulmonary arterial hypertension. Circulation 128, 512–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbert C.L., Allen J.M., and Miller A.D. (2001). Adeno-associated virus type 6 (AAV6) vectors mediate efficient transduction of airway epithelial cells in mouse lungs compared to that of AAV2 vectors. J. Virol. 75, 6615–6624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- International PPH Consortium, Lane K.B., Machado R.D., et al. (2000). Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat. Genet. 26, 81–84 [DOI] [PubMed] [Google Scholar]
- Ito T., Okada T., Mimuro J., et al. (2007). Adenoassociated virus-mediated prostacyclin synthase expression prevents pulmonary arterial hypertension in rats. Hypertension 50, 531–536 [DOI] [PubMed] [Google Scholar]
- Johnson L.G., Vanhook M.K., Coyne C.B., et al. (2003). Safety and efficiency of modulating paracellular permeability to enhance airway epithelial gene transfer in vivo. Hum. Gene Ther. 14, 729–747 [DOI] [PubMed] [Google Scholar]
- Kataoka M., Kawakami T., Tamura Y., et al. (2013). Gene transfer therapy by either type 1 or type 2 adeno-associated virus expressing human prostaglandin I2 synthase gene is effective for treatment of pulmonary arterial hypertension. J. Cardiovasc. Pharmacol. Ther. 18, 54–59 [DOI] [PubMed] [Google Scholar]
- Kawakami T., Kanazawa H., Satoh T., et al. (2007). AAV-PGIS gene transfer improves hypoxia-induced pulmonary hypertension in mice. Biochem. Biophys. Res. Commun. 363, 656–661 [DOI] [PubMed] [Google Scholar]
- Kido M., Du L., Sullivan C.C., et al. (2005). Gene transfer of a TIE2 receptor antagonist prevents pulmonary hypertension in rodents. J. Thorac. Cardiovasc. Surg. 129, 268–276 [DOI] [PubMed] [Google Scholar]
- Limberis M.P., and Wilson J.M. (2006). Adeno-associated virus serotype 9 vectors transduce murine alveolar and nasal epithelia and can be readministered. Proc. Natl. Acad. Sci. USA 103, 12993–12998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limberis M.P., Vandenberghe L.H., Zhang L., et al. (2009). Transduction efficiencies of novel AAV vectors in mouse airway epithelium in vivo and human ciliated airway epithelium in vitro. Mol. Ther. 17, 294–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mclaughlin V. (2013). Managing pulmonary arterial hypertension and optimizing treatment options: prognosis of pulmonary artery hypertension. Am. J. Cardiol. 111, 10C–15C [DOI] [PubMed] [Google Scholar]
- Mclaughlin V.V., Shillington A., and Rich S. (2002). Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation 106, 1477–1482 [DOI] [PubMed] [Google Scholar]
- Mclaughlin V.V., Sitbon O., Badesch D.B., et al. (2005). Survival with first-line bosentan in patients with primary pulmonary hypertension. Eur. Respir. J. 25, 244–249 [DOI] [PubMed] [Google Scholar]
- McMurtry M.S., Archer S.L., Altieri D.C., et al. (2005). Gene therapy targeting survivin selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. J. Clin. Invest. 115, 1479–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss R.B., Rodman D., Spencer L.T., et al. (2004). Repeated adeno-associated virus serotype 2 aerosol-mediated cystic fibrosis transmembrane regulator gene transfer to the lungs of patients with cystic fibrosis: a multicenter, double-blind, placebo-controlled trial. Chest 125, 509–521 [DOI] [PubMed] [Google Scholar]
- Nagaya N., Yokoyama C., Kyotani S., et al. (2000). Gene transfer of human prostacyclin synthase ameliorates monocrotaline-induced pulmonary hypertension in rats. Circulation 102, 2005–2010 [DOI] [PubMed] [Google Scholar]
- Ono M., Sawa Y., Fukushima N., et al. (2004). Gene transfer of hepatocyte growth factor with prostacyclin synthase in severe pulmonary hypertension of rats. Eur. J. Cardiothorac. Surg. 26, 1092–1097 [DOI] [PubMed] [Google Scholar]
- Pan L.C., Wilson D.W., and Segall H.J. (1993). Strain differences in the response of Fischer 344 and Sprague-Dawley rats to monocrotaline induced pulmonary vascular disease. Toxicology 79, 21–35 [DOI] [PubMed] [Google Scholar]
- Rubin L.J., Badesch D.B., Barst R.J., et al. (2002). Bosentan therapy for pulmonary arterial hypertension. N. Engl. J. Med. 346, 896–903 [DOI] [PubMed] [Google Scholar]
- Shintani M., Yagi H., Nakayama T., et al. (2009). A new nonsense mutation of SMAD8 associated with pulmonary arterial hypertension. J. Med. Genet. 46, 331–337 [DOI] [PubMed] [Google Scholar]
- Sitbon O., Mclaughlin V.V., Badesch D.B., et al. (2005). Survival in patients with class III idiopathic pulmonary arterial hypertension treated with first line oral bosentan compared with an historical cohort of patients started on intravenous epoprostenol. Thorax 60, 1025–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S., Morgan M., Ellis T., et al. (1998). Sustained secretion of human alpha-1-antitrypsin from murine muscle transduced with adeno-associated virus vectors. Proc. Natl. Acad. Sci. USA 95, 14384–14388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stripp B.R., Sawaya P.L., Luse D.S., et al. (1992). Cis-acting elements that confer lung epithelial cell expression of the CC10 gene. J. Biol. Chem. 267, 14703–14712 [PubMed] [Google Scholar]
- Zabner J., Seiler M., Walters R., et al. (2000). Adeno-associated virus type 5 (AAV5) but not AAV2 binds to the apical surfaces of airway epithelia and facilitates gene transfer. J. Virol. 74, 3852–3858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.D., Courtman D.W., Deng Y., et al. (2005). Rescue of monocrotaline-induced pulmonary arterial hypertension using bone marrow-derived endothelial-like progenitor cells: efficacy of combined cell and eNOS gene therapy in established disease. Circ. Res. 96, 442–450 [DOI] [PubMed] [Google Scholar]