

Abstract
Background
Autosomal dominant polycystic kidney disease (ADPKD) is a common genetic disorder characterized by the progressive development of renal and hepatic cysts. Follicle-stimulating hormone (FSH) has been demonstrated to be a trophic factor for biliary cells in normal rats and experimental cholestasis induced by bile duct ligation (BDL).
Aims
To assess the effect of FSH on cholangiocyte proliferation during ADPKD using both in vivo and in vitro models.
Methods
Evaluation of FSH receptor (FSHR), FSH, phospho-extracellular-regulated kinase (pERK) and c-myc expression in liver fragments from normal patients and patients with ADPKD. In vitro, we studied proliferating cell nuclear antigen (PCNA) and cAMP levels in a human immortalized, non-malignant cholangiocyte cell line (H69) and in an immortalized cell line obtained from the epithelium lining the hepatic cysts from the patients with ADPKD (LCDE) with or without transient silencing of the FSH gene.
Results
Follicle-stimulating hormone is linked to the active proliferation of the cystic wall and to the localization of p-ERK and c-myc. This hormone sustains the biliary growth by activation of the cAMP/ERK signalling pathway.
Conclusion
These results showed that FSH has an important function in cystic growth acting on the cAMP pathway, demonstrating that it provides a target for medical therapy of hepatic cysts during ADPKD.
Keywords: autosomal dominant polycystic kidney disease, biliary epithelium, follicle, stimulating hormone, immunohistochemistry
Polycystic liver disease phenotypes arise from two distinct inherited diseases, autosomal dominant polycystic kidney disease (ADPKD) and polycystic liver disease (PCLD). ADPKD, caused by mutations in PKD1 or PKD2 genes, is characterized by polycystic kidneys (1). In many patients with ADPKD, there is the development of a polycystic liver manifestation. On the other hand, PCLD is caused by mutations in PRKCSH or SEC63 genes and is characterized by the presence of an isolated polycystic liver without the kidney phenotype (2, 3).
The diagnosis of polycystic liver is usually made during the third or fourth decade of life with hepatic capacity preserved in the great majority of patients (4, 5). This disease is usually asymptomatic, but the progressive growth of the liver cysts may cause dyspnoea, gastrooesophageal reflux, nausea and mechanical low back pain arise because of the mass effect of the polycystic liver (6). Severe ADPKD primarily affects women and is characterized by the massive cystic liver disease. The number and size of hepatic cysts correlate with the occurrence of pregnancy, female gender, increased age and severity of the renal lesion (7). Treatment is initiated only in those with the symptoms and all interventional procedures are aimed to reduce liver volume (5). In the last few years, the number of studies to discover viable medical options has increased with indications that somatostatin analogues or mTOR inhibitors may slow cyst growth (8–10).
Many experimental and clinical studies have demonstrated that cholangiocytes respond to hormones, growth factors, neuropeptides and cytokines increasing their proliferative capacity (11–13). In particular, oestrogens play a key role in sustaining cholangiocyte growth, cyst formation and progression in ADPKD patients. Oestrogens act not only directly, but also by promoting the synthesis and release of other growth factors from the cystic epithelium (14). Additional sex hormones such as prolactin (15), progesterone (16) and follicle-stimulating hormone (FSH) (17) regulate biliary function.
Many events in the adult ovary are controlled by two hormones, FSH and luteinizing hormone (LH) secreted from the anterior pituitary gland under the control of gonadotropin-releasing hormone (GnRH) from the hypothalamus. FSH is required for granulosa cell differentiation and facilitates the follicular growth (18). In the classical cascade, occupancy of FSH receptor (FSHR) causes activation of the heterotrimeric GS protein, which stimulates the effector adenylyl cyclase with the consequent increase in the synthesis of the second messenger cAMP (19, 20).
One of the most characterized components of the MAPK family is the extracellular-regulated kinase (ERK). The ERK pathway regulates cell proliferation, differentiation and cell survival (21). C-myc represents a key downstream target of this mechanism (22). Others have demonstrated that c-myc participates in the progression of the G1-cell cycle phase by enhancing cyclin expression (23) and CDK/cyclin complex activities (24). Lastly, both c-myc and ERK, as a consequence of their marked capacity to promote proliferation, play a pivotal role in the control of the differentiation programme in several cell types (25–27). We have previously shown that the cAMP/ERK-dependent signalling mechanism is activated in proliferating cholangiocytes (13, 28). In particular, in the hyperplastic BDL model, cholangiocyte proliferation is closely associated with increased cAMP levels (29–32). It has been demonstrated that FSH plays an important role in stimulating rat cholangiocyte proliferation through an autocrine mechanism that is associated with increased cAMP-dependent phosphorylation of ERK1/2 and Elk-1 both in vivo and in vitro (17). However, no information exists regarding the role of FSH and its receptors in the regulation of epithelial cell growth in the hepatic cysts. The aim of this study was to evaluate the hypothesis that FSH regulates hepatic cysts growth during the course of ADPKD.
Materials and methods
Materials
All reagents were purchased from Sigma Chemical (St. Louis, MO, USA) unless otherwise indicated. The FSH primers for real-time PCR were purchased from SABio-sciences (Frederick, MD, USA). The RNeasy Mini Kit to purify total cholangiocyte RNA was purchased from Qiagen Inc. (Valencia, CA, USA). The RIA kits for the measurement of intracellular cAMP [cAMP (125I) Biotrak Assay System, RPA509] were purchased from GE Healthcare (Arlington Heights, IL, USA). All the antibodies used for immunohistochemistry, immunofluorescence and western blots were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), Invitrogen srl (Milan, Italy), Dako Italia S.p.A. (Milan, Italy) or from Abcam (Cambridge, UK).
Human liver samples
We studied eight patients (six females and two males, 61–78 years of age) with a diagnosis of ADPKD based on the standard international criteria (33). Liver cysts were subdivided on the basis of their size in large (>3 cm maximum diameter) or small cysts (<3 cm maximum diameter) as previously showed (14). As controls, we evaluated liver biopsies with a normal histology from patients submitted to laparotomy (4 fragments, 2 from female and 2 from male, 59–75 years of age). This study protocol was approved by the institutional committee and abided by the ethical guidelines of the 1975 Declaration of Helsinki.
Immunohistochemistry
Immunohistochemistry was performed in 3–4 μm sections. Sections were deparaffinized and endogenous peroxidase activity was blocked by a 30-min incubation in methanolic hydrogen peroxide (2.5%). Later, the endogenous biotin was blocked by a biotin blocking system (code X0590; Dako, Copenhagen, Denmark) according to the instructions supplied by the vendor. Sections were then hydrated in graded alcohol and rinsed in 1× phosphate-buffered saline (PBS, pH 7.4) before applying the selected primary antibody. Sections were incubated overnight at 4°C with polyclonal antibodies for CK-19 (M0888; Dako Italia S.p.A.), FSHR (sc-7798; Santa Cruz), FSHβ (sc-7797; Santa Cruz), pERK (sc-7383; Santa Cruz) or c-myc (ab39688; Abcam). The following day, samples were rinsed with PBS for 5 min, incubated for 20 min at room temperature (RT) with secondary biotinylated antibody (LSAB Plus system; Dako, Milan, Italy), then with Dako ABC (LSAB Plus system), and finally developed with 3,3′-diaminobenzidine. To confirm the specificity of immunoreaction, negative controls were performed for all immunoreactions.
Sections were examined with a Leica Microsystems DM 4500 B Microscopy (Weltzlar, Germany) equipped with a Jenoptik Prog Res C10 Plus Videocam (Jena, Germany). Observations were processed with an Image Analysis System (IAS; Delta Sistemi, Rome, Italy) and were independently performed by two researchers in a blinded fashion. The number of positive cells was counted in six non-overlapping fields (magnification ×20) for each slide. The data are expressed as per cent positive cells (34).
Immunofluorescence
For double immunofluorescence, sections were hydrated in graded alcohol and rinsed in 1× PBS with 0.1% Triton X (PBS-T) for 15 min and then incubated with 10% normal blocking serum in 1× PBS for 30 min at RT. After washing, slides were incubated overnight at 4°C with FSHR (sc-7798; goat polyclonal; Santa Cruz) primary antibodies and proliferating cell nuclear antigen (PCNA) (PC10, sc-7907; rabbit polyclonal; Santa Cruz) or with the same FSHR and pERK (sc-7383; mouse monoclonal; Santa Cruz) diluted in PBS with 1.5% normal blocking serum. Samples were rinsed in PBS-T with three changes and incubated for 45 min at RT with the specific secondary antibodies conjugated with Alexa fluorochrome (488 or 594) diluted in 1× PBS with 1.5% normal blocking serum. Then the samples were washed in buffer and mounted with Ultra-Cruz mounting medium (sc-24941; Santa Cruz). Images were taken by DM4500B light microscopy (Leica).
Regarding cellular staining, cholangiocytes from cell lines were seeded on coverslip in a six-well-plate (500 000 per well) and allowed to adhere overnight. Immunofluorescence was performed by fixing cells in 4% paraformaldehyde for 5 min and following washes and incubation in 4% bovine serum albumin (BSA), in PBS-T the cells were incubated with the selected primary antibody (FSH, FSHR or pERK). After 1 h at RT, the cells were washed three times in PBS-T and then placed in the specific Alexa Fluor 594 secondary antibody in a dark room for 45 min. Finally, cholangiocytes were rinsed and the coverslip was put onto slide with a drop of DAPI. In the same manner of immunohistochemistry, to demonstrate the specificity of the immunoreaction, negative controls were performed without the incubation with primary antibody.
H69 and LCDE cell lines
The in vitro studies were performed utilizing a human immortalized non-malignant cholangiocyte cell line (H69) and an immortalized cell line obtained from the epithelium lining the hepatic cysts from patients with ADPKD (LCDE). Cells were maintained in hormonally supplemented medium consisting of Dulbecco’s modified Eagle’s medium-Ham’s F-12 nutrient mixture (3:1) (Cambrex Bio Science, Walkersville, MD, USA) supplemented with 1.8 × 104 mol/L adenine (LKT; Santa Cruz Biotechnology, Santa Cruz, CA, USA), 5 g/ml insulin, 5 g/ml transferrin (Calbiochem Biochemicals, Darmstadt, Germany), 2 × 109 mol/L triiodothyronine, 1.1 × 106 mol/L hydrocortisone, 1.64 × 106 human epidermal growth factor, 5.5 × 106 epinephrine, 10% foetal bovine serum (Gibco/BRL, Life Technologies, Italia srl., Milan, Italy), 100 U/ml of penicillin and 100 g/ml of streptomycin in a 5% CO2 atmosphere at 37°C. To evaluate the effect of FSH on proliferation, H69 and LCDE cells following culture in the appropriate medium containing 10% foetal bovine serum were deprived of serum for 24 h. Cells were then maintained in serum-deprived conditions for an additional 24 h for MTS proliferation assay (controls) or exposed to serum, FSH (1–100 μg/ml) with or without PD98059 (10 nM), a MEK/ERK inhibitor. In detail, cell medium was replaced with a fresh serum-free medium without hormone supplementation, but added with the tested agent. We used a commercially available colorimetric cell proliferation assay (CellTiter 96 aqueous nonradioactive cell proliferation assay, MTS Kit; Promega, Madison, WI, USA), following the manufacturer’s instructions. Proliferation index was calculated as the ratio (multiplied ×100) between cell numbers in both unstimulated and stimulated cultures.
In addition, we measured intracellular cAMP levels. After incubation for 1 h at 37°C, cholangiocytes (1 × 105 cells) were stimulated at RT for 5 min with 0.2% BSA (basal), or FSH (100 μg/ml in 0.2% BSA) in the absence or presence of PD98059 or an anti-FSHR antibody (150 pg/ml) (17). Intracellular cAMP levels were measured with a commercially available kit [cAMP (125I) Biotrak Assay System, RPA509].
FSH silencing
To evaluate the effects of FSH on LCDE, we used an available silencer small interfering RNA (siRNA) to knock down the expression of FSH before evaluating: (i) cholangiocyte proliferation by PCNA and biliary apoptosis by Bax protein expression using immunoblotting analysis; and (ii) intracellular cAMP levels. LCDE were plated into six-well plates and allowed to adhere overnight. siRNA transfection (0.25–1 μg of FSH siRNA was used) was carried out according to the instructions provided by Santa Cruz. The extent of FSH silencing was evaluated by measuring the expression of total FSH in transfected vs. control LCDE cells by real-time PCR and western blots for FSH expression. Cellular growth was investigated by western blots for PCNA, whereas biliary apoptosis was evaluated by Bax protein expression. PCNA and Bax expression was performed in protein (10 μg) from whole cell lysates from LCDE cholangiocytes. Blots were normalized by β-actin immunoblots. The intensity of the bands was determined by scanning video densitometry using the phospho-imager, Storm 860 (GE Healthcare, Piscataway, NJ, USA) and the ImageQuant TL software version 2003.02 (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Finally, spontaneous and secretin-stimulated intracellular cAMP levels were determined. Transfected and control cholangiocytes were incubated for 2 h at 37°C to restore secretin receptor that may be damaged with the treatment of proteolytic enzymes (35). Cells were stimulated with 10_7 M secretin in 1% BSA or 1% BSA alone for 5 min at 22°C (36). After extraction with ethanol, cAMP levels were determined by a commercially available kit (cAMP [125I] Biotrak Assay System, RPA509) according to the instructions of the vendor.
Statistical analysis
Data are presented as arithmetic mean ± standard deviation. The Student’s t-test or Mann–Whitney U-test was used to determine differences between groups for normally or not normally distributed data respectively. A P-value of <0.05 was considered statistically significant. Statistical analyses were performed using SPSS statistical software (SPSS Inc., Chicago, IL, USA).
Results
FSHR and FSH cholangiocyte expression
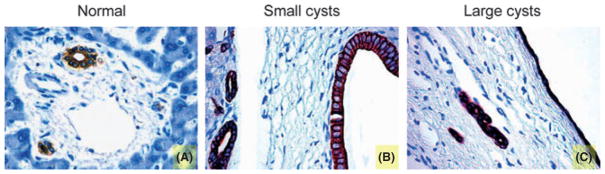
Hepatic cysts are lined by epithelial cells and these cysts bud from interlobular or smaller biliary ducts with phenotypical and functional characteristics of biliary epithelium as shown in Fig. 1 (immunohistochemistry for cytokeratin 19, a specific marker of cholangiocytes). Biliary epithelium also displays immunopositivity for FSHR and FSH hormone in liver sections from normal patients and patients affected with ADPKD (Fig. 2). The immunohistochemistry for FSHR appears negative in cholangiocytes lining interlobular bile ducts in normal livers (Fig. 2A), whereas FSH is faintly positive (Fig. 2D). In contrast, FSHR and FSH were more positive in the epithelial cells lining the smallest hepatic cysts (Fig. 2B, E) and strongly expressed in the largest cysts (Fig. 2C, F). The expression of FSH and FSHR is related to the cyst size. We found that the percentage of FSHR-positive cholangiocytes is 47 ± 25.1% in small cysts (diameter <3 cm) vs. 72.3 ± 26.2% (P < 0.05) in large cysts (diameter >3 cm). Similarly, the expression of the hormone FSH is higher in cholangiocytes lining large cysts (73.8 ± 19.8%) in comparison with small cysts (39.6 ± 19.4%; P < 0.05) (Fig. 2).
Fig. 1.
Immunohistochemistry for CK-19, a specific marker of the biliary epithelium. (A) Normal liver demonstrates a characteristic portal space, with some biliary ducts (stained in brown for the immunoreaction), a branch of the portal vein (the largest blood vessel) and a terminal ramification of hepatic artery (the smallest blood vessel). (B) A small cyst with a diameter <1 cm lined by a columnar biliary epithelium. (C) A large cyst with a diameter >3 cm is enclosed with cholangiocytes that are flatter in appearance compared with the non-cystic biliary cells, because of the enlargement of the cyst and the resulting epithelium strain. CK-19 cholangiocytes are stained brown. Original magnification, 40×.
Fig. 2.

Representative immunohistochemistry for FSH receptor (FSHR) and follicle-stimulating hormone (FSH) in liver sections (3–4 μm thick) from normal patients or patients with autosomal dominant polycystic kidney disease (ADPKD). In particular, the staining showed a negativity for FSHR in liver from normal patient (A) and higher expression in small (B) and in large (C) cysts. The hormone FSH presents a low positivity in normal liver (D) that increases in cysts of small (E) and large diameter (F). Original magnification, 40×. The table contains the percentage of FSHR and FSH-positive cholangiocytes in normal biliary epithelium and in both small and large cysts. The positivity for the hormone and its receptor enhances with the cyst size, suggesting an important effect of FSH in the growth of the hepatic cysts.
Intracellular mechanisms of FSH regulation of cholangiocyte growth
As we have previously shown (14), the cystic epithelium showed a marked proliferative index. Normal cholangiocytes have a low expression of pERK and c-myc, two key proteins of the intracellular cAMP mechanism (Fig. 3A, D). In pathological cholangiocytes, the presence of the two cAMP mediators increases in both small and large cysts (Fig. 3B, C, E, F). The presence of pERK, the positivity for FSHR and the intense cholangiocyte proliferation in the course of ADPKD was confirmed by immunofluorescence, where we initially co-localized FSHR with PCNA (Fig. 4A) and then FSHR with pERK (Fig. 4B). In cystic cholangiocytes, FSHR presence may be associated with a paracrine action, but in some cells it can co-localize with PCNA thus sustaining an autocrine mechanism (Fig. 4A). FSHR expression has also been linked to the expression of pERK (Fig. 4B). For this reason, the phosphorylation of ERK is associated with the activation of the intracellular cAMP pathway and many cells simultaneously express FSHR with PCNA and pERK with FSHR supporting the concept that FSH induces cholangiocyte proliferation via ERK (37).
Fig. 3.
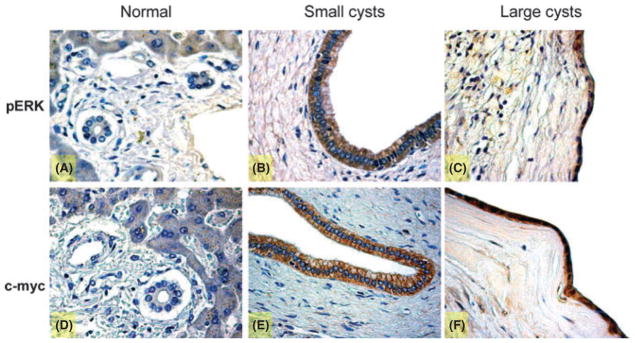
Immunohistochemistry for phospho-extracellular-regulated kinase (p-ERK) and c-myc in liver sections (3–4 μm thick) from normal patients or with autosomal dominant polycystic kidney disease (ADPKD). In particular, the staining showed a positive reaction for pERK in liver from normal patient (A) and both in small (B) and in large (C) cysts. The same profile for c-myc in normal liver (D) and in cysts of small (E) and large diameter (F) was found. The staining demonstrated that the phosphorylation of ERK and the expression of c-myc increase in course of polycystic disease. Original magnification, 40×.
Fig. 4.

Immunofluorescences in liver sections from normal and autosomal dominant polycystic kidney disease (ADPKD) patients (3–4 μm thick) indicating the colocalization of two specific immunoreactions to co-staining: (A) proliferating cell nuclear antigen (PCNA) (a marker of cellular proliferation) and FSH receptor (FSHR). Normal biliary epithelium does not show proliferative activity [nuclei are negative for proliferating cell nuclear antigen (PCNA)] and it is negative for FSHR. In small cysts, cholangiocyte proliferation increases, as it is evident with more PCNA positive nuclei, and they start to express also FSHR. In large cysts, the number of PCNA positive nuclei enhances and FSHR is present almost in all cells. (B) Phospho-extracellular-regulated kinase (p-ERK) and FSHR expression. A normal bile duct shows negativity for both FSHR and pERK. In a small cyst, the biliary epithelium starts to express FSHR and pERK that co-localize in the same cells. In the end, in the largest cysts we find a higher presence of the receptor and the phosphorylated protein. Co-localization of PCNA and FSHR was associated with increased cellular growth in cholangiocytes expressing FSHR. Simultaneously, FSHR expression is apparently linked to the phosphorylation of ERK. Original magnification, 40×.
Evaluation of the role of FSH in human cell lines
Both H69 and LCDE express FSHR and FSH (Fig. 5). These cells were starved without serum for 24 h and then exposed to FSH with or without PD98059. The addition of FSH increased the cholangiocyte proliferative index (tested by MTS proliferation assay and western blots for PCNA protein expression) whereas pre-incubation with PD98059 partially blocked this effect (Fig. 6A, B). To measure the intracellular levels of cAMP, we treated normal and pathological cholangiocytes with a basal solution of BSA or FSH in the absence or presence of PD98059 or an anti-FHSR antibody. Similar to that shown for secretin (37), we found that FSH increases cAMP levels, an increase that was prevented by pre-incubation with PD98059 or with the antibody anti-FSHR (Fig. 6C). Immunofluorescence for pERK in basal conditions and after treatment with the highest dose of FSH (100 μg/ml) demonstrates that the hormone increases the phosphorylation of ERK to a higher extent in LCDE cells compared with H69 cultured cells supporting enhanced cell proliferation (Fig. 6D).
Fig. 5.

Representative immunofluorescence for (A, B) FSH receptor FSHR and (C, D) follicle-stimulating hormone FSH in H69 and LCDE human cell lines. In the LCDE cells, the expression of FSHR and FSH is higher when compared with the non-malignant cholangiocytes (red), whereas in blue are stained nuclei (DAPI). No staining was visible when primary antibodies were replaced with non-immune serum. Bar = 200 μm.
Fig. 6.

Evaluation of cholangiocyte proliferation by MTS assay (A) and by proliferating cell nuclear antigen (PCNA) protein expression (B) in H69 (white bar) and in LCDE (black bar) treated with 0.2% BSA (basal) or follicle-stimulating hormone (FSH) (1–100 ng/ml with 0.2% BSA) in the absence or presence of PD98059 for 24 h. Intracellular cAMP levels (C) were measured in H69 (white bar) and in LCDE (black bar) treated with 0.2% BSA (basal) or FSH (100 ng/ml with 0.2% BSA) in the absence or presence of PD98059 or an anti-FSHR antibody. FSH significantly increased the growth of LCDE in a dose-dependent manner and the levels of cAMP inside the cell. Effects were blocked by pre-incubation with the inhibitor PD98059 and the anti-FSHR antibody. (D) Immunofluorescence for phospho-extracellular-regulated kinase p-ERK in basal conditions and after treatment with FSH (100 μg/ml) in H69 and LCDE cells. Images show that the hormone increases the phosphorylation of ERK to a higher extent in LCDE cells compared with H69 cultured cells. Data are expressed as mean ± SD of 6 experiments. *P < 0.05 vs. its corresponding basal value.
To confirm the evidence that FSH is a critical factor for sustaining cholangiocyte growth, we specifically knocked down the expression of FSH in LCDE cells by transient transfection (siRNA) (Fig. 7A, B). Real-time PCR for FSH showed that the most efficient siRNA-FSH concentration was 1 μg, which results in the largest reduction in FSH message expression (Fig. 7A). Furthermore, the FSH siRNA cell line exhibited reduced PCNA protein expression compared with mock-transfected cells, indicating that decreasing FSH expression impairs the proliferative capacity of cholangiocytes (Fig. 8A). These cells manifest a higher apoptotic degree compared with mock-transfected cholangiocytes as demonstrated by increased Bax protein expression (Fig. 8B). Lastly, we found that in the knocked-down cells, the intracellular secretin-stimulated cAMP levels as well as cholangiocyte proliferation decrease (Fig. 8C). This supports the concept that FSH sustains biliary growth via a cAMP-dependent signalling pathway. In general, the modifications of cAMP levels after stimulation with secretin are considered to be a reliable test to evaluate the effects of secretin on cholangiocyte proliferation as extensively demonstrated in the experimental models of cholangiocyte proliferation (37–39).
Fig. 7.

Evaluation of transient transfection with follicle-stimulating hormone (FSH) siRNA in LCDE cells. FSH silencing was investigated by real-time PCR to evaluate the message of the protein (A) and by western blot to assess the protein expression of FSH (B). Using increasing amounts of siRNA FSH (0.25–1 μg), there was a significant reduction in FSH mRNA and protein expression.
Fig. 8.

The effects of follicle-stimulating hormone (FSH) silencing in proliferation of LCDE cells were evaluated by proliferating cell nuclear antigen (PCNA) protein expression (A), Bax protein expression (B), and secretin-stimulated cAMP levels (C). Knockdown of FSH blocked the stimulatory effects of FSH on (A) PCNA protein expression, and (C) secretin-stimulated cAMP levels, and increased cholangiocyte apoptosis as evaluated by blots for Bax protein expression (B). Data are expressed as mean ± SD of 5 experiments. *P < 0.05 vs. the corresponding basal value.
Discussion
Our in vivo results show that: (i) the biliary epithelium that lines hepatic cysts stains positive for FSHR and FSH, whose expression is in relationship with the cyst size; (ii) FSH sustains cellular growth; and (iii) FSHR co-localizes with pERK in larger cysts. Regarding the in vitro studies, we demonstrated that: (i) both H69 and LCDE cells express FSHR and FSH; (ii) FSH stimulation of cholangiocyte proliferation is associated with increased cAMP levels; and (iii) knocking down FSH expression by siRNA decreases cholangiocyte proliferation and cAMP levels while increasing apoptosis. Cyst fragments were obtained from patients with ADPKD who underwent liver resection. ADPKD is caused by mutation in the PKD1 gene (85%) or PKD2 gene (10–15%) (40), which encodes the polycystin 1 (Pc-1) and polycystin 2 (Pc-2) proteins (41) respectively. The Pc-1/Pc-2 complex is located in the primary cilium at the apical pole of cholangiocytes (42). Recently, the key role of hormones such as oestrogens in this pathology has been studied in detail. Indeed, 1 year of oestrogen use in post-menopausal ADPKD patients selectively increases total liver volume by 7%, whereas total kidney volume remains unaffected (43). In addition, oestrogens sustain the enhanced proliferative and secretory activities of biliary epithelium, as experimentally shown in BDL rats, by acting either directly with growth factors or potentiating their effects (11, 44–46). Studies have shown that the epithelial surface of hepatic cysts of ADPKD patients displays a marked and diffuse immunoreaction for oestrogen receptors (14).
According to these recent findings, we hypothesized that the hepatic cyst epithelium of ADPKD patients could be considered as a hormone-responsive tissue. Hence, we have studied the role of FSH in the pathophysiology of hepatic cysts. FSH stimulates preovulatory follicles of the ovaries and is related to steroidogenesis (47). FSH induces cell proliferation and DNA synthesis by acting on its receptor (FSHR) (48). The human FSHR belongs to the superfamily of G proteincoupled receptors (49). Agonist binding to the FSHR triggers the rapid activation of multiple signalling cascades, mainly the cAMP–adenylyl cyclase–proteinkinase A cascade (50). We have already demonstrated that the FSH induces cholangiocyte proliferation in normal rats by acting on the cAMP-dependent ERK1/2–Elk-1 signalling pathway (17). This increase was partially blocked by treatment with Antide (a GnRH antagonist) or by a neutralizing FSH antibody (17). In general, FSH represents the major stimulator and regulator of oestrogen production. In particular, FSH determines the aromatization of androgens into oestrogens via the activation of the cAMP/protein kinase A (PKA)-dependent transcription factor, leading to the transcription of the aromatase enzyme (51, 52).
In this study, we found that normal human cholangiocytes from interlobular bile ducts and those derived from biliary epithelium of hepatic cysts express FSHR and FSH. The increasing presence of this hormone is correlated with a higher proliferation index, most likely because of the effect of FSH on the cAMP/ERK-dependent signalling pathway, which is one of the most critical intracellular mechanisms regulating cholangiocyte proliferation and phosphorylation of ERK (20, 28, 53–55). This role of FSH may also be because of the effects of this hormone on the transcriptional activation of ER-responsive genes that are under the regulation of the cAMP/PKA/ERK-signalling pathway (56, 57). Presumably, in this case FSH cooperates with oestrogens and other hormones to increase the proliferative response of the biliary epithelium (58, 59). A direct link was not found in these preliminary studies, which will be the aim of further studies. However, the combination of FSH and the activation of other hormones may result in a synergistic effect on cell proliferation that may be attenuated using anti-oestrogens. To support our in vivo findings, the in vitro studies were expanded in two ways: (i) ablation of the proliferative effect of FSH with an inhibitor of the MEK/ERK pathway and (ii) silencing FSH by siRNA. FSH stimulates the increase in cholangiocyte proliferation predominantly in LCDE cells, together with the enhanced cAMP levels, which were blocked by PD98059. As conclusive evidence that FSH plays a key role in sustaining cyst growth acting on the cAMP pathway, the knock down of FSH expression in LCDE cells demonstrates that lack of this hormone decreases the proliferative index of cholangiocyte and impairs cellular levels of cAMP.
Parallel to our findings, others have shown that the effects of FSH are mediated by the activation of a cAMP-dependent mechanism, such as in granulosa cells, where FSH stimulates mTOR signalling through the ERK-rather than the Akt-dependent pathway (60). The mTOR signalling pathway regulates growth and proliferation of cells from yeast to mammals in response to growth factors, hormones and nutrient availability (61). Inhibition of mTOR has been shown to cause G1 phase arrest of the cell cycle (62, 63). Hence the mTOR pathway may be involved in the mediation of the cyst progression in an orthologous animal model of human ARPKD (64).
In addition, several studies investigated the role of resident progenitor cells in liver pathophysiology (65, 66); in polycystic liver disease, the implication of the liver regenerative compartment and its potential role in generating liver cysts have not been elucidated. Interestingly, ADPKD and ARPLD are associated with a characteristic cholangiopathy, which is considered to be a prototypic example of ductal plate malformation (DPM) (67). DPM are congenital diseases of the intrahepatic bile ducts caused by the failure of the physiologic ductal plate remodelling during embryonic development of the biliary system. Human hepatic stem cells (hHpSC) are considered to be the remnant of the ductal plate in the adult liver (68). Moreover, epithelial cells lining the liver cysts show signs of immaturity, express adhesion molecules and a number of vascular growth factors that are reminiscent of ductal plate cells (67–70).
In addition, Qian et al. demonstrated that liver cysts arise from peribiliary glands (PBGs) located in the large intrahepatic bile ducts (71). The intrahepatic cysts are within the liver parenchyma, but not in contact with the larger portal triads, whereas the peribiliary cysts are adjacent to the larger portal triads or in the hepatic hilum (71). Recently, the presence of biliary tree stem cells (BTSC) has been demonstrated in PBGs (72); these cells represent the remnant of the fetal bilio-pancreatic precursors (73, 74). The role of BTSCs in generating liver cysts is unknown. Our preliminary observations indicate that the hHpSC and BTSC compartments are expanded in liver parenchyma adjacent to liver cysts and that these cells are able to express FSH (data not shown). Probably, the expansion of liver regenerative compartments may be related to the compression because of the cysts, but their role in cyst formation needs to be better investigated. However, this concept will need to be evaluated in depth in human pathology. Similar to other studies, we have determined that an additional hormone, FSH, exerts a fundamental effect to sustain cholangiocyte growth during the course of polycystic liver disease via the cAMP/ERK-dependent signalling pathway. These data support the main role of cAMP that causes cholangiocyte hyperproliferation, abnormal cell–matrix interactions and other cellular condition can lead to cystogenesis. Thus, further studies are necessary to elucidate therapeutic approaches that target this signalling pathway. Finally, additional studies are needed to determine other factors that may interact in the cAMP-dependent signalling mechanism during the course of autosomal dominant polycystic liver disease.
Acknowledgments
Thanks to Mrs Liliana Domizi for her skilful technical assistance.
Funding: This work was funded by the Sapienza University funds and PRIN 2009 to E. Gaudio, and Dr Nicholas C. Hightower Centennial Chair of Gastroenterology from Scott & White and the NIH grant DK062975 to Dr Alpini.
References
- 1.Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76:149–68. doi: 10.1038/ki.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drenth JP, Tahvanainen E, Te Morsche RH, et al. Abnormal hepatocystin caused by truncating PRKCSH mutations leads to autosomal dominant polycystic liver disease. Hepatology. 2004;39:924–31. doi: 10.1002/hep.20141. [DOI] [PubMed] [Google Scholar]
- 3.Davila S, Furu L, Gharavi AG, et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet. 2004;36:575–7. doi: 10.1038/ng1357. [DOI] [PubMed] [Google Scholar]
- 4.Russell RT, Pinson CW. Surgical management of polycystic liver disease. World J Gastroenterol. 2007;13:5052–9. doi: 10.3748/wjg.v13.i38.5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Keimpema L, Hockerstedt K. Treatment of polycystic liver disease. Br J Surg. 2009;96:1379–80. doi: 10.1002/bjs.6738. [DOI] [PubMed] [Google Scholar]
- 6.Onori P, Franchitto A, Mancinelli R, et al. Polycystic liver diseases. Dig Liver Dis. 2010;42:261–71. doi: 10.1016/j.dld.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gabow PA, Johnson AM, Kaehny WD, et al. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology. 1990;11:1033–7. doi: 10.1002/hep.1840110619. [DOI] [PubMed] [Google Scholar]
- 8.Masyuk TV, Masyuk AI, Torres VE, Harris PC, Larusso NF. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3′,5′-cyclic monophosphate. Gastroenterology. 2007;132:1104–16. doi: 10.1053/j.gastro.2006.12.039. [DOI] [PubMed] [Google Scholar]
- 9.Van Keimpema L, De Man RA, Drenth JP. Somatostatin analogues reduce liver volume in polycystic liver disease. Gut. 2008;57:1338–9. doi: 10.1136/gut.2008.155721. [DOI] [PubMed] [Google Scholar]
- 10.Drenth JP, Chrispijn M, Nagorney DM, Kamath PS, Torres VE. Medical and surgical treatment options for polycystic liver disease. Hepatology. 2010;52:2223–30. doi: 10.1002/hep.24036. [DOI] [PubMed] [Google Scholar]
- 11.Alvaro D, Barbaro B, Franchitto A, et al. Estrogens and insulin-like growth factor 1 modulate neoplastic cell growth in human cholangiocarcinoma. Am J Pathol. 2006;169:877–88. doi: 10.2353/ajpath.2006.050464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alvaro D, Mancino MG, Onori P, et al. Estrogens and the pathophysiology of the biliary tree. World J Gastroenterol. 2006;12:3537–45. doi: 10.3748/wjg.v12.i22.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glaser SS, Onori P, Wise C, et al. Recent advances in the regulation of cholangiocyte proliferation and function during extrahepatic cholestasis. Dig Liver Dis. 2010;42:245–52. doi: 10.1016/j.dld.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alvaro D, Onori P, Alpini G, et al. Morphological and functional features of hepatic cyst epithelium in autosomal dominant polycystic kidney disease. Am J Pathol. 2008;172:321–32. doi: 10.2353/ajpath.2008.070293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taffetani S, Glaser S, Francis H, et al. Prolactin stimulates the proliferation of normal female cholangiocytes by differential regulation of Ca2+-dependent PKC isoforms. BMC Physiol. 2007;7:6. doi: 10.1186/1472-6793-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glaser S, Demorrow S, Francis H, et al. Progesterone stimulates the proliferation of female and male cholangiocytes via autocrine/paracrine mechanisms. Am J Physiol Gastrointest Liver Physiol. 2008;295:G124–36. doi: 10.1152/ajpgi.00536.2007. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Mancinelli R, Onori P, Gaudio E, et al. Follicle-stimulating hormone increases cholangiocyte proliferation by an autocrine mechanism via cAMP-dependent phosphorylation of ERK1/2 and Elk-1. Am J Physiol Gastrointest Liver Physiol. 2009;297:G11–26. doi: 10.1152/ajpgi.00025.2009. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Richards JS, Pangas SA. The ovary: basic biology and clinical implications. J Clin Invest. 2010;120:963–72. doi: 10.1172/JCI41350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hunzicker-Dunn M, Maizels ET. FSH signaling pathways in immature granulosa cells that regulate target gene expression: branching out from protein kinase A. Cell Signal. 2006;18:1351–9. doi: 10.1016/j.cellsig.2006.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mancinelli R, Franchitto A, Gaudio E, et al. After damage of large bile ducts by gamma-aminobutyric acid, small ducts replenish the biliary tree by amplification of calcium-dependent signaling and de novo acquisition of large cholangiocyte phenotypes. Am J Pathol. 2010;176:1790–800. doi: 10.2353/ajpath.2010.090677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryan KE, Casey SM, Canty MJ, et al. Akt and Erk signal transduction pathways are early markers of differentiation in dominant and subordinate ovarian follicles in cattle. Reproduction. 2007;133:617–26. doi: 10.1530/REP-06-0130. [DOI] [PubMed] [Google Scholar]
- 22.Sears R, Nuckolls F, Haura E, et al. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501–14. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ussar S, Voss T. MEK1 and MEK2, different regulators of the G1/S transition. J Biol Chem. 2004;279:43861–9. doi: 10.1074/jbc.M406240200. [DOI] [PubMed] [Google Scholar]
- 24.Berns K, Hijmans EM, Bernards R. Repression of c-Myc responsive genes in cycling cells causes G1 arrest through reduction of cyclin E/CDK2 kinase activity. Oncogene. 1997;15:1347–56. doi: 10.1038/sj.onc.1201280. [DOI] [PubMed] [Google Scholar]
- 25.Oster SK, Ho CS, Soucie EL, Penn LZ. The myc oncogene: MarvelouslY Complex. Adv Cancer Res. 2002;84:81–154. doi: 10.1016/s0065-230x(02)84004-0. [DOI] [PubMed] [Google Scholar]
- 26.Johnston LA, Prober DA, Edgar BA, Eisenman RN, Gallant P. Drosophila myc regulates cellular growth during development. Cell. 1999;98:779–90. doi: 10.1016/s0092-8674(00)81512-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kohno M, Pouyssegur J. Pharmacological inhibitors of the ERK signaling pathway: application as anticancer drugs. Prog Cell Cycle Res. 2003;5:219–24. [PubMed] [Google Scholar]
- 28.Glaser SS, Gaudio E, Miller T, Alvaro D, Alpini G. Cholangiocyte proliferation and liver fibrosis. Expert Rev Mol Med. 2009;11:e7. doi: 10.1017/S1462399409000994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alpini G, Ulrich CD, 2nd, Phillips JO, et al. Upregulation of secretin receptor gene expression in rat cholangiocytes after bile duct ligation. Am J Physiol. 1994;266(5 Pt 1):G922–8. doi: 10.1152/ajpgi.1994.266.5.G922. [DOI] [PubMed] [Google Scholar]
- 30.Lesage G, Glaser SS, Gubba S, et al. Regrowth of the rat biliary tree after 70% partial hepatectomy is coupled to increased secretin-induced ductal secretion. Gastroenterology. 1996;111:1633–44. doi: 10.1016/s0016-5085(96)70027-6. [DOI] [PubMed] [Google Scholar]
- 31.Lesage G, Glaser S, Alpini G. Regulation of cholangiocyte proliferation. Liver. 2001;21:73–80. doi: 10.1034/j.1600-0676.2001.021002073.x. [DOI] [PubMed] [Google Scholar]
- 32.Lesage G, Glaser S, Ueno Y, et al. Regression of cholangiocyte proliferation after cessation of ANIT feeding is coupled with increased apoptosis. Am J Physiol Gastrointest Liver Physiol. 2001;281:G182–90. doi: 10.1152/ajpgi.2001.281.1.G182. [DOI] [PubMed] [Google Scholar]
- 33.Ravine D, Gibson RN, Walker RG, et al. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet. 1994;343:824–7. doi: 10.1016/s0140-6736(94)92026-5. [DOI] [PubMed] [Google Scholar]
- 34.Carpino G, Cardinale V, Onori P, et al. Biliary tree stem/ progenitor cells in glands of extrahepatic and intraheptic bile ducts: an anatomical in situ study yielding evidence of maturational lineages. J Anat. 2012;220:186–99. doi: 10.1111/j.1469-7580.2011.01462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kato A, Gores GJ, Larusso NF. Secretin stimulates exocytosis in isolated bile duct epithelial cells by a cyclic AMPmediated mechanism. J Biol Chem. 1992;267:15523–9. [PubMed] [Google Scholar]
- 36.Lenzen R, Alpini G, Tavoloni N. Secretin stimulates bile ductular secretory activity through the cAMP system. Am J Physiol. 1992;263(4 Pt 1):G527–32. doi: 10.1152/ajpgi.1992.263.4.G527. [DOI] [PubMed] [Google Scholar]
- 37.Francis H, Glaser S, Ueno Y, et al. cAMP stimulates the secretory and proliferative capacity of the rat intrahepatic biliary epithelium through changes in the PKA/Src/MEK/ ERK1/2 pathway. J Hepatol. 2004;41:528–37. doi: 10.1016/j.jhep.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 38.Lesage GD, Benedetti A, Glaser S, et al. Acute carbon tetrachloride feeding selectively damages large, but not small, cholangiocytes from normal rat liver. Hepatology. 1999;29:307–19. doi: 10.1002/hep.510290242. [DOI] [PubMed] [Google Scholar]
- 39.Alpini G, Ulrich C, Roberts S, et al. Molecular and functional heterogeneity of cholangiocytes from rat liver after bile duct ligation. Am J Physiol. 1997;272(2 Pt 1):G289–97. doi: 10.1152/ajpgi.1997.272.2.G289. [DOI] [PubMed] [Google Scholar]
- 40.Mochizuki T, Wu G, Hayashi T, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339–42. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- 41.Hanaoka K, Qian F, Boletta A, et al. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature. 2000;408:990–4. doi: 10.1038/35050128. [DOI] [PubMed] [Google Scholar]
- 42.Nauli SM, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–37. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 43.Van Keimpema L, De Koning DB, Van Hoek B, et al. Patients with isolated polycystic liver disease referred to liver centres: clinical characterization of 137 cases. Liver Int. 2011;31:92–8. doi: 10.1111/j.1478-3231.2010.02247.x. [DOI] [PubMed] [Google Scholar]
- 44.Alvaro D, Alpini G, Onori P, et al. Effect of ovariectomy on the proliferative capacity of intrahepatic rat cholangiocytes. Gastroenterology. 2002;123:336–44. doi: 10.1053/gast.2002.34169. [DOI] [PubMed] [Google Scholar]
- 45.Alvaro D, Metalli VD, Alpini G, et al. The intrahepatic biliary epithelium is a target of the growth hormone/insulin-like growth factor 1 axis. J Hepatol. 2005;43:875–83. doi: 10.1016/j.jhep.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 46.Alvaro D, Alpini G, Onori P, et al. Alfa and beta estrogen receptors and the biliary tree. Mol Cell Endocrinol. 2002;193:105–8. doi: 10.1016/s0303-7207(02)00103-x. [DOI] [PubMed] [Google Scholar]
- 47.Park YH, Kim SJ, Jeong BH, et al. Follicular stimulating hormone enhances Notch 1 expression in SK-OV-3 ovarian cancer cells. J Gynecol Oncol. 2010;21:119–24. doi: 10.3802/jgo.2010.21.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu HY, Zeng WD, Cao AL, Zhang CQ. Follicle-stimulating hormone promotes proliferation of cultured chicken ovarian germ cells through protein kinases A and C activation. J Zhejiang Univ Sci B. 2010;11:952–7. doi: 10.1631/jzus.B1000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dias JA, Cohen BD, Lindau-Shepard B, et al. Molecular, structural, and cellular biology of follitropin and follitropin receptor. Vitam Horm. 2002;64:249–322. doi: 10.1016/s0083-6729(02)64008-7. [DOI] [PubMed] [Google Scholar]
- 50.Ulloa-Aguirre A, Zarinan T, Pasapera AM, Casas-Gonzalez P, Dias JA. Multiple facets of follicle-stimulating hormone receptor function. Endocrine. 2007;32:251–63. doi: 10.1007/s12020-008-9041-6. [DOI] [PubMed] [Google Scholar]
- 51.Chen H, Guo JH, Lu YC, et al. Impaired CFTR-dependent amplification of FSH-stimulated estrogen production in cystic fibrosis and PCOS. J Clin Endocrinol Metab. 2012;97:923–32. doi: 10.1210/jc.2011-1363. [DOI] [PubMed] [Google Scholar]
- 52.Stocco C. Aromatase expression in the ovary: hormonal and molecular regulation. Steroids. 2008;73:473–87. doi: 10.1016/j.steroids.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Francis H, Lesage G, Demorrow S, et al. The alpha2-adrenergic receptor agonist UK 14,304 inhibits secretin-stimulated ductal secretion by downregulation of the cAMP system in bile duct-ligated rats. Am J Physiol Cell Physiol. 2007;293:C1252–62. doi: 10.1152/ajpcell.00031.2007. [DOI] [PubMed] [Google Scholar]
- 54.Alpini G, Franchitto A, Demorrow S, et al. Activation of alpha(1)-adrenergic receptors stimulate the growth of small mouse cholangiocytes via calcium-dependent activation of nuclear factor of activated T cells 2 and specificity protein 1. Hepatology. 2011;53:628–39. doi: 10.1002/hep.24041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Francis H, Glaser S, Demorrow S, et al. Small mouse cholangiocytes proliferate in response to H1 histamine receptor stimulation by activation of the IP3/CaMK I/CREB pathway. Am J Physiol Cell Physiol. 2008;295:C499–513. doi: 10.1152/ajpcell.00369.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Onori P, Wise C, Gaudio E, et al. Secretin inhibits cholangiocarcinoma growth via dysregulation of the cAMP-dependent signaling mechanisms of secretin receptor. Int J Cancer. 2010;127:43–54. doi: 10.1002/ijc.25028. [DOI] [PubMed] [Google Scholar]
- 57.Pasapera AM, del Jimenez-Aguilera MP, Chauchereau A, et al. Effects of FSH and 17beta-estradiol on the transactivation of estrogen-regulated promoters and cell proliferation in L cells. J Steroid Biochem Mol Biol. 2005;94:289–302. doi: 10.1016/j.jsbmb.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 58.Gaudio E, Barbaro B, Alvaro D, et al. Administration of r-VEGF-A prevents hepatic artery ligation-induced bile duct damage in bile duct ligated rats. Am J Physiol Gastrointest Liver Physiol. 2006;291:G307–17. doi: 10.1152/ajpgi.00507.2005. [DOI] [PubMed] [Google Scholar]
- 59.Gaudio E, Barbaro B, Alvaro D, et al. Vascular endothelial growth factor stimulates rat cholangiocyte proliferation via an autocrine mechanism. Gastroenterology. 2006;130:1270–82. doi: 10.1053/j.gastro.2005.12.034. [DOI] [PubMed] [Google Scholar]
- 60.Kayampilly PP, Menon KM. Follicle-stimulating hormone increases tuberin phosphorylation and mammalian target of rapamycin signaling through an extracellular signal-regulated kinase-dependent pathway in rat granulosa cells. Endocrinology. 2007;148:3950–7. doi: 10.1210/en.2007-0202. [DOI] [PubMed] [Google Scholar]
- 61.Harris TE, Lawrence JC., Jr TOR signaling. Sci STKE. 2003;212:re15. doi: 10.1126/stke.2122003re15. [DOI] [PubMed] [Google Scholar]
- 62.Crespo JL, Hall MN. Elucidating TOR signaling and rapamycin action: lessons from Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2002;66:579–91. doi: 10.1128/MMBR.66.4.579-591.2002. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oldham S, Hafen E. Insulin/IGF and target of rapamycin signaling: a TOR de force in growth control. Trends Cell Biol. 2003;13:79–85. doi: 10.1016/s0962-8924(02)00042-9. [DOI] [PubMed] [Google Scholar]
- 64.Renken C, Fischer DC, Kundt G, Gretz N, Haffner D. Inhibition of mTOR with sirolimus does not attenuate progression of liver and kidney disease in PCK rats. Nephrol Dial Transplant. 2011;26:92–100. doi: 10.1093/ndt/gfq384. [DOI] [PubMed] [Google Scholar]
- 65.Spee B, Carpino G, Schotanus BA, et al. Characterisation of the liver progenitor cell niche in liver diseases: potential involvement of Wnt and Notch signalling. Gut. 2010;59:247–57. doi: 10.1136/gut.2009.188367. [DOI] [PubMed] [Google Scholar]
- 66.Gaudio E, Carpino G, Cardinale V, et al. New insights into liver stem cells. Dig Liver Dis. 2009;41:455–62. doi: 10.1016/j.dld.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 67.Raynaud P, Tate J, Callens C, et al. A classification of ductal plate malformations based on distinct pathogenic mechanisms of biliary dysmorphogenesis. Hepatology. 2011;53:1959–66. doi: 10.1002/hep.24292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Turner R, Lozoya O, Wang Y, et al. Human hepatic stem cell and maturational liver lineage biology. Hepatology. 2011;53:1035–45. doi: 10.1002/hep.24157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kida T, Nakanuma Y, Terada T. Cystic dilatation of peribiliary glands in livers with adult polycystic disease and livers with solitary nonparasitic cysts: an autopsy study. Hepatology. 1992;16:334–40. doi: 10.1002/hep.1840160209. [DOI] [PubMed] [Google Scholar]
- 70.Ramos A, Torres VE, Holley KE, et al. The liver in autosomal dominant polycystic kidney disease. Implications for pathogenesis. Arch Pathol Lab Med. 1990;114:180–4. [PubMed] [Google Scholar]
- 71.Qian Q, Li A, King BF, et al. Clinical profile of autosomal dominant polycystic liver disease. Hepatology. 2003;37:164–71. doi: 10.1053/jhep.2003.50006. [DOI] [PubMed] [Google Scholar]
- 72.Cardinale V, Wang Y, Carpino G, et al. Multipotent stem/ progenitor cells in human biliary tree give rise to hepatocytes, cholangiocytes, and pancreatic islets. Hepatology. 2011;54:2159–72. doi: 10.1002/hep.24590. [DOI] [PubMed] [Google Scholar]
- 73.Cardinale V, Wang Y, Carpino G, et al. The biliary tree – a reservoir of multipotent stem cells. Nat Rev Gastroenterol Hepatol. 2012;9:231–40. doi: 10.1038/nrgastro.2012.23. [DOI] [PubMed] [Google Scholar]
- 74.Semeraro R, Carpino G, Cardinale V, et al. Multipotent stem/progenitor cells in the human foetal biliary tree. J Hepatol. 2012;57:987–94. doi: 10.1016/j.jhep.2012.07.013. [DOI] [PubMed] [Google Scholar]