

Abstract
The treatment of central nervous system (CNS) disease has long been difficult due to the ineffectiveness of drug delivery across the blood-brain barrier (BBB). This review summarizes important concepts of the BBB in normal versus pathophysiology and how this physical, enzymatic, and efflux barrier provides necessary protection to the CNS during drug delivery, and consequently treatment challenging. Small molecules account for the vast majority of available CNS drugs primarily due to their ability to penetrate the phospholipid membrane of the BBB by passive or carrier-mediated mechanisms. Physiochemical and biological factors relevant for designing small molecules with optimal capabilities for BBB permeability are discussed, as well as the most promising classes of transporters suitable for small-molecule drug delivery. Clinically translatable imaging methodologies for detecting and quantifying drug uptake and targeting in the brain are discussed as a means of further understanding and refining delivery parameters for both drugs and imaging probes in preclinical and clinical domains. This information can be used as a guide to design drugs with preserved drug action and better delivery profiles for improved treatment outcomes over existing therapeutic approaches.
Keywords: brain delivery, CNS drug, blood-brain barrier (BBB), efflux pumps, passive diffusion, facilitated transport
Introduction
The blood-brain barrier (BBB) has long been the rate-limiting step for successful translation of central nervous system (CNS)-targeted drugs for a variety of neurological-based pathologies. CNS diseases range from psychiatric disorders to neuroinflammatory and neurodegenerative diseases, and primary and metastatic brain cancer. The BBB serves to protect the brain by maintaining CNS homeostasis and keeping toxic materials out of this privileged compartment. Achieving effective drug concentrations within the CNS depends on multiple factors, including the bioavailability of the drug for CNS delivery, brain penetrability of the drug, the extent to which the agent is actively transported out of the brain compartment, and its volume of distribution in the brain parenchyma at the desired site of action.
From a medicinal chemistry perspective, the ability to design drugs capable of crossing the BBB and eliciting the desirable biological response is a formidable challenge. The normal barrier itself is essentially impenetrable to large macromolecules such as peptides and immunoglobulin (IgG) antibody proteins,1 whereas less than 2% of all US Food and Drug Agency (FDA)-approved small-molecule drugs cross the intact BBB to varying degrees. The limited success in CNS drug delivery is disconcerting considering a recent World Health Organization (WHO) report detailing that brain disorders affect nearly one billion people worldwide.2 Although this report specifically excludes psychiatric disorders, a recent report highlights that more than 450 million people suffer from mental illnesses.3 This represents an enormous societal burden in terms of human suffering and economic cost. Hence, there is a great need to develop novel disease-targeted neurotherapeutics optimized for brain drug delivery as well as repurpose/redesign existing drugs, originally discarded for insufficient brain delivery, to improve their brain delivery profile.
This review seeks to characterize the physical, enzymatic, and transport barriers created by the BBB and how these barriers are altered by disease. Examples and strategies for overcoming the BBB with small-molecule CNS-acting drugs are presented, as well as the physiochemical and structural attributes to consider in designing effective neurotherapeutics are presented. Finally, noninvasive imaging is presented as a translational tool for guiding and quantifying the optimal delivery of CNS-targeted agents to improve our knowledge and management of neurological disorders.
Components of the BBB
Structural barrier
Before examining what properties of molecules cause them to permeate the BBB, a working knowledge of the structural, enzymatic, and transport barrier itself is necessary. The BBB is composed of a network of blood vessels that form a physical and chemical barrier between the brain parenchyma and systemic circulation. The surface of a polarized cell facing the lumen is referred to as the apical membrane; the side facing the brain parenchyma is called the abluminal/basolateral side. The primary physical barrier of this neurovascular unit is the monolayer of brain capillary endothelial cells (BCECs) (Fig. 1A). BCECs lack fenestrae for rapid exchange of molecules between tissues and vessels, have limited pinocytotic vesicles to minimize uptake of extracellular substances for transcellular transport, and are characterized by high-resistance tight junctions (TJs) that severely restrict paracellular permeability (Fig. 1B).4 Occludin, claudins, and junctional adhesion molecules (JAM)-A are key transmembrane components of the TJ complex. Accessory proteins, such as zonula occludens (ZO)-1 and ZO-2, anchor the junctional proteins to the BCEC cytoskeleton.5,6 The integrin class of adhesion molecules is responsible for tethering and stabilizing the abluminal side of the BBB endothelial monolayer to the mechanical support offered by the extracellular basal lamina matrix.7,8
Figure 1.

The barriers to drug delivery across the BBB. The brain is composed of an extensive vascular network, and the BBB is at the interface between the systemic circulation and the brain parenchyma to control access of drugs to the brain by multiple means. (A) The neurovascular unit (NVU) of the BBB consists of BCEC monolayer surrounding the vascular lumen of the extensive vascular network of the brain. The endothelium, with its highly resistant TJs, is surrounded by a basement membrane and is further supported by pericytes, astrocytes, neurons, and microglia. (B) Expansion of the TJ of the NVU shows the various proteins in the molecular complex involved in creating the physically impenetrable barrier of the TJ. Intercellular proteins (occludin, claudin, JAM) are anchored to the BCEC cytoplasm by anchoring proteins (eg, ZO-1, ZO-2) and actin cytoskeleton. Figure adapted from Hawkins et al.25 (C) Further expansion of the BCEC shows the relative location of drug efflux pumps located on the BBB. MRP1, MRP3, MRP5, and MRP6 are all located on the abluminal plasma membrane of BCEC and serve to transport materials from within the brain to within BCECs. P-gp, MRP2, and MRP-4 are all located on the apical plasma membrane and serve to transport drugs and conjugates of the BCECs and into the systemic circulation. All efflux pumps require ATP to transport substrates against their concentration gradient and have unique substrates/inhibitors. Table 1 shows the specific inhibitors denoted by (i)–(vi) in panel C.
Pericytes cover 20–30% of the capillary surface and serve to stabilize and monitor vessel wall stability through the basal lamina supporting the BCECs.7,8 The last major cell type comprising the BBB is astrocytes, whose feet wrap around >99% of BBB endothelium.8,9 Astrocytes are responsible for maintaining homeostasis through water transport, free radical scavenging, nutrient uptake/excretion, and ion buffering, for example.10 They also serve as scaffolds, guiding neurons to their proper place during development and direct vessels of the BBB. Under normal physiological conditions, these components of the BBB together form a distinct physical barrier that tightly controls blood-borne molecules access to the brain.
Enzymatic barrier
For those compounds bypassing the structural barrier, there exists an enzymatic barrier at the BBB that metabolizes drugs and nutrients to further restrict the transfer or retention of materials across the BBB. This enzymatic surveillance system includes phase-I (eg, γ-glutamyl transpeptidase (γ-GTP),11 alkaline phosphatase,12 cytochrome p450,13 and aromatic acid decarboxylase14) and phase-II (eg, glutathione S-transferase (GST)15 and epoxide hydrolase/UDP-glucuronosyl-transferase16) metabolizing enzymes which modify drugs to make them substrates for efflux pumps that are further described below. These enzymes also convert numerous compounds to inactive metabolites. Arachidonic acid, testosterone, progesterone, and the tricyclic antidepressant desipramine are just a few substrates of cytochrome p450 that once metabolized, become inactive.17 These enzymes are highly expressed in cerebral vessels and are often polarized between the luminal and abluminal membrane surface of BCECs, where they typically co-localize to a large extent with drug efflux pumps.
Efflux barrier
Evading drug efflux mechanisms in the BBB to maintain relevant drug concentrations in the brain is another tremendous challenge in CNS drug delivery. Several different energy-dependent efflux transporters are present in the BBB, both on the luminal and abluminal sides of BCECs, functioning as clearance systems for metabolites and catabolites produced in the brain. The BBB efflux pumps are transmembrane P-glycoprotein (P-gp) and multidrug resistance-associated protein (MRP) 1–5, both classified as ATP-binding cassette (ABC) transporters that use ATP hydrolysis to translocate substrates across their concentration gradient. These transporters recognize a wide diversity of xenobiotics with some degrees of overlap. Various compounds, as listed in Table 1, have been shown to inhibit efflux transporter expression and thus can significantly alter drug concentration in the brain.18–20
Table 1.
| ABC TRANSPORTERS ON BBB | ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| P-gp | MRP1 | MRP2 | MRP3 | MRP4 | MRP5 | |||
| Analgesics | Antibiotics | Anticancer Drugs | Anticancer Drugs | Anticancer Drugs | cGMP | |||
| Asimadoline | Erythomycin | Actinomycin D | Daunorubicine | Methotrexate | cGMP | |||
| Methadone | Valinomycin | Daunorubicin | Doxorubicin | 6-Mercaptopurine | Fluorescein | |||
| Doxorubicin | Etoposide | Thioguanine | 6-Mercaptopurine | |||||
| Paclitaxel | Methotrexate | Thioguanine | ||||||
| Vinblastine | Teniposide | |||||||
| Vincristine | ||||||||
| Antifungals | Antihistamines | Antihypertensives | ||||||
| Ketoconazole | Cetirizine | Atorvastatin | ||||||
| Itraconazole | Fexofenadine | Losartan | ||||||
| Substrates | Antipsychotics | β-Blockers | Cardioactive Drugs | Drug-Conjugates | Similar to MRP1 Substrates | |||
| Risperidone | Talinolol | Amiodarone | Glutathione | |||||
| Digoxin | Glucuronide | |||||||
| Quinidine | Sulfate | |||||||
| Verapamil | ||||||||
| Carticosteroids | HIV Protease | Immunosuppressants | Unconjugated Ligands | |||||
| Corticosterone | Inhibitors | Cyclosporine A | Fluorescein | |||||
| Dexamethasone | Nelfinavir | Tacrolimus | ||||||
| Hydrocortisone | Ritonavir | |||||||
|
| ||||||||
| Inhibitors (Notation in Fig 1C) | 1st Generation | 2nd Generation | 3rd Generation | Cyclosporine A | Same as | Indomethacin | Celecoxib | Probenecid |
| Cyclosporine A | Dexverapamil | Elacridar | Probenecid | MRP1 | Probenecid | Didofenac | Sildenafil | |
| Quinidine | Emopamil | Laniquidar | Verapamil | Inhibitors | Sulfinpyrazone | Trequensin | ||
| Verapamil | Valspodar | Zosuquidar | ||||||
| (i) | (ii) | (iii) | (iv) | (v) | (vi) | |||
P-gp is encoded in humans by the multidrug resistance gene MDR1 and it is the primary drug efflux mechanism of the BBB. The P-gp transporter is promiscuous in that it transports many structurally diverse classes of drugs out of the brain (Table 1). Its expression is localized to the luminal (apical) membrane of the BBB.21 The functional significance of P-gp was highlighted in Mdr1a(−/−) knockout mice; having complete absence of P-gp in BCEC these mice displayed 100-fold greater sensitivity to the neurotoxic effects of the antiparasitic ivermectin.22 As recently reviewed, in addition to genetic modulation, pharmacological inhibition of P-gp-mediated transport significantly increases brain drug levels in mouse models and in non-human primates compared to control groups up to ∼150-fold.20 The immunosuppressant cyclosporine A (CsA) inhibits P-gp transport to increase CNS delivery of the selective serotonin reuptake inhibitor (SSRI) escitalopram and augments anti-depressant effects in rodents.23,24 Vascular endothelial growth factor (VEGF) and interleukin (IL)-6 are two endogenous compounds that downregulate P-gp expression, with the response to IL-6 likely being part of the brain’s innate neuroprotective response to an inflammatory insult.25,26 Tumor necrosis factor (TNF)-α and IL-1β are endogenous inflammatory cytokines shown to upregulate P-gp activity and increase drug efflux across the BBB.26 Drug exposure is thought to cause overexpression of P-gp by both selection of resistant cells and induction of P-gp expression at the level of the MDR1 promoter.27,28
The role of the breast cancer resistance protein (BCRP, ABCG2) in drug efflux at the BBB is still not clearly understood and has been reviewed elsewhere.29 However, recent discoveries highlight that dual Mdr1a/b(−/−)Bcrp1(−/−) knockdown in mice or simultaneous P-gp and BCRP inhibition with Elacridar results in dramatic brain uptake levels of Dasatinib, a tyrosine kinase inhibitor used to treat Gleevec-resistant chronic myelogenous leukemia (CML).30 Studies like these demonstrate the complexity of the BBB and how P-gp works in concert with other transporter types to form an effective barrier to alter the BBB permeability of small-molecule drugs.
MRPs 1–5 are differentially expressed on the luminal and/or abluminal side of the BBB and are also found on astrocytes and microglia.10 Similar to P-gp, MRP inhibition has been shown to increase drug levels in the brain. Each MRP has its own substrate selectivity and localization on the BBB. MRP1, expressed on the basolateral side of the BCEC, transports organic anionic compounds. Drugs conjugated to acidic ligands such as glucoronate or sulfate are preferred MRP1 substrates. MRP1 is also one of the glutathione-S-conjugate pumps, able to transport glutathione (GSH)-drug conjugates out of the cell. MRP2 and MRP3 have similar substrate overlap with MRP1; however, they are expressed on the luminal and basolateral side of BCECs, respectively. MRP4 and MRP5 both efflux nucleoside analogs such as anti-HIV drugs and chemotherapeutics. However, both have different sets of inhibitors, and MRP5 has a smaller set of substrates.10,31 There are no definitive rules to designing molecular structures that are not substrates for efflux transporter expulsion which makes preventing efflux transporter recognition very challenging. And, while modulating transporter function has the potential to increase drug concentrations in the brain and enhance neuroprotective effects, increased brain drug levels can also result in significant CNS toxicity.
Altered BBB Function in Neurological Disease
The BBB is traditionally viewed simply as a barrier to treatment, ie, getting drugs into the brain and keeping them there. In truth, the BBB also plays an important role in the pathology and progression of a broad spectrum of CNS disorders. While the BBB maintains CNS homeostasis and preserves ever-important neuronal viability, pathological conditions, ranging for systemic inflammation to neurotrauma, can alter the physically restrictive functions of the BBB to allow drug entry into the brain by the paracellular route (Fig. 2 iv).8,32 These molecules can also include potentially CNS-neurotoxic plasma constituents, which can affect injury progression, cause neuronal injury, and loss of CNS functions.33,34 Disruption in BBB integrity and concomitant increase in BBB permeability can be due to exposure of BBB to pro-inflammatory cytokines TNF-α and IL-1β, which induces expression of matrix metalloproteases (MMPs).35 Upregulation and activation of MMPs degrade endothelium basement membranes, disrupting BBB stability.36 Pro-inflammatory cytokines can induce JAM-A shedding from BCECs,37 although this does not necessarily correlate with a loss in BBB function alone.38 CNS tumors associated with angiogenesis can also disrupt the interaction between astrocytes and BCECs and destabilize the BBB to enhance permeability.39,40
Figure 2.

Schematic depicting various methods for small molecules to cross the BBB. There are four routes for small molecules to cross the BBB and reach the parenchyma. (i) Paracellular diffusion of agents is primarily impacted by size and requires small molecules to pass through TJs between BCECs. (ii) Transcellular free diffusion of small molecules is governed by specific physiochemical parameters that favor diffusion across the phospholipid membrane of BCECs. Parameters to consider include size, lipophilicity, polar surface area, hydrogen bonding potential, and molecular charge. (iii) Carrier-mediated transport is an energy-independent process involving small molecules crossing the BBB along their concentration gradient with the assistance of suitable transporters. Each transporter class (Glut1, LAT1, ENT1, and MCT1) exhibits substrate specificity and saturable transport capacity (see Table 2). (iv) BBB disruption of the BCEC TJs due to pharmacological/physical/pathophysiological mechanisms causes an increase in BBB permeability by permitting otherwise limited paracellular diffusion. Small molecules not capable of diffusing through the phospholipid membrane of BCECs can easily pass from vessel lumen to brain parenchyma through these disrupted TJs.
In Alzheimer’s Disease (AD), the deposition of neurotoxic aggregates of β-amyloid peptide (Aβ) and neurofibrillary tangles of hyperphosphorylated Tau protein are postulated to precipitate loss of neuronal synapses, and ultimately neuronal cell death, and widespread brain damage.41,42 Aβ peptide is transported bidirectionally across the BBB using different transport mechanisms. Blood-to-brain influx is mediated by the receptor for advanced glycation end products (RAGE), whereas low-density lipoprotein receptor-related protein-1 (LRP-1)43 and P-gp mediate brain-to-blood transport.44 In AD, decreased P-gp expression at the BBB is associated with disease progression due to impaired efflux of Aβ.45 In this scenario, P-gp inducers could be beneficial to pump out CNS-derived Aβ to control the toxic effect of Aβ, including altering the permeability of BBB TJ.46 Tau proteins also disrupt the BBB through cytokine release from activated microglia.46,47
Loss of BBB integrity in epilepsy is speculated to be a causal mechanism of this well-characterized CNS disorder; BBB disruption would permit detrimental serum proteins into the brain, altering available nutrients, inducing astrocyte changes, causing increasing neuronal activity, and causing synaptic remodeling culminating in an epileptic episode.48,49 P-gp and MRP pumps are overexpressed, resulting in antiepileptic agents being readily expelled from the brain.50
Parkinson’s disease (PD) is another neurodegenerative disease of the CNS where death of dopaminergic neurons in the midbrain underlies motor symptoms. PD is characterized by decreased TJ protein expression and decreased P-gp functionality, which is a potential route for neurotoxin entry into the brain.48,51 Similar to AD, decreased P-gp efflux function could likely underlie the accumulation of the protein alpha-synuclein into Lewy body inclusions found in neurons of PD patients. l-DOPA (l-3,4-dihydroxyphenylalanine) is a precursor of the neurotransmitter dopamine, which itself does not cross the BBB. Thus, l-DOPA is used as a prodrug for dopamine replacement therapy in PD. As described in a subsequent section, l-DOPA is a small molecule that crosses the BBB via the neutral amino acid transporter where it is converted to dopamine and relieves the dopamine-related symptoms in PD. There are many tractable CNS disease targets to which novel small-molecule drugs can be directed toward. It is also worthwhile to consider that the BBB itself, in its compromised state as seen in many brain diseases, is an underappreciated target for therapeutic “BBB normalization” strategies.
Brain Uptake Mechanisms
Certain physiochemical and molecular properties dramatically affect a drugs entry into the brain. To improve the brain penetration of small molecules, numerous chemistry-and biology-based drug delivery strategies have been developed and explored (Fig. 2). These strategies fall into three main categories: (1) optimizing physicochemical properties to facilitate passive paracellular and transcellular diffusion, (2) utilizing endogenous protein carrier-mediated transport (CMT) mechanisms expressed on the BBB, and (3) employing BBB disruption strategies to allow paracellular drug transport. The subsequent section will explore approaches for small-molecule brain drug delivery across an intact BBB.
Passive diffusion
Most small-molecule CNS drugs reach the brain via passive diffusion across the BBB. As concluded by Lipinski, and supported by experimental and computational Quantitative Structure Activity Relationship (QSAR) approaches, a few key physiochemical parameters require consideration to predict BBB permeability by passive diffusion.52 Lipinski’s “rule of five” relates BBB permeability to molecular weight, lipophilicity, polar surface area, hydrogen bonding, and charge.
Molecular weight
CNS active drugs have significantly lower molecular weight cutoffs compared with other systemic therapeutics. Small molecules with molecular mass under 400 Da to 500 Da undergo significant free diffusion through the BBB.53,54 Albeit, BBB permeability decreases 100-fold going from 200 Da to 450 Da. Space-filling models of the phospholipid bilayer of the plasma membrane reveal that transient pores formed by kinks in their unsaturated fatty acid tails can be filled by small molecules with a limited size range.55,56 This observation aligns with the notion that free diffusion across the BBB is more a function of molecular volume57 than molecular weight per se.
Lipophilicity
For small molecules in particular, lipophilicity, as measured by log P, can be an excellent indicator of BBB permeability. To cross the hydrophobic phospholipid bilayer of a cell membrane by passive diffusion, a molecule must be lipophilic. Hydrophilic substances do not possess the ability to penetrate such membranes. Initial thought was that the higher the log P, the higher the BBB permeability.54 However, given that log P values range for most drugs between −0.05 and 6.0,58 the ideal range for BBB permeability has been found to be 1.5–2.5.53 The classic example of increased BBB permeability by increasing log P is that comparing morphine, codeine, and heroin. As shown in Figure 3A, converting the 3-hydroxy moiety of morphine (log P 0.99) to a methoxy to give codeine, the log P is increased to 1.2. Heroin is produced by acetylation of both the 3- and 6-hydroxy groups of morphine. This results in a log P value of 2.3 for heroin, resulting in a >100 fold higher lipophilicity than morphine. This increased lipophilicity results in 100 times more rat brain uptake for 14C-labeled heroin compared to 14C-morphine using BBB-impermeable 3H-mannitol as a reference control (Fig. 3B).59 While measuring the partition coefficient of a drug between n-octanol and water phases is the long-standing approach to determining log P as an experimental predictor of lipophilicity, there are currently a variety of readily available computational procedures that have been developed for log P prediction.60
Figure 3.
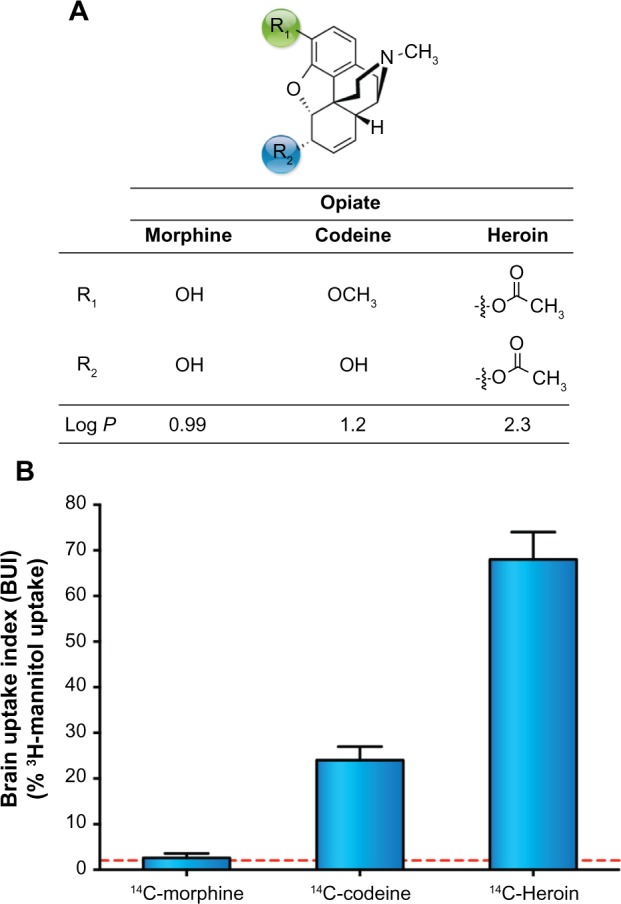
(A) Chemical structures of morphine, codeine, and heroin with their respective log P. (B) Relative rat brain uptake index (BUI) of 14C-morphine, 14C-codeine, and 14C-heroin in rats following a single brain passage after carotid injection. The greater uptake of codeine and heroin relative to morphine can be explained on the basis of their greater lipid solubility (as reflected by log P) relative to morphine. 3H-mannitol was used as a reference ligand for its poor BBB permeability. For each mean and standard deviation, n = 6.59
Polar surface area, charge, hydrogen bonding, and molecular flexibility
Although log P is a reliable indicator of permeability, other chemical properties of small molecules greatly influence BBB permeability. Generally, molecules with a large polar surface area do not readily diffuse through the BBB, with the upper limit estimated between 60 and 90 Å2.58,60 For example, isosteric replacement of an aromatic methine group in the tetracyclic psychoactive mianserin ((±)-2-methyl-1,2,3,4,10,14b-hexahydrodibenzo[c,f]pyrazino[1,2-a]azepine) with an sp2 nitrogen atom in mirtazapine (ie, 6-aza-mianserin) increases the polar surface area and decreases the lipophilicity resulting in a decreased ratio of mirtazapine concentration in the brain relative to its concentration in blood (Fig. 4).61
Figure 4.

(A) 2D Chemical structure and (B) 3D QuteMol space-filling rendering of mianserin (X = C) versus mirtazapine (X = N) with respective physiochemical parameters (C) calculated in plugins/extensions offered in ChemBioDraw Ultra 13.0.2 (Cambridgesoft).
In addition to polar surface area, molecules with significant electrostatic charge will not passively diffuse across the BBB. The net movement of a compound with the same anionic charge as the hydrophobic BCEC membrane is thermodynamically unfavorable, due to mutual repulsion of negative electrostatic charge. Interestingly, lipophilic compounds containing a tertiary nitrogen have been shown to have increased BBB permeation,62 revealing that at physiological pH, a slight positive charge allows passive diffusion.
Predictions about BBB permeability can also be made by examination of the hydrogen bonding capacity of the drug with water. An increasing number of hydrogen bonds (H-bonds) negatively impacts passive diffusion across the BBB, with <5 H-bond donors and <10 H-bond acceptors being acceptable for a CNS drug candidate.60 Drugs with total H-bond acceptors >10 have minimal BBB transport. The greater the drug solvation in water, the more hydrophilic the drug, resulting in less interaction with the lipophilic cell membrane and thus limited drug transport by passive diffusion. The addition of functional groups to mask hydrogen bonding has been explored as a prodrug approach. This is in part exemplified with morphine, codeine, and heroin, where masking the polar hydroxyl moiety (an H-bond donor) of morphine with H-bond acceptor groups such as methoxy (codeine) or acetyl groups (morphine) dramatically increases brain uptake (Fig. 3A).
Molecular flexibility, as quantified by the number of rotatable single bonds not involved in a ring structure or connected to a non-terminal heavy atom, is now a widely used filter for predicting BBB permeability. Compared with other drug classes, CNS drugs have significantly fewer rotatable bonds (five or less) suggesting that the conformational range for BBB permeability is limited.63 An extended conformation could roll up into a spherical and rather bulky shape. In other words, owing to its geometry, a molecule could potentially permeate the BBB to a lesser extent than its molecular weight would indicate. Thus, the number of rotatable bonds could be more reflective of potential conformation changes in molecular shape, ie, an increase in drug bulkiness. More research is needed to establish more general and quantitative measurements of molecular flexibility.
Pharmacokinetic rule
The rule of five is a good starting point for determining a molecule’s ability to cross the BBB by passive diffusion. However, for any drug or candidate to achieve optimum therapeutic efficacy, it must possess a high degree of potency and selectivity for interactions with its molecular target as well as an ability to attain target tissue concentrations that are above a certain threshold. Regardless of the route of drug administration, drug absorption, distribution, metabolism, and excretion (ADME) processes play a pivotal role in defining the bioavailability of the drug for brain uptake, and thus its therapeutic efficacy. Hence, altering the structure of a small molecule to optimize physiochemical properties for enhanced BBB permeability cannot be done without considering the consequences on ADME properties of the drug.
In many cases, increasing the lipophilicity of a drug via the prodrug approach to enhance brain uptake have no dramatic effects on CNS concentrations. As the drug or prodrug enters the blood, it interacts with blood components such as serum proteins and distributes to all tissue compartments in the body. Since the brain receives only 15% of the blood flow on first pass during each cardiac cycle, a drug has equal likelihood of penetrating the lipophilic cell membranes in all other tissue compartments. Hence, plasma concentration of the drug decreases because of enhanced serum protein binding and increased distribution to peripheral tissues. This is illustrated with lipidization of the chemotherapeutic chlorambucil to its more lipophilic prodrug.9 Despite increased brain-to-plasma ratios, chlorambucil prodrugs do not demonstrate superior anticancer activity in disease models when compared with equimolar parent chlorambucil administration, due to increased distribution in peripheral tissues.
Any small molecule that preferentially binds to serum proteins in the blood effectively increases their size and will not cross the BBB.60 Serum proteins have net negative charge, and the 67 kDa human serum albumin (HSA) is by far the most abundant with a reference range of 3.5–5.5 g/dL64 and therefore the most important to consider. HSA has been shown to bind in excess of 70% of all drugs via its two binding domains, warfarin site I, and indole/benzodiazepine site II. Drug binding to HSA is difficult to predict and often only estimated via modeling.65,66 And, while this binding is reversible, it significantly limits the amount of free drug available for transport to the brain, because it exceeds the size limitation for the BBB when bound to HSA. Metabolism of a drug by serum enzymes can further limit free drug concentrations available for brain delivery.
Carrier-mediated transport
Despite being the most common way to enter the brain, passive diffusion is not the only means for small molecules to enter the brain. Endogenous protein-mediated transport processes are potential portals of entry to the brain for circulating drugs. CMT systems facilitate the passive diffusion (ie, energy independent) of small-molecule hexoses, amino acids, nucleoside, vitamins, and hormones, and operate on the order of milliseconds.67,68 In contrast, receptor-mediated transcytosis (RMT) mediates the BBB transport of circulating peptides and plasma proteins, such as transferrin and insulin, via energy-dependent endocytotic mechanisms that occur on the order of minutes.
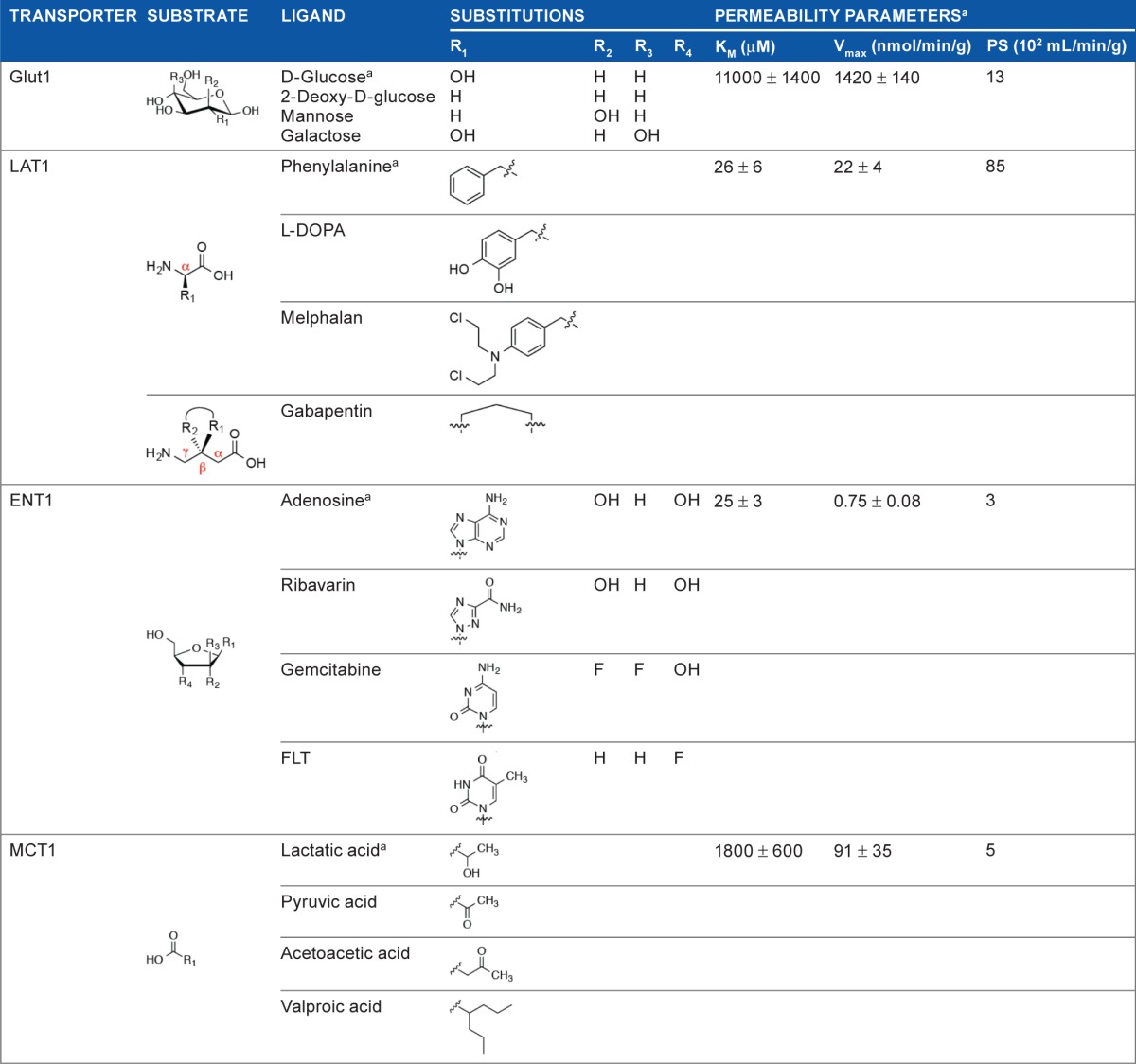
More than 20 carrier-mediated transporter proteins have been identified on the luminal or abluminal side of BCECs. Transmembrane domains of transporter proteins form pores across the membrane bilayer and are characterized by their substrate affinity, selectivity, stereoselectivity, and saturability. Brain permeability of a molecule across a transporter is often described kinetically as the BBB permeability surface (PS) area product, or the Vmax/Km ratio.69–71 Maximal transport capacity is indicated by Vmax, whereas Km is the substrate affinity for the transporter. In general, as substrate affinity to the transporter increases, transport capacity decreases. Estimates of maximal PS are shown in Table 2 for representative substrates of key BBB nutrient transporters.72 Targeting carriers with sufficiently high transport capacity and low affinity has long been an attractive strategy for controlling the delivery and retention of drugs in the brain. To maximize small-molecular delivery by this route, drugs and prodrugs are designed as structural analogs of endogenous substrates for a specific carrier system for efficient binding and transport across the BBB.73
Table 2.
Key BBB transporters that facilitate drug diffusion into the brain. Chemical structures of ligands of pharmacological significance, with permeability parameters indicated for representative substrates.72
|
Note:
representative substrate.
Possibly, the most well-studied transporter on the BBB is the hexose uniporter Glut1, which constitutes more than 90% of all the BBB glucose transporters. Glut1 is highly expressed on the luminal and abluminal sides of BCECs of the BBB (6 × 106 molecules per brain endothelial cell), and selectively transports the sugar nutrients d-glucose, 2-deoxy-d-glucose, mannose, and galactose, but not l-glucose, with their solute gradient.68 Combined with its high expression and high capacity for CMT at the BBB, Glut1 has become an attractive target for prodrug delivery. However, the BBB transport of glucose–drug conjugates is significantly restricted due to prodrug size and loss of Glut1 affinity.74 Limited examples for this approach exist, and they include Glut1-mediated transport of chlorambucil-glucose conjugates across the BBB for treatment of chronic lymphocytic leukemia.75 Further studies have revealed “guidelines” for what prodrugs will retain affinity for Glut1 when attached to glucose: (i) hydrophilic drugs may be conjugated to the hydroxyl group at the C-6 position of d-glucose and be transported by Glut1, (ii) the attached drug must be small and linked through a biodegradable bond, but stable enough to make it to cross the BBB.76 The limitations on prodrug delivery through Glut1 have made drug design difficult, and in the best case it only applies to a small subset of molecules. All of these factors combine to leave the clinical relevance of Glut1 for drug delivery up for debate.
The large neutral amino acid carrier LAT1, expressed both on the luminal and abluminal membrane of BCECs, is another heavily studied transporter system. LAT1 is fairly stereospecific to l-amino acids and can also efficiently shuttle substrates that contain relatively large and lipophilic substituents on the α-carbon (eg, l-phenylalanine).77 The most notable LAT1 substrate of pharmacological significance for the past 30 years is the prodrug l-DOPA for dopamine replacement therapy in PD. Interestingly, while 95% of l-DOPA is decarboxylated to dopamine in the peripheral tissues following systemic administration, the <1% orally dosed l-DOPA brain uptake is sufficient for therapeutic benefit.78 Other examples include α-methyl DOPA for hypertension treatment, and the chemotherapeutic melphalan.58 Even the γ-amino acid anti-epileptic gabapentin can cross the BBB via LAT1 because its cyclic structure mimics that of an α-amino acid. A recent report describing the LAT1-mediated rat brain uptake of ketoprofen, an l-lysine conjugate, anti-inflammatory drug, which itself is impenetrable, further exemplifies the prodrug approach for CNS drug delivery.79
Other carrier systems employed for drug delivery include the equilibrative nucleoside transporter (ENT) 1. This lower capacity transporter mediates the brain uptake of drugs that are both pyrimidine and purine nucleoside analogs, such as the cancer therapeutic gemcitabine and the antiviral agent ribavirin, respectively,80 as well as the cellular proliferation PET imaging agent 18F-FLT (18F-3′-fluoro-3′-deoxythymidine).81 Future drug development must consider purine analogs that are resistant to adenosine deaminase metabolism, which presents a significant enzymatic barrier at the BBB.
Monocarboxylate transporter (MCT) 1 exhibits broad specificity for short-chain monocarboxylates and is responsible for the brain uptake of circulating lactate, short-chain fatty acids (eg, pyruvate), and ketone bodies that can serve as an alternative energy source (eg, acetoacetic acid).82 The anticonvulsant drug, valproic acid, also recently approved by the FDA for treating age-related memory loss, is purported to be taken up by the brain via MCT1.83
Other CMT systems are being explored for small-molecule delivery, as drugs and prodrugs. However, challenges to exploiting this approach include targeting transporters without diminishing affinity, and staying within size limitations. Furthermore, for transporters with lower to mid transporter capacity, potential inhibition of drug uptake by high plasma levels of endogenous substrates needs to be considered. Stability to enzymatic degradation at the BBB interface, as seen with some nucleoside analogs, can be a barrier to achieving therapeutic levels of drug in the brain. Finally, as discussed above, efflux mechanisms for these and other drugs must be considered.
Noninvasive Imaging of Drug Delivery to the Brain
Although a number of analytical methods and in silico techniques have been used to predict the potential for brain drug delivery, many questions remain, such as exactly how much drug can cross the BBB? And, where is it located within the brain? Many methods have been developed for quantifying these parameters,84,85 including radioactive biodistribution and PK studies,53,86 brain microdialysis,87,88 and quantitative autoradiography in small animal models.89 A recent review highlights the use of model organisms, such as the fruit fly (Drosophila melanogaster) and zebrafish (Danio rerio), as a high-throughput screening approach for assessing BBB permeability at reduced cost.90 Although useful in preclinical studies, these are highly invasive or terminal procedures in models that are not necessarily clinically translatable for understanding and quantifying drug delivery to the CNS in humans.
Real-time and noninvasive imaging techniques such as magnetic resonance imaging (MRI), positron emission tomography (PET), and single photon emission computed tomography (SPECT) have achieved significant gains in drug discovery and brain drug targeting, particularly in pharmacokinetic modeling, receptor occupancy studies, and quantification of therapeutic response. MRI exploits the magnetic properties of atomic nuclei to induce and detect radiant energy, typically from irradiated protons in water. The clinical value of MRI depends not only on the strength of signals received, but also on how quickly they relax to their base energy level. The relaxation properties (T1 and T2) of excited nuclei can be influenced by water concentration in soft tissue and by pathological changes in tissue. Ligands conjugated to paramagnetic materials like gadolinium (Gd3+)-chelates (T1 contrast agent) or iron oxides (T2 contrast agent) are often used as MRI contrast agents. This atomic information images with high spatial resolution (sub-millimeter), temporal resolution, and excellent soft tissue contrast. Traditional contrast-enhanced MRI is often used to measure BBB permeability,48 and targeted probes can be used to directly measure brain delivery in some instances.91 While MRI boasts high sub-millimeter spatial resolution due to excellent soft tissue contrast, it can suffer from poor detection sensitivity (10−3 to 10−9 M), which would be a challenge in trying to quantify drug delivery of contrast agents with already limited brain uptake.
The nuclear imaging modalities, PET and SPECT, have exquisite sensitivity which is especially useful for detecting radiolabeled probes in nanomolar to picomolar (10−9 to 10−12 M) concentrations in the brain, for example. PET detects coincident 511 keV gamma rays produced from the annihilation event of an electron with a positron emitted from a radiolabeled PET probe. SPECT relies on directly detecting low-energy gamma rays emitted by the radiopharmaceutical. The challenge with classical short-lived PET isotopes (eg, 11C, t1/2 = 20.3 minutes; 18F, t1/2 = 110 minutes) is the need for an on-site or nearby cyclotron for isotope production and efficient radiochemistry. However, the increasing availability of nonstandard radioisotopes with longer half-lives (eg, 64Cu, t1/2 = 12.7 hours; 89Zr, t1/2 = 3.25 days; 124I, t1/2 = 4.2 days) expands the utility of PET imaging for those ligands that have a protracted pharmacokinetic profile. Many SPECT isotopes are used routinely in the clinic (eg, 99 mTc, t1/2 = 6 hours; 123I, t1/2 = 13.2 hours; 111In, t1/2 = 2.8 days). While both modalities posses’ extremely high sensitivity relative to MRI, PET has two to three orders of magnitude higher sensitivity and faster temporal resolution compared with SPECT.92 Improvements in nuclear imaging technology have brought their spatial resolution to around 4 mm, which can be sufficient for mapping tracer distribution in the brain and can be further improved when co-registered with MRI brain scans for anatomical or functional information.93
A wide variety of CNS-targeted imaging probes for SPECT or PET are available for preclinical and clinical assessments of BBB permeability, neurotransmitter receptor, and transporter targeting as diagnostic biomarkers of neuropathophysiology.93,94 These imaging modalities have also proven to be a tremendous tool in examining the kinetics of small-molecule brain uptake. 11C-Verapamil, a P-gP substrate, has been used for PET imaging of human response to P-gP inhibition with cyclosporine.95 18F-florbetapir, which freely diffuses across the BBB, is a recently FDA-approved PET imaging agent that binds Aβ-plaques and can be used as a noninvasive predicative biomarker of plaque burden in AD in various regions of the brain.47 With no effective therapeutic approved for AD, Aβ-plaque-targeted PET agents are being used in hopes of measuring changes in plaque burden in response to therapy. As shown in Figure 5, most MRI and PET imaging studies in brain tumor patients is limited to detecting anatomic features in addition to a small subset of functional and biological processes, such as tumor enhancement/BBB permeability (Fig. 5A), glucose metabolism (Fig. 5B), and amino acid metabolism (Fig. 5C, 5D). In a similar vein, molecularly targeted small-molecule ligands, developed as diagnostics or therapeutics, can be radiolabeled and administered in vivo to confirm and quantify the specificity of brain uptake values in targeted regions of the brain. Should a radioligand to a specific molecular target in the brain be available, nonlabeled ligands can be administered in vivo to study how well the drug can displace radioligand binding. A recent description of this approach is for the evaluation of a second-generation antipsychotic drug Lurasidone in non-human primates.96 PET imaging pre- and post-drug administration confirms drug occupancy of D2 and 5-HT2 A receptors with 11C-raclopride and 11C-R-(+)-α-(2,3-dimethoxyphenyl)-1-[2-(4-fluorophenylethyl)]-4-piperidine-methanol, respectively.
Figure 5.
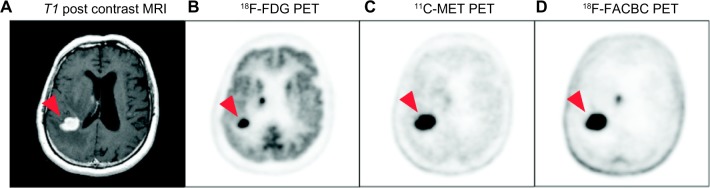
MRI and PET brain scans in a patient with primary CNS lymphoma (red arrowhead). (A) Postcontrast MRI shows tumor enhancement indicative of BBB disruption. (B) 18F-FDG shows high glucose metabolism throughout the gray matter with focally increased uptake in sites of disease. (C) The natural amino acid 11C-MET also shows the high amino acid metabolism in the disease by PET. (D) 18F-FACBC is a synthetic L-amino acid analog showing high amino acid metabolism in the disease and very low background utilization in normal brain parenchyma as compared to 11C-MET. Images courtesy of Department of Radiology, Memorial Sloan-Kettering Cancer Center. Figure adapted from Chacko et al.1
Abbreviations: FACBC: 1-Amino-3-fluorocyclobutane-1-carboxylic acid; FDG: fluorodeoxyglucose; MET: L-methionine.
On combining complementary imaging modalities into one system, as with the recently developed PET/MRI systems, MRI-derived measures of both anatomy and function correlated with SPECT or PET-derived quantitation of disease-specific molecular processes can inform different phases of the drug discovery process. This includes validating the mechanism and specificity of drug action in various animal models, quantifying neuropharmacokinetics and neuropharmacodynamics across species, screening and selecting subjects for clinical trials, and monitoring the clinical response to drug treatment via predictive biomarker imaging. In the era of personalized medicine and image-guided therapy, it is clear that as a translational tool, real-time noninvasive imaging technologies can facilitate and perhaps shorten the time between CNS drug discovery and clinical approval.
Conclusions
Our understanding of the BBB has advanced considerably in recent years, and now the BBB is appreciated as not just a physical cellular barrier, but an actively regulated regulatory interface, with transport, secretory, and enzymatic activities. This review has described how small molecules cross the BBB, what properties affect diffusion and how natural active transport process can be harnessed, all of which permit development of more permeable and effective small molecules. However, the BBB has been proven to be difficult for scientists and clinicians to circumvent, as shown by the limited treatment options and poor outcomes of individuals suffering from CNS disorders. Ultimately, the overall goal of future CNS research needs to expand beyond the current drug formulary of mostly small, lipophilic molecules. Exploiting pathways to deliver small molecules via protein-facilitated routes or small molecule-antibody conjugates for RMT will allow BBB permeation of a greater range of pharmaceuticals. Nanocarrier systems are another exciting route for systemic brain delivery owing to their high drug loading capacity, and ability to load diverse agents, from small hydrophilic drugs to larger gene vectors, enzymes, and peptides.97,98 This latter approach may allow for the repurposing of many CNS-active agents that could not pass through initial ADME testing. Alternatives to focusing on the drug delivery system are to modulate the permeability of the BBB transiently to enhance paracellular delivery of small and large materials into the brain or to evaluate alternative routes of drug administration. Intracarotid, intranasal, intraretinal, and intracerebral delivery of drugs are all explored with interesting results. With a cross-disciplinary approach, integrating biologists, medicinal chemists, molecular modelers, pharmaceutical scientists, bioengineers, and imaging scientists, the field can move toward a more informed and rational approach to optimizing drug design for delivery to the CNS.
Footnotes
Author Contributions
Wrote the first draft of the manuscript: JM, AMC. Contributed to the writing of the manuscript: JM, AMC. Agree with manuscript results and conclusions: JM, AMC. Jointly developed the structure and arguments for the paper: JM, AMC. Made critical revisions and approved final version: AMC. All authors reviewed and approved of the final manuscript.
ACADEMIC EDITOR: Yitzhak Tor, Editor in Chief
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
FUNDING: The authors are supported by the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant KL2TR000139 (AMC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
This paper was subject to independent, expert peer review by a minimum of two blind peer reviewers. All editorial decisions were made by the independent academic editor. All authors have provided signed confirmation of their compliance with ethical and legal obligations including (but not limited to) use of any copyrighted material, compliance with ICMJE authorship and competing interests disclosure guidelines and, where applicable, compliance with legal and ethical guidelines on human and animal research participants. Provenance: the authors were invited to submit this paper.
REFERENCES
- 1.Chacko AM, Li C, Pryma DA, Brem S, Coukos G, Muzykantov V. Targeted delivery of antibody-based therapeutic and imaging agents to CNS tumors: crossing the blood-brain barrier divide. Expert Opin Drug Deliv. 2013;10(7):907–26. doi: 10.1517/17425247.2013.808184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WHO . Neurological Disorders: Public Health Challenges. Geneva, Switzerland: World Health Organization, WHO Press; 2006. [Google Scholar]
- 3.Funk M, Drew N, Freeman M, Faydi E. Mental Health and Development: Targeting People with Mental Health Conditions as A Vulnerable Group. Geneva, Switzerland: World Health Organization; 2010. [Google Scholar]
- 4.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37(1):13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 5.Furuse M, Itoh M, Hirase T, et al. Direct association of occludin with ZO-1 and its possible involvement in the localization of occludin at tight junctions. J Cell Biol. 1994;127(6 pt 1):1617–26. doi: 10.1083/jcb.127.6.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fanning AS, Anderson JM. Protein-protein interactions: PDZ domain networks. Curr Biol. 1996;6(11):1385–8. doi: 10.1016/s0960-9822(96)00737-3. [DOI] [PubMed] [Google Scholar]
- 7.Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16(1):1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 8.Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol. 2006;1(3):223–36. doi: 10.1007/s11481-006-9025-3. [DOI] [PubMed] [Google Scholar]
- 9.Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7(1):41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 10.Dallas S, Miller DS, Bendayan R. Multidrug resistance-associated proteins: expression and function in the central nervous system. Pharmacol Rev. 2006;58(2):140–61. doi: 10.1124/pr.58.2.3. [DOI] [PubMed] [Google Scholar]
- 11.Roux F, Durieu-Trautmann O, Chaverot N, et al. Regulation of gamma-glutamyl transpeptidase and alkaline phosphatase activities in immortalized rat brain microvessel endothelial cells. J Cell Physiol. 1994;159(1):101–13. doi: 10.1002/jcp.1041590114. [DOI] [PubMed] [Google Scholar]
- 12.Meyer J, Rauh J, Galla HJ. The susceptibility of cerebral endothelial cells to astroglial induction of blood-brain barrier enzymes depends on their prolifera-tive state. J Neurochem. 1991;57(6):1971–7. doi: 10.1111/j.1471-4159.1991.tb06411.x. [DOI] [PubMed] [Google Scholar]
- 13.Ghersi-Egea JF, Perrin R, Leininger-Muller B, et al. Subcellular localization of cytochrome P450, and activities of several enzymes responsible for drug metabolism in the human brain. Biochem Pharmacol. 1993;45(3):647–58. doi: 10.1016/0006-2952(93)90139-n. [DOI] [PubMed] [Google Scholar]
- 14.Yee RE, Huang SC, Stout DB, et al. Nigrostriatal reduction of aromatic l-amino acid decarboxylase activity in MPTP-treated squirrel monkeys: in vivo and in vitro investigations. J Neurochem. 2000;74(3):1147–57. doi: 10.1046/j.1471-4159.2000.741147.x. [DOI] [PubMed] [Google Scholar]
- 15.Bauer B, Hartz AM, Lucking JR, Yang X, Pollack GM, Miller DS. Coordinated nuclear receptor regulation of the efflux transporter, Mrp2, and the phase-II metabolizing enzyme, GSTpi, at the blood-brain barrier. J Cereb Blood Flow Metab. 2008;28(6):1222–34. doi: 10.1038/jcbfm.2008.16. [DOI] [PubMed] [Google Scholar]
- 16.Ghersi-Egea JF, Minn A, Siest G. A new aspect of the protective functions of the blood-brain barrier: activities of four drug-metabolizing enzymes in isolated rat brain microvessels. Life Sci. 1988;42(24):2515–23. doi: 10.1016/0024-3205(88)90351-7. [DOI] [PubMed] [Google Scholar]
- 17.Miksys S, Tyndale RF. Cytochrome P450-mediated drug metabolism in the brain. J Psychiatry Neurosci. 2013;38(3):152–63. doi: 10.1503/jpn.120133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci. 2005;6(8):591–602. doi: 10.1038/nrn1728. [DOI] [PubMed] [Google Scholar]
- 19.Loscher W, Potschka H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx. 2005;2(1):86–98. doi: 10.1602/neurorx.2.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eyal S, Hsiao P, Unadkat JD. Drug interactions at the blood-brain barrier: fact or fantasy? Pharmacol Ther. 2009;123(1):80–104. doi: 10.1016/j.pharmthera.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bendayan R, Ronaldson PT, Gingras D, Bendayan M. J Histochem Cytochem; In situ localization of P-glycoprotein (ABCB1) in human and rat brain. ; 2006. pp. 1159–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schinkel AH, Smit JJ, van Tellingen O, et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 1994;77(4):491–502. doi: 10.1016/0092-8674(94)90212-7. [DOI] [PubMed] [Google Scholar]
- 23.O’Brien FE, O’Connor RM, Clarke G, Dinan TG, Griffin BT, Cryan JF. P-glycoprotein inhibition increases the brain distribution and antidepressant-like activity of escitalopram in rodents. Neuropsychopharmacology. 2013;38(11):2209–19. doi: 10.1038/npp.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Brien FE, O’Connor RM, Clarke G, et al. The P-glycoprotein inhibitor cyclosporin A differentially influences behavioural and neurochemical responses to the antidepressant escitalopram. Behav Brain Res. 2014;261:17–25. doi: 10.1016/j.bbr.2013.11.027. [DOI] [PubMed] [Google Scholar]
- 25.Hawkins BT, Sykes DB, Miller DS. Rapid, reversible modulation of blood-brain barrier P-glycoprotein transport activity by vascular endothelial growth factor. J Neurosci. 2010;30(4):1417–25. doi: 10.1523/JNEUROSCI.5103-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ronaldson PT, Bendayan R. HIV-1 viral envelope glycoprotein gp120 triggers an inflammatory response in cultured rat astrocytes and regulates the functional expression of P-glycoprotein. Mol Pharmacol. 2006;70(3):1087–98. doi: 10.1124/mol.106.025973. [DOI] [PubMed] [Google Scholar]
- 27.Endicott JA, Ling V. The biochemistry of P-glycoprotein-mediated multidrug resistance. Annu Rev Biochem. 1989;58:137–71. doi: 10.1146/annurev.bi.58.070189.001033. [DOI] [PubMed] [Google Scholar]
- 28.Morrow CS, Cowan KH, Frei E., III . Drug resistance and its clinical circumvention. In: Holland JF, Bast RC, Morton DL, Kufe DW, Weichselbaum RR, editors. Cancer Medicine. 4th ed. Baltimore, MD: The Williams & Wilkins Co.; 1997. pp. 799–815. [Google Scholar]
- 29.Nicolazzo JA, Katneni K. Drug transport across the blood-brain barrier and the impact of breast cancer resistance protein (ABCG2) Curr Top Med Chem. 2009;9(2):130–47. doi: 10.2174/156802609787521580. [DOI] [PubMed] [Google Scholar]
- 30.Chen Y, Agarwal S, Shaik NM, Chen C, Yang Z, Elmquist WF. P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. J Pharmacol Exp Ther. 2009;330(3):956–63. doi: 10.1124/jpet.109.154781. [DOI] [PubMed] [Google Scholar]
- 31.Tian Q, Zhang J, Tan TM, et al. Human multidrug resistance associated protein 4 confers resistance to camptothecins. Pharm Res. 2005;22(11):1837–53. doi: 10.1007/s11095-005-7595-z. [DOI] [PubMed] [Google Scholar]
- 32.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57(2):178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 33.Nordborg C, Sokrab TE, Johansson BB. The relationship between plasma protein extravasation and remote tissue changes after experimental brain infarction. Acta Neuropathol. 1991;82(2):118–26. doi: 10.1007/BF00293954. [DOI] [PubMed] [Google Scholar]
- 34.Wagner KR, Packard BA, Hall CL, et al. Protein oxidation and heme oxygenase-1 induction in porcine white matter following intracerebral infusions of whole blood or plasma. Dev Neurosci. 2002;24:2–3. 154–60. doi: 10.1159/000065703. [DOI] [PubMed] [Google Scholar]
- 35.Ravindran J, Agrawal M, Gupta N, Rao PV. Alteration of blood brain barrier permeability by T-2 toxin: role of MMP-9 and inflammatory cytokines. Toxicology. 2011;280:1–2. 44–52. doi: 10.1016/j.tox.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 36.Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998;29(10):2189–95. doi: 10.1161/01.str.29.10.2189. [DOI] [PubMed] [Google Scholar]
- 37.Yeung D, Manias JL, Stewart DJ, Nag S. Decreased junctional adhesion molecule-A expression during blood-brain barrier breakdown. Acta Neuropathol. 2008;115(6):635–42. doi: 10.1007/s00401-008-0364-4. [DOI] [PubMed] [Google Scholar]
- 38.Haarmann A, Deiss A, Prochaska J, et al. Evaluation of soluble junctional adhesion molecule-A as a biomarker of human brain endothelial barrier breakdown. PLoS One. 2010;5(10):e13568. doi: 10.1371/journal.pone.0013568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eichler AF, Chung E, Kodack DP, Loeffler JS, Fukumura D, Jain RK. The biology of brain metastases-translation to new therapies. Nat Rev Clin Oncol. 2011;8(6):344–56. doi: 10.1038/nrclinonc.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang RD, Price JE, Fujimaki T, Bucana CD, Fidler IJ. Differential permeability of the blood-brain barrier in experimental brain metastases produced by human neoplasms implanted into nude mice. Am J Pathol. 1992;141(5):1115–24. [PMC free article] [PubMed] [Google Scholar]
- 41.Mudher A, Lovestone S. Alzheimer’s disease-do tauists and baptists finally shake hands? Trends Neurosci. 2002;25(1):22–6. doi: 10.1016/s0166-2236(00)02031-2. [DOI] [PubMed] [Google Scholar]
- 42.Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology. 2004;62(11):1984–9. doi: 10.1212/01.wnl.0000129697.01779.0a. [DOI] [PubMed] [Google Scholar]
- 43.Deane R, Wu Z, Zlokovic BV. RAGE (yin) versus LRP (yang) balance regulates Alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke. 2004;35(11 suppl 1):2628–31. doi: 10.1161/01.STR.0000143452.85382.d1. [DOI] [PubMed] [Google Scholar]
- 44.Vogelgesang S, Jedlitschky G, Brenn A, Walker LC. The role of the ATP-binding cassette transporter P-glycoprotein in the transport of beta-amyloid across the blood-brain barrier. Curr Pharm Des. 2011;17(26):2778–86. doi: 10.2174/138161211797440168. [DOI] [PubMed] [Google Scholar]
- 45.Cirrito JR, Deane R, Fagan AM, et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest. 2005;115(11):3285–90. doi: 10.1172/JCI25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kovac A, Zilkova M, Deli MA, Zilka N, Novak M. Human truncated tau is using a different mechanism from amyloid-beta to damage the blood-brain barrier. J Alzheimers Dis. 2009;18(4):897–906. doi: 10.3233/JAD-2009-1197. [DOI] [PubMed] [Google Scholar]
- 47.Clark CM, Schneider JA, Bedell BJ, et al. Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA. 2011;305(3):275–83. doi: 10.1001/jama.2010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deo AK, Theil FP, Nicolas JM. Confounding parameters in preclinical assessment of blood-brain barrier permeation: an overview with emphasis on species differences and effect of disease states. Mol Pharm. 2013;10(5):1581–95. doi: 10.1021/mp300570z. [DOI] [PubMed] [Google Scholar]
- 49.Friedman A. Blood-brain barrier dysfunction, status epilepticus, seizures, and epilepsy: a puzzle of a chicken and egg? Epilepsia. 2011;52(suppl 8):19–20. doi: 10.1111/j.1528-1167.2011.03227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ghosh C, Gonzalez-Martinez J, Hossain M, et al. Pattern of P450 expression at the human blood-brain barrier: roles of epileptic condition and laminar flow. Epilepsia. 2010;51(8):1408–17. doi: 10.1111/j.1528-1167.2009.02428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vautier S, Fernandez C. ABCB1: the role in Parkinson’s disease and phar-macokinetics of antiparkinsonian drugs. Expert Opin Drug Metab Toxicol. 2009;5(11):1349–58. doi: 10.1517/17425250903193079. [DOI] [PubMed] [Google Scholar]
- 52.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:1–3. 3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 53.Misra A, Ganesh S, Shahiwala A, Shah SP. Drug delivery to the central nervous system: a review. J Pharm Pharm Sci. 2003;6(2):252–73. [PubMed] [Google Scholar]
- 54.Levin VA, Patlak CS, Landahl HD. Heuristic modeling of drug delivery to malignant brain tumors. J Pharmacokinet Biopharm. 1980;8(3):257–96. doi: 10.1007/BF01059646. [DOI] [PubMed] [Google Scholar]
- 55.Trauble H. The movement of molecules across lipid membranes: a molecular theory. J Membr Biol. 1971;4(1):193–208. doi: 10.1007/BF02431971. [DOI] [PubMed] [Google Scholar]
- 56.Seelig A, Seelig J. The dynamic structure of fatty acyl chains in a phos-pholipid bilayer measured by deuterium magnetic resonance. Biochemistry. 1974;13(23):4839–45. doi: 10.1021/bi00720a024. [DOI] [PubMed] [Google Scholar]
- 57.Durchschlag H, Zipper P. Calculation of the partial volume of organic compounds and polymers. Ultracentrifugation. 1994;94:20–39. [Google Scholar]
- 58.Begley DJ. Delivery of therapeutic agents to the central nervous system: the problems and the possibilities. Pharmacol Ther. 2004;104(1):29–45. doi: 10.1016/j.pharmthera.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 59.Oldendorf WH, Hyman S, Braun L, Oldendorf SZ. Blood-brain barrier: penetration of morphine, codeine, heroin, and methadone after carotid injection. Science. 1972;178(4064):984–6. doi: 10.1126/science.178.4064.984. [DOI] [PubMed] [Google Scholar]
- 60.Pajouhesh H, Lenz GR. Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005;2(4):541–53. doi: 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kelder J, Grootenhuis PD, Bayada DM, Delbressine LP, Ploemen JP. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm Res. 1999;16(10):1514–9. doi: 10.1023/a:1015040217741. [DOI] [PubMed] [Google Scholar]
- 62.Fischer H, Gottschlich R, Seelig A. Blood-brain barrier permeation: molecular parameters governing passive diffusion. J Membr Biol. 1998;165(3):201–11. doi: 10.1007/s002329900434. [DOI] [PubMed] [Google Scholar]
- 63.Leeson PD, Davis AM. Time-related differences in the physical property profiles of oral drugs. J Med Chem. 2004;47(25):6338–48. doi: 10.1021/jm049717d. [DOI] [PubMed] [Google Scholar]
- 64.Kratz A, Ferraro M, Sluss PM, Lewandrowski KB. Case records of the Mas-sachusetts General Hospital. Weekly clinicopathological exercises. Laboratory reference values. N Engl J Med. 2004;351(15):1548–63. doi: 10.1056/NEJMcpc049016. [DOI] [PubMed] [Google Scholar]
- 65.Kratochwil NA, Huber W, Muller F, Kansy M, Gerber PR. Predicting plasma protein binding of drugs: a new approach. Biochem Pharmacol. 2002;64(9):1355–74. doi: 10.1016/s0006-2952(02)01074-2. [DOI] [PubMed] [Google Scholar]
- 66.Fehske KJ, Muller WE, Wollert U. The location of drug binding sites in human serum albumin. Biochem Pharmacol. 1981;30(7):687–92. doi: 10.1016/0006-2952(81)90151-9. [DOI] [PubMed] [Google Scholar]
- 67.Pardridge WM. Transport of small molecules through the blood-brain barrier: biology and methodology. Adv Drug Deliv Rev. 1995;15:1–3. 5–36. [PubMed] [Google Scholar]
- 68.Tsuji A, Tamai II. Carrier-mediated or specialized transport of drugs across the blood-brain barrier. Adv Drug Deliv Rev. 1999;36:2–3. 277–90. doi: 10.1016/s0169-409x(98)00084-2. [DOI] [PubMed] [Google Scholar]
- 69.Pardridge WM, Oldendorf WH. Kinetics of blood-brain transport of hexoses. Biochim Biophys Acta. 1975;382(3):377–92. doi: 10.1016/0005-2736(75)90279-5. [DOI] [PubMed] [Google Scholar]
- 70.Pardridge WM, Oldendorf WH. Kinetic analysis of blood-brain barrier transport of amino acids. Biochim Biophys Acta. 1975;401(1):128–36. doi: 10.1016/0005-2736(75)90347-8. [DOI] [PubMed] [Google Scholar]
- 71.Pardridge WM. Blood-brain barrier carrier-mediated transport and brain metabolism of amino acids. Neurochem Res. 1998;23(5):635–44. doi: 10.1023/a:1022482604276. [DOI] [PubMed] [Google Scholar]
- 72.Pardridge WM. Peptide Drug Delivery to the Brain. New York, NY: Raven Press; 2001. [Google Scholar]
- 73.Yang C, Tirucherai GS, Mitra AK. Prodrug based optimal drug delivery via membrane transporter/receptor. Expert Opin Biol Ther. 2001;1(2):159–75. doi: 10.1517/14712598.1.2.159. [DOI] [PubMed] [Google Scholar]
- 74.Pardridge WM. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32(11):1959–72. doi: 10.1038/jcbfm.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Halmos T, Santarromana M, Antonakis K, Scherman D. Synthesis of glucose-chlorambucil derivatives and their recognition by the human GLUT1 glucose transporter. Eur J Pharmacol. 1996;318:2–3. 477–84. doi: 10.1016/s0014-2999(96)00796-0. [DOI] [PubMed] [Google Scholar]
- 76.Gynther M, Ropponen J, Laine K, et al. Glucose promoiety enables glucose transporter mediated brain uptake of ketoprofen and indomethacin prodrugs in rats. J Med Chem. 2009;52(10):3348–53. doi: 10.1021/jm8015409. [DOI] [PubMed] [Google Scholar]
- 77.Smith QR, Momma S, Aoyagi M, Rapoport SI. Kinetics of neutral amino acid transport across the blood-brain barrier. J Neurochem. 1987;49(5):1651–8. doi: 10.1111/j.1471-4159.1987.tb01039.x. [DOI] [PubMed] [Google Scholar]
- 78.Vangilder RL, Rosen CL, Barr TL, Huber JD. Targeting the neurovascular unit for treatment of neurological disorders. Pharmacol Ther. 2011;130(3):239–47. doi: 10.1016/j.pharmthera.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gynther M, Jalkanen A, Lehtonen M, et al. Brain uptake of ketoprofen-lysine prodrug in rats. Int J Pharm. 2010;399:1–2. 121–8. doi: 10.1016/j.ijpharm.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 80.Parkinson FE, Damaraju VL, Graham K, et al. Molecular biology of nucleoside transporters and their distributions and functions in the brain. Curr Top Med Chem. 2011;11(8):948–72. doi: 10.2174/156802611795347582. [DOI] [PubMed] [Google Scholar]
- 81.Paproski RJ, Wuest M, Jans HS, et al. Biodistribution and uptake of 3’-deoxy-3’-fluorothymidine in ENT1-knockout mice and in an ENT1-knockdown tumor model. J Nucl Med. 2010;51(9):1447–55. doi: 10.2967/jnumed.110.076356. [DOI] [PubMed] [Google Scholar]
- 82.Oldendorf WH. Carrier-mediated blood-brain barrier transport of short-chain monocarboxylic organic acids. Am J Physiol. 1973;224(6):1450–3. doi: 10.1152/ajplegacy.1973.224.6.1450. [DOI] [PubMed] [Google Scholar]
- 83.Fischer W, Praetor K, Metzner L, Neubert RH, Brandsch M. Transport of val-proate at intestinal epithelial (Caco-2) and brain endothelial (RBE4) cells: mechanism and substrate specificity. Eur J Pharm Biopharm. 2008;70(2):486–92. doi: 10.1016/j.ejpb.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 84.Nag S, editor. The Blood-Brain Barrier: Biology and Research Protocols. Totowa, NJ: Humana Press; 2003. Part III. Blood-brain barrier transport techniques; pp. 193–304. [Google Scholar]
- 85.Pardridge WM, editor. Introduction to the Blood-Brain Barrier. Cambridge: Cambridge University Press; 1998. [Google Scholar]
- 86.Pardridge WM, Connor JD, Crawford IL. Permeability changes in the blood-brain barrier: causes and consequences. CRC Crit Rev Toxicol. 1975;3(2):159–99. doi: 10.3109/10408447509079857. [DOI] [PubMed] [Google Scholar]
- 87.de Lange EC, Danhof M, de Boer AG, Breimer DD. Methodological considerations of intracerebral microdialysis in pharmacokinetic studies on drug transport across the blood-brain barrier. Brain Res Brain Res Rev. 1997;25(1):27–49. doi: 10.1016/s0165-0173(97)00014-3. [DOI] [PubMed] [Google Scholar]
- 88.Tao R, Hjorth S. Differences in the in vitro and in vivo 5-hydroxytryptamine extraction performance among three common microdialysis membranes. J Neurochem. 1992;59(5):1778–85. doi: 10.1111/j.1471-4159.1992.tb11010.x. [DOI] [PubMed] [Google Scholar]
- 89.Benveniste H, Drejer J, Schousboe A, Diemer NH. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem. 1984;43(5):1369–74. doi: 10.1111/j.1471-4159.1984.tb05396.x. [DOI] [PubMed] [Google Scholar]
- 90.Geldenhuys WJ, Allen DD, Bloomquist JR. Novel models for assessing blood-brain barrier drug permeation. Expert Opin Drug Metab Toxicol. 2012;8(6):647–53. doi: 10.1517/17425255.2012.677433. [DOI] [PubMed] [Google Scholar]
- 91.Liu HL, Hua MY, Yang HW, et al. Magnetic resonance monitoring of focused ultrasound/magnetic nanoparticle targeting delivery of therapeutic agents to the brain. Proc Natl Acad Sci USA. 2010;107(34):15205–10. doi: 10.1073/pnas.1003388107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rahmim A, Zaidi H. PET versus SPECT: strengths, limitations and challenges. Nucl Med Commun. 2008;29(3):193–207. doi: 10.1097/MNM.0b013e3282f3a515. [DOI] [PubMed] [Google Scholar]
- 93.Lee CM, Farde L. Using positron emission tomography to facilitate CNS drug development. Trends Pharmacol Sci. 2006;27(6):310–6. doi: 10.1016/j.tips.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 94.Kung HF, Kung MP, Choi SR. Radiopharmaceuticals for single-photon emission computed tomography brain imaging. Semin Nucl Med. 2003;33(1):2–13. doi: 10.1053/snuc.2003.127296. [DOI] [PubMed] [Google Scholar]
- 95.Sasongko L, Link JM, Muzi M, et al. Imaging P-glycoprotein transport activity at the human blood-brain barrier with positron emission tomography. Clin Pharmacol Ther. 2005;77(6):503–14. doi: 10.1016/j.clpt.2005.01.022. [DOI] [PubMed] [Google Scholar]
- 96.Nakazawa S, Yokoyama C, Nishimura N, et al. Evaluation of dopamine D2/D3 and serotonin 5-HT2A receptor occupancy for a novel antipsychotic, lurasidone, in conscious common marmosets using small-animal positron emission tomography. Psychopharmacology (Berl) 2013;225(2):329–39. doi: 10.1007/s00213-012-2815-9. [DOI] [PubMed] [Google Scholar]
- 97.Chen Y, Liu L. Modern methods for delivery of drugs across the blood-brain barrier. Adv Drug Deliv Rev. 2012;64(7):640–65. doi: 10.1016/j.addr.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 98.Alyautdin R, Khalin I, Nafeeza MI, Haron MH, Kuznetsov D. Nano-scale drug delivery systems and the blood-brain barrier. Int J Nanomedicine. 2014;9:795–811. doi: 10.2147/IJN.S52236. [DOI] [PMC free article] [PubMed] [Google Scholar]