

Abstract
Background
The explosion in biological information creates the need for databases that are easy to develop, easy to maintain and can be easily manipulated by annotators who are most likely to be biologists. However, deployment of scalable and extensible databases is not an easy task and generally requires substantial expertise in database development.
Results
BioBuilder is a Zope-based software tool that was developed to facilitate intuitive creation of protein databases. Protein data can be entered and annotated through web forms along with the flexibility to add customized annotation features to protein entries. A built-in review system permits a global team of scientists to coordinate their annotation efforts. We have already used BioBuilder to develop Human Protein Reference Database http://www.hprd.org, a comprehensive annotated repository of the human proteome. The data can be exported in the extensible markup language (XML) format, which is rapidly becoming as the standard format for data exchange.
Conclusions
As the proteomic data for several organisms begins to accumulate, BioBuilder will prove to be an invaluable platform for functional annotation and development of customizable protein centric databases. BioBuilder is open source and is available under the terms of LGPL.
Background
To manage the large amount of proteomic data being generated by the scientific community, a comprehensive protein database solution is necessary, in particular to store the proteomic data together with all other relevant annotations. Designing a protein database, which can store highly complex and heterogeneous information and provide the end users with key features such as data interoperability, portability and a user-friendly query system, is a challenging prospect. We have developed BioBuilder as a platform that allows biologists to create custom databases rapidly for dealing with manually curated data or those derived from high-throughput experiments. The data that is entered can be visualized, retrieved and edited through a web browser without practically any technical intervention from the biologist. Such data can be made available through XML-RPC web service or through proteomics standards initiative molecular interaction (PSI-MI) format, the recently standardized format for protein-protein interaction data [1]. This facilitates the synchronization of information with other databases or applications in real-time. Significantly, BioBuilder was successfully used by us recently to develop a novel database of human proteins, Human Protein Reference Database (HPRD) [2,3].
Implementation
BioBuilder was built as a component on top of Zope, an open source web application server written in Python [4,5] (Figure 1). In BioBuilder, every protein annotated by the user is an instance of a 'protein' object. BioBuilder provides an object constructor for 'protein' object and 'interaction' object – the two main units in BioBuilder. These units are themselves composed of sub-objects that represent features such as protein domain architecture, expression, molecular function, biological process, cellular component and post-translational modifications. The object tree is stored in a true object oriented database, Zope Object Data Base (ZODB).
Figure 1.
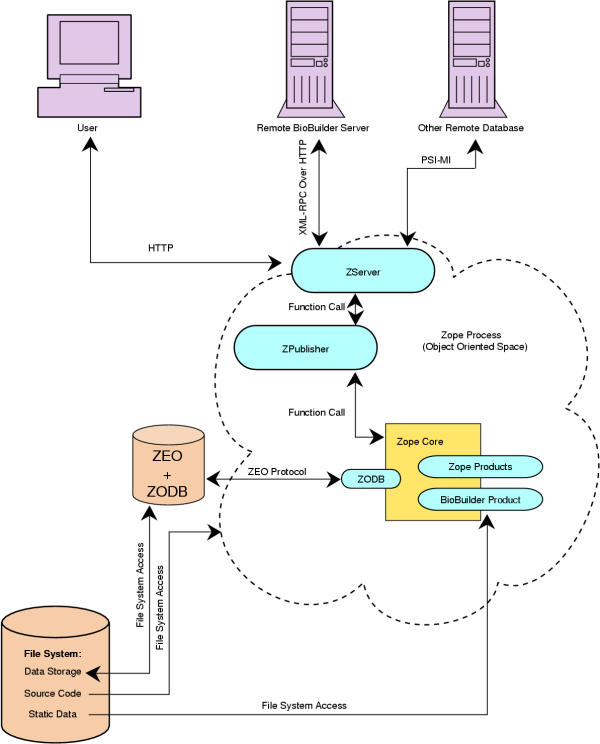
A schematic showing BioBuilder in the context of Zope environment. When a client or any other remote database calls Zope, the ZServer processes the request and sends it to ZPublisher. The ZPublisher acts as an object request broker, finding the requested object, and delivering the objects back to the ZServer in their requested form. BioBuilder was built on top of the Zope core application code. Part of the application code is stored in the file system and other parts are in object database (ZODB).
ZODB allows replaceable storage back-ends. The most popular storage back-ends are FileStorage, which stores all data in a single file, and BerkeleyStorage, which uses Sleepy Cat Software's Berkeley DB database. The FileStorage backend is append-only: changes are always written to the end of the file, nothing is ever overwritten, leaving no chance for data corruption at the database level. Several other third-party storages are also available, such as DirectoryStorage, which stores each object as a separate file. It is possible to write a storage back-end that stores in a relational database such as MySQL or Oracle, but no mature product is currently available.
Although purely SQL systems are in widespread use, an object oriented database simplifies the design of a protein database since proteins and their interactions naturally fit the concept of object than a relational table [6]. Having a protein in an object database allows flexible redesign of the schema, even during the implementation phase. This feature makes BioBuilder a highly flexible and scalable platform.
The objects forming the protein definition can each have their own attributes. For example, the amino acid residue in a protein that undergoes a phosphorylation event can be specified. The attributes in most of the cases can be simple variables belonging to an object (e.g. tyrosine at position 77 is phosphorylated), references to other internal objects (e.g. tyrosine at position 77 is phosphorylated by Fyn kinase) or references to external resources (e.g. PubMed). In an object database, cross-referencing between attributes or even instances is quite straightforward. Adding an attribute to any class (and thus reflecting this attribute for all the existing objects) just needs an addition of this attribute listing its name and type in the properties section of that particular class.
Retrieval of information
BioBuilder incorporates an automated mechanism for cataloging every data structure stored in the object database into ZCatalog [7]. ZCatalog is a flexible indexing system used with ZODB that maintains multiple indexes of full-text, field, date or path types and makes every data structure searchable via a query interface. The search scripts use high-level methods provided by ZCatalog instead of querying the object database directly. The ZCatalog methods are optimized to handle a large dataset by using relatively less memory. ZCatalog maintains a vocabulary of words used in the database and maintains forward and reverse indexes linking the vocabulary to specific fields in the database. This makes it possible to not just search for individual words, but also use Boolean operators, search for words appearing near each other, and search for words similar to the specified words (useful for spell correction). As a consequence, the search scripts are short and intuitive giving the possibility of performing powerful custom searches with simple Python scripts and, importantly, with objects in mind instead of tables. An important aspect is that any new object inserted with BioBuilder is automatically indexed in the database without any intervention from the user. Other functionalities such as using ZCatalog outside of Zope can be done easily. The software can be broken into a Python Catalog that is wrapped by a ZCatalog. This Python Catalog can then be used in any Python program by providing ZODB and indexing.
Security
BioBuilder depends heavily on Zope's internal security mechanisms. Zope provides a very fine-grained access control mechanism. Performing any operation in Zope, including viewing a web page, requires a specific Permission. Permissions are granted to Roles, and Roles are assigned to Users. Permissions, Roles and Users can be redefined at any point in the object hierarchy, and these definitions are automatically acquired by objects further down the hierarchy, unless explicitly overridden. The object that manages users' passwords and roles can be replaced with a more advanced version that reads from an external LDAP or SQL database and provides better permission management functionality.
Web interface
An efficient visual interface over the web is crucial for the success of biological databases. In BioBuilder, the data needs to be retrieved from the object database, rendered into HTML and served. Depending on the type of interface by which an instance is accessed, the HTML page is differently rendered. BioBuilder makes use of Zope page templates (ZPT) for generation of dynamic HTML pages [8]. ZPT is an XML namespaces based markup language for creating dynamic web page content for Zope web applications. To provide dynamic data, the values of pertinent objects are dynamically passed to the corresponding ZPT, which uses them to fill a static template. The separate localization of HTML creation – ZPT – and internal code-writing of methods or attributes allows a clear delineation of web design aspects from application programming.
Platform compatibility
BioBuilder was developed and tested on the Linux on x86 and Mac OS X on PowerPC platforms. We expect BioBuilder to be compatible with Windows and the various BSD variants as well.
Results and discussion
We have used BioBuilder to develop a novel protein data resource called Human Protein Reference Database (HPRD) [2,3] that was developed as a large international effort.
Use of BioBuilder to create Human Protein Reference Database
The installation of BioBuilder provides an empty functional database with the default settings indicated in Appendix 1. Two choices are available to populate BioBuilder with data. The manual method involves entering of data through the web forms created for BioBuilder and the automatic method permits importing of data through a BioBuilder native parser.
Manual method
BioBuilder was primarily designed to be a tool for creating a protein database manually. Therefore, it features a complete repertoire of web forms for editing and managing data from a web browser. Figure 2 depicts some the fields that can be used to enter general features of proteins including alternate names, gene symbol and links to other major database entries. Typographical and logical errors are automatically recognized and reported when the page is uploaded e.g. listing subcellular localization without the 'source' attribute will not be accepted by BioBuilder. Figure 3 shows the fields for annotating protein-protein interactions that permit annotation of features such as interacting regions, species specificity, type of interaction (e.g. in vitro or in vivo) and links to the literature. As we used BioBuilder to develop HPRD [2,3], it comes with the standard vocabulary that was used in the development of HPRD by default. Wherever applicable, the web forms for data annotation restrict the choice of terms to the standard vocabulary. It is also straightforward to add, delete or otherwise edit this standardized vocabulary list.
Figure 2.
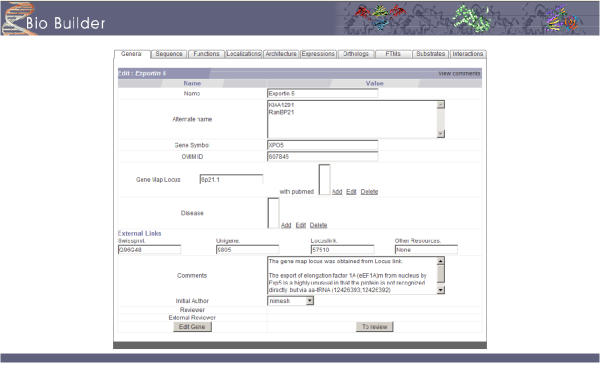
A screenshot showing a web form to enter the attributes for a 'protein' instance. The tabs facilitate entering of features of proteins. When the annotation of a protein is complete, it is submitted for review. An external reviewer can then assess the quality by using the 'Edit Gene' option.
Figure 3.
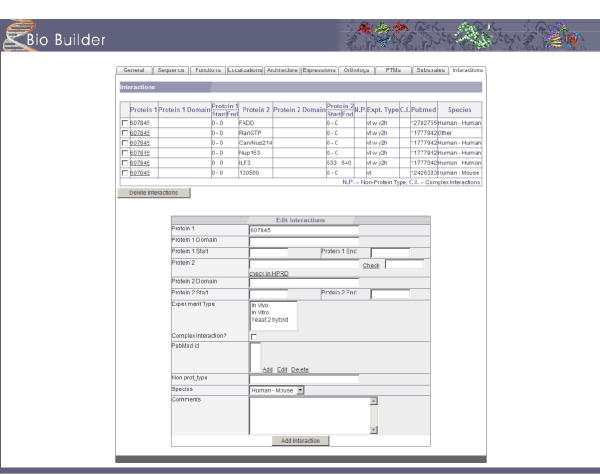
A screenshot showing the 'Interactions' tab of BioBuilder. While reviewing the interactions for a given protein, one can intuitively add or delete interactions. Here protein 1 indicates the protein that is being annotated and protein 2 is the interacting protein. The 'Check' function assists if protein 2 already exists in the database. This feature avoids the redundancy that would arise from entering the same interactions from different proteins. An option to annotate non-protein types of interacting molecules such as drugs, DNA or carbohydrates is also available.
A major advantage of being able to administer the data easily through a web browser is the possibility of sharing the annotation process globally. In the case of HPRD, the annotations were indeed performed by several different groups worldwide on the same server. To maintain consistency among annotators and for quality control, BioBuilder incorporates a review system. The review system serves two purposes: first, it allows reviewers from anywhere in the world to review the protein annotations and approve entries for committing to the database, and second, it allows the annotators to maintain data standards. When a protein is first annotated, it is assigned to a queue that is not publicly available until a person designated as reviewer, validates the correctness of the data and approves for such protein to leave the review queue and become public. If the entry is disapproved, the comments submitted by the reviewer are automatically sent to the original annotator for further changes. Figure 4 shows how this system can also be used to suggest a new domain name and shape for approval.
Figure 4.
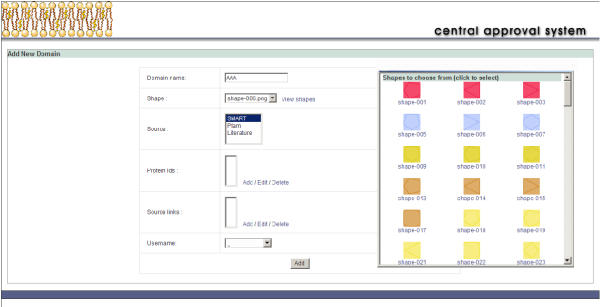
Review system for features in BioBuilder. The central approval system helps in choosing domain and motif shapes from a library of shapes and colors as shown in the screenshot.
Automatic method
BioBuilder can be used for quick presentation of large sets of protein and interaction data. By default, the generic parser accepts data in a BioBuilder native XML format. Whenever it is necessary to import data from other sources, the data can be easily transformed to the BioBuilder native format quite simply. The second format that is supported by BioBuilder is the proteomics standards initiative molecular interactions format (PSI-MI), a recently established standard for protein interaction data [1]. The interaction data can be exported in an XML file that is compliant with the PSI-MI format, a standard for representation of data in proteomics and to facilitate data comparison, exchange and verification that allows users to retrieve all relevant data from different sites and perform comparative analysis of different data sets much more easily than is currently possible [1,9]. Users can also submit their own set of interactions and can dynamically generate the PSI-MI complaint XML file which is readily portable across different protein interaction databases.
Interoperability
There are many heterogeneous, distributed data sources containing related information in biology. There is an immense need to integrate these systems by providing a unified data system where all these distributed data sources can be used and accessed. Common Object Broker Architecture (CORBA) and XML are two favorable mechanisms that make interconnecting heterogeneous biological data possible. EBI-EMBL has implemented CORBA infrastructure to integrate to provide an easy access to the EMBL data [10]. WILMA is one platform for automated annotation of proteins [11]. This system integrates bioinformatic tools and data retrieval for implementation of automatic annotation. BioBuilder provides several options that permit interoperability with other databases. On the other hand XML has become a powerful standard data format [12] for many biological standardization projects such as PSI-MI. One other excellent mechanism that has been made possible with the use of XML specification is Distributed Annotation System (DAS) that allows several groups scattered around the globe annotate the same centralized entry in a server [13].
It is highly unlikely that any single biological database resource can be totally comprehensive and address the needs of all scientists. Therefore, it is almost inevitable that a large number of highly specific databases will continue to coexist with a few really large ones. As long as the data formats are compatible, one can merge datasets obtained from different databases as per specific user requirements. The first level is XML input/output. Every object in BioBuilder can be dumped into an XML file, and conversely, a database instance can be populated with the XML files as described above. These XML files can then be easily exported to any database systems such as MySQL, and PostgreSQL. By default, BioBuilder comes with its own lightweight XML format which can be changed to the PSI-MI format where required. The second level of interoperability is at the 'application' level. Every object in BioBuilder automatically exposes an XML-RPC interface over HTTP. This enables the possibility of writing web services that call, in real-time, practically any data structure or method provided by the database. This means that a database created with BioBuilder is ready for supporting client applications and other potential bioinformatics web services [14]. Lastly, it is also possible to exchange data by using Zope's own import and export support, or by transferring the single file that contains the object database between Zope systems.
BioBuilder is released under the LGPL license and can be downloaded from http://biobuilder.hprd.org. We anticipate that BioBuilder will assist both biologists and non-biologists alike in developing their own protein databases.
Conclusions
BioBuilder facilitates manual annotation of protein data and development of databases that can be served through the World Wide Web. In fact, we have already used it to create a comprehensive annotated repository of the human proteome called HPRD. The scalable nature of the BioBuilder annotation tool will allow us to rapidly deploy additional features pertaining to proteins in HPRD. The data in BioBuilder can be easily exported to any relational database systems using XML files. BioBuilder is open source software and can be obtained under the terms of LGPL. The object oriented framework on which BioBuilder has been developed permitted easy cataloging and handling of complex proteomic data. We feel that that incorporating proteomic data with other data types such as microarray data will require object-relational models as gene expression microarray data is best suited for relational databases. Therefore, in the future, we hope to engineer BioBuilder tool on an object-relational model for developing even more complex databases encompassing DNA, mRNA and protein information.
Availability and requirements
Project name
BioBuilder
Project homepage
Operating systems
Linux, Mac OS X
Programming language
Python
Other requirements
Python Imaging Library (PIL)
License
LGPL
List of abbreviations used
SQL: Structured Query Language
XML-RPC: Extended Markup Language – Remote Procedure Call
PSI-MI: Proteomics Standards Initiative – Molecular Interchange format
HTTP: HyperText Transfer Protocol
ZPT: Zope Page Templates
HTML: HyperText Markup Language
ZOPE: Z Object Publishing Environment
ZODB: Zope Object DataBase
Authors' contributions
JDN was responsible for project design and sharing domain knowledge and NT worked on project design and implementation. SP interacted with the design team to share domain knowledge, BMV worked on the user interface design, BPR and NP shared domain knowledge, VS assisted in the development of BioBuilder, CKJ made HPRD compatible with PSI-MI and was involved in the BioBuilder development. PSK worked on implementation of CAS and ND and KS were responsible for PSI-MI implementation. AP was responsible for the overall direction of the project and cowrote the manuscript with JDN and SP. All authors read and approved the final manuscript.
Appendix 1
Default parameters in BioBuilder
1. Name and related database entries: The name of the protein along with alternate names, gene name and accession numbers in other databases.
2. Protein-protein interactions: Type of experiment (e.g. in vitro or in vivo) and the region/domain of the protein that mediates the interaction with other proteins. Options are also provided for the type of interaction such as a direct interaction or interaction in a complex.
3. Post-translational modifications: Type of modification, site of modification and the enzyme involved in the modification. The graphical representation to display each modification can be chosen from a number of available options.
4. Enzyme-substrate relationships: Upstream enzymes, downstream substrates and type and position of the modified residue(s) can be entered.
5. Domains and motifs: Range of amino acids for any domain or motif and link to information source. The geometrical shapes and colors can be chosen from a number of available options.
6. Standardized vocabulary: molecular function, biological process, cellular component and tissue of expression.
7. Diseases: Disease in which the protein is implicated and links to the source of information.
Acknowledgments
Acknowledgments
AP is supported by a Career Development Award from the Breast Cancer SPORE (CA 88843) at the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins. AP serves as the Chief Scientific Advisor to the Institute of Bioinformatics. The terms of this arrangement are being managed by the Johns Hopkins University in accordance with its conflict of interest policies.
Contributor Information
J Daniel Navarro, Email: dnavarro@jhmi.edu.
Naveen Talreja, Email: naveen@ibioinformatics.org.
Suraj Peri, Email: peri@jhmi.edu.
BM Vrushabendra, Email: vrushbi@ibioinformatics.org.
BP Rashmi, Email: rashmi@ibioinformatics.org.
N Padma, Email: padma@ibioinformatics.org.
Vineeth Surendranath, Email: vineeth@ibioinformatics.org.
Chandra Kiran Jonnalagadda, Email: jace@pobox.com.
PS Kousthub, Email: kousthub@ibioinformatics.org.
Nandan Deshpande, Email: nandan@ibioinformatics.org.
K Shanker, Email: kalyan@ibioinformatics.org.
Akhilesh Pandey, Email: pandey@jhmi.edu.
References
- Hermjakob H, Montecchi-Palazzi L, Bader G, Wojcik J, Salwinski L, Ceol A, Moore S, Orchard S, Sarkans U, von Mering C, Roechert B, Poux S, Jung E, Mersch H, Kersey P, Lappe M, Li Y, Zeng R, Rana D, Nikolski M, Husi H, Brun C, Shanker K, Grant SG, Sander C, Bork P, Zhu W, Pandey A, Brazma A, Jacq B, Vidal M, Sherman D, Legrain P, Cesareni G, Xenarios I, Eisenberg D, Steipe B, Hogue C, Apweiler R. The HUPO PSI Molecular Interaction Format – A community standard for the representation of protein interaction data. Nature Biotechnol. 2004;22:177–183. doi: 10.1038/nbt926. [DOI] [PubMed] [Google Scholar]
- Peri S, Navarro JD, Amanchy R, Kristiansen TZ, Jonnalagadda CK, Surendranath V, Niranjan V, Muthusamy B, Gandhi TK, Gronborg M, Ibarrola N, Deshpande N, Shanker K, Shivashankar HN, Rashmi BP, Ramya MA, Zhao Z, Chandrika KN, Padma N, Harsha HC, Yatish AJ, Kavitha MP, Menezes M, Choudhury DR, Suresh S, Ghosh N, Saravana R, Chandran S, Krishna S, Joy M, Anand SK, Madavan V, Joseph A, Wong GW, Schiemann WP, Constantinescu SN, Huang L, Khosravi-Far R, Steen H, Tewari M, Ghaffari S, Blobe GC, Dang CV, Garcia JG, Pevsner J, Jensen ON, Roepstorff P, Deshpande KS, Chinnaiyan AM, Hamosh A, Chakravarti A, Pandey A. Development of Human Protein Reference Database as an initial platform for approaching systems biology in humans. Genome Res. 2003;13:2363–2371. doi: 10.1101/gr.1680803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Protein Reference Database http://www.hprd.org
- Zope Homepage http://www.zope.org
- Python Home page http://www.python.org
- Navarro JD, Niranjan V, Peri S, Jonnalagadda CK, Pandey A. From biological databases to platforms for biomedical discovery. Trends Biotechnol. 2003;21:263–268. doi: 10.1016/S0167-7799(03)00108-2. [DOI] [PubMed] [Google Scholar]
- ZCatalog tutorial http://www.zope.org/Documentation/How-To/ZCatalogTutorial
- Using Zope Page Templates http://www.zope.org/Documentation/Books/ZopeBook/2_6Edition/ZPT.stx
- Web services for bioinformatics http://webservices.xml.com/pub/a/ws/2002/05/14/biows.html?page=1
- Orchard S, Kersey P, Zhu W, Montecchi-Palazzi L, Hermjakob H, Apweiler R. Progress in establishing common standards for exchanging proteomics data The second meeting of the HUPO Proteomics Standards Initiative. Comp Func Genomics. 2003;4:203–206. doi: 10.1002/cfg.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Rodriguez-Tome P, Redaschi N, McNeil P, Robinson A, Lijnzaad P. Accessing and distributing EMBL data using CORBA (common object request broker architecture) Genome Biol. 2000;1:RESEARCH0010. doi: 10.1186/gb-2000-1-5-research0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achard F, Vaysseix G, Barillot E. XML, bioinformatics and data integration. Bioinformatics. 2001;17:115–125. doi: 10.1093/bioinformatics/17.2.115. [DOI] [PubMed] [Google Scholar]
- Dowell RD, Jokerst RM, Day A, Eddy SR, Stein L. The Distribution Annotation System. BMC Bioinformatics. 2001;2:7. doi: 10.1186/1471-2105-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prlic A, Domingues FS, Lackner P, Sippl MJ. WILMA-automated annotation of protein sequences. Bioinformatics. 2004;20:127–128. doi: 10.1093/bioinformatics/btg380. [DOI] [PubMed] [Google Scholar]