

Presenilin 1 mutations are associated with synucleinopathies, but their contribution to pathology is unclear. Winslow et al. reveal an interaction between presenilin 1 and α-synuclein in cognitively normal individuals, which is increased in patients with dementia with Lewy bodies, and familial Alzheimer’s disease. The interaction may regulate α-synuclein levels and localization.
Keywords: presenilin, α-synuclein, Lewy body, Alzheimer’s disease, Parkinson’s disease, dementia, ageing
Abstract
A growing number of PSEN1 mutations have been associated with dementia with Lewy bodies and familial Alzheimer’s disease with concomitant α-synuclein pathology. The objective of this study was to determine if PSEN1 plays a direct role in the development of α-synuclein pathology in these diseases. Using mass spectrometry, immunoelectron microscopy and fluorescence lifetime image microscopy based on Forster resonance energy transfer (FLIM-FRET) we identified α-synuclein as a novel interactor of PSEN1 in wild-type mouse brain tissue. The interaction of α-synuclein with PSEN1 was detected in post-mortem brain tissue from cognitively normal cases and was significantly increased in tissue from cases with dementia with Lewy bodies and familial Alzheimer’s disease associated with known PSEN1 mutations. We confirmed an increased interaction of PSEN1 and α-synuclein in cell lines expressing well characterized familial Alzheimer’s disease PSEN1 mutations, L166P and delta exon 9, and demonstrated that PSEN1 mutations associate with increased membrane association and accumulation of α-synuclein. Our data provides evidence of a molecular interaction of PSEN1 and α-synuclein that may explain the clinical and pathophysiological overlap seen in synucleinopathies, including Parkinson’s disease, dementia with Lewy bodies, and some forms of Alzheimer’s disease.
Introduction
Although dementia with Lewy bodies is the second-leading cause of dementia after Alzheimer’s disease, little is known about the molecular causes of the disease. Dementia with Lewy bodies is pathologically characterized by the deposition of α-synuclein in Lewy bodies and Lewy neurites in the brainstem, limbic, and cortical areas (McKeith et al., 2004), often accompanied by hippocampal amyloid-β plaque deposition. Clinically, the loss of cholinergic neurons causes a dramatic loss in cognitive function, whereas many patients with dementia with Lewy bodies have concomitant loss of dopaminergic neurons in the substantia nigra that causes diminished motor control. Although familial dementia with Lewy bodies is rare, families with a mixed clinical presentation of dementia with Lewy bodies and Alzheimer’s disease-associated dementia with parkinsonism have been identified, suggestive of genetic links to a heterogenous presentation of disease (Waters and Miller, 1994; Denson et al., 1997; Ohara et al., 1999; Brett et al., 2002; Bonner et al., 2003; Tsuang et al., 2004; Ishikawa et al., 2005; Nervi et al., 2011). More recently, a candidate gene study of known Alzheimer’s disease and Parkinson’s disease genes in cases of familial and sporadic dementia with Lewy bodies and Parkinson’s disease with dementia revealed a striking association of known and novel mutations in presenilin 1 (PSEN1), presenilin 2 (PSEN2), microtubule-associated protein tau (MAPT), α-synuclein (SNCA), and leucine-rich repeat kinase 2 (LRRK2) in these disease groups (Meeus et al., 2012). Although PSEN1 mutations are most commonly associated with familial forms of Alzheimer’s disease, a growing number of studies have linked PSEN1 mutations to the development of significant α-synuclein pathology in some forms of familial Alzheimer’s disease and dementia with Lewy bodies (Denson et al., 1997; Snider et al., 2005; Leverenz et al., 2006). In a study of familial Alzheimer’s disease cases with known PSEN1 and PSEN2 mutations, Lewy body pathology in the amygdala was found in 96% of the cases, with higher frequency of Lewy body deposition in the cases with PSEN1 mutations compared to those with PSEN2 mutations (Leverenz et al., 2006).
PSEN1 is a multi-pass transmembrane protein primarily known as the catalytic core of the γ-secretase complex. Alternative functions of PSEN1 include regulation of intracellular calcium levels, cell-to-cell adhesion, presynaptic neurotransmitter release, and autophagy (Tu et al., 2006; Green et al., 2008; Lee et al., 2010; Ohta et al., 2010). Interestingly, although the primary cellular function of α-synuclein is unknown, many known functions of PSEN1 overlap with cellular processes involving α-synuclein including early secretory pathway events, vesicle formation and trafficking, SNARE complex formation, and synaptic vesicle regulation (Chandra et al., 2005; Cooper et al., 2006; Gitler et al., 2008; Burre et al., 2010; Pranke et al., 2011; Scott and Roy, 2012; Chen et al., 2013; Choi et al., 2013). α-Synuclein is a small (14 kDa, 140 amino acids) protein that is composed of an amphipathic N-terminal domain, a central hydrophobic region, and an acidic C-terminal domain. It is considered a natively unfolded protein in the cytosol that readily adopts an alpha-helical structure upon membrane binding. Despite years of research, there is little consensus on which α-synuclein conformation or subcellular association contributes to toxic events within the cell (Lee et al., 2002; Chandra et al., 2005; Vamvaca et al., 2009; Leftin et al., 2013).
The functional convergence of PSEN1 and α-synuclein in normal cellular functions and in disease suggests a molecular interaction of these proteins. In this study we investigate the molecular convergence of PSEN1 and α-synuclein. Using complementary molecular techniques including co-immunoprecipitation, immunoelectron microscopy, and fluorescence lifetime image microscopy based on Forster resonance energy transfer (FLIM-FRET) we find that α-synuclein and PSEN1 interact in cell lines, primary neuronal cultures, mouse brain tissue, and human brain tissue. Furthermore, the interaction in human brain is increased in the presence of PSEN1 familial Alzheimer’s disease linked mutations and in dementia with Lewy bodies amygdala. Our data suggest that this endogenous complex regulates α-synuclein membrane association and overall accumulation.
Materials and methods
Human tissue
Human post-mortem brain tissue was provided by the brain bank at the Mayo Clinic in Jacksonville. For this study amygdala tissue from cognitively normal controls (n = 4, mean age ± SD, 59.5 ± 4.7 years), familial Alzheimer’s disease cases with PSEN1 mutations (n = 4, 52.3 ± 8.4 years), and dementia with Lewy bodies (n = 4, 62.5 ± 5.2 years) cases was studied.
Cell lines
Chinese hamster ovary and PSEN1 knockout mouse embryonic fibroblast (Herreman et al., 2000) cell lines were maintained in Opti MEM® (Invitrogen) medium supplemented with 5% foetal bovine serum (Invitrogen) and stable expression of wild-type, L166P and dE9 PSEN1 was maintained in the presence of 5 µg/ml of puromycin. All cells were grown and incubated at 37°C and 5% CO2.
Glutathione S-transferase protein cloning and purification
N-terminal, loop 1–2, and loop 6–7 PSEN1 GST fusion fragments were created using the glutathione S-transferase (GST) expression vector pGEX-6P-2 (GE Lifesciences). Fusion plasmids were transformed into chemically competent BL21 cells (Escherichia coli) and grown at 37°C until the culture OD600 reached 0.6–1.0, at which point expression was induced by the addition of isopropyl-d-thio-galactoside (IPTG) (Sigma) to 1 mM for 5 h. Cultures were collected and lysed in Tris-buffered saline with 0.1% Tween-20 (TBST) and the peptides were isolated by precipitation with glutathione coated Sepharose beads (GE). Peptides were maintained in TBST before use. Cortical brain homogenates were incubated with GST-PSEN1 fragments or recombinant GST as a control. A control set of beads for each condition without mouse brain lysate was also incubated. After an overnight incubation, samples were eluted with 2× SDS NuPAGE® sample buffer (Life Technologies) then boiled at 95°C before running on a 4–12% 1.5 mm Bis-Tris NuPAGE® gel (Life Technologies). The gel was stained with Coomassie blue stain (Life Technologies) per kit instructions before samples were excised. Samples were sent to Harvard University’s Taplin Biological Mass Spectrometry Facility for analysis. https://taplin.med.harvard.edu/
Brain homogenate
CD1 mice (Charles River Laboratories) were anaesthetized using CO2 asphyxiation. Brain cortices were dissected and homogenized in 50 mM HEPES, 125 mM NaCl, 0.5 mM EDTA, 100 mM sucrose, 1% Triton™ X-100, pH 7.4 lysis buffer with protease inhibitors (Roche) using a motor driven homogenizer. Brains were solubilized for 1 h at 4°C then centrifuged at 14 000g. The soluble fraction was used for mass spectrometry. All studies were performed with the approval of the Massachusetts General Hospital Animal Care and Use Committee and in compliance with the National Institute of Health guidelines for the use of experimental animals.
Co-immunoprecipitation
CD1 mice (Charles River Laboratories) were anaesthetized using CO2 asphyxiation. Brains were mechanically homogenized in a 1% CHAPS lysis buffer [50 mM Tris-HCl, 50 mM NaCl, and EDTA-free inhibitor (Roche), 1% w/v CHAPS] by 10 passes through a p200 pipette tip followed by a 27-gauge needle. Homogenates were then incubated for 1 h at 4°C on a rocker. Homogenates were spun at 1000 rpm at 4°C for 5 min and the supernatant protein concentrations were calculated and equalized for all samples before the addition of antibody. Samples were incubated with primary α-synuclein antibody [syn-1 (BD) 1/250] overnight on a rocker at 4°C. Magnetic IgG beads (Invitrogen) were washed twice in 1% CHAPS lysis then added to each sample. Samples and IgG beads were incubated for 2 h on a rocker at 4°C and then washed five times in 1% CHAPS lysis buffer. Bound proteins were eluted by resuspending the magnetic beads and bound material in NuPAGE® sample buffer with reducing agent and incubating at 37°C for 10 min with intermittent shaking. Immunoprecipitates were analysed by standard SDS-PAGE using the NuPAGE® system (Invitrogen). PSEN1 and α-synuclein were detected separately by sequential detection with first PSEN1 antibody and then α-synuclein antibody. Due to this sequential process, detection with α-synuclein antibody sometimes shows two bands, one at 14 kD corresponding to α-synuclein and one at 17 kD, which corresponds to the previously detected CTF-PSEN1. For co-immunoprecipitation from the PSEN1 MEF cell lines we avoided artefacts caused by difference in cell line transfection rates by pulling down α-synuclein and then quantifying the amount of PSEN1 as a ratio of immunoprecipitated PSEN1 to immunoprecipitated α-synuclein).
Crosslinking
For immunoprecipitation from crosslinked wild-type murine brain, crosslinking was performed using an adapted version of the ReCLIP method (reversible cross-link immuno-precipitation; Smith et al., 2011). Briefly, a gross 10% (w/v) brain homogenate was made in ice cold PBS by rapid mechanical homogenization. Lysine residues of interacting proteins were cross-linked by incubation with 10 mM dithiobissuccinimidyl propionate (22585, Thermoscientific) for 30 min at 4°C. Thereafter, an equal volume of lysis buffer (100 mM NaCl, 50 mM Tris HCl, 5 mM EDTA, 0.3% Triton™ X-100 with protease inhibitor (11836153, Roche), pH 7.4, was added. Samples were incubated for 30 min on ice with frequent vortexing. Finally the homogenate was centrifuged at 9000g for 10 min at 4°C and the supernatant was transferred to a new tube. After following the standard immunoprecipitation protocol, dithiothreitol (43816, Sigma) was added at a final concentration of 50 mM to de-crosslink proteins before loading onto a gel.
Immunoelectron microscopy
Adult (3–4 month old) CD1 mice were deeply anaesthetized and perfused with 0.1% glutaraldehyde and 4% paraformaldehyde in PBS. After 48 h fixation, cortices were cut in 100 µm sections using a Vibratome and then post-fixed in 0.1% osmium tetroxide for 1 h. Post-fixation was followed by sequential dehydration in ethanol solutions and propylene oxide. Samples were embedded in Araldite/DDSA resin (Electron Microscopy Sciences), sectioned to 70 nm using an ultracut microtome (Reichart-Jung), and mounted on grids for immunolabelling with gold particles. Samples were incubated with primary antibodies for 24 h at 4°C in blocking solution. PSEN1 and α-synuclein co-localization was confirmed by two antibodies for each protein: PSEN1 (NT 14–33 aa Millipore, and loop region Epitomics), α-synuclein (syn-1 BD, and PA1-18264 Pierce). PSEN1 was labelled with 15 nm gold particles and α-synuclein was labelled with 6 nm gold particles for 3 h. Immunogold-labelled tissue was negatively stained with 2% uranyl acetate prepared in 50% ethanol. Grids were observed on a JEOL JEM-1011 transmission electron microscope with a Hamamatsu ORCA digital camera.
Primary neuronal cell culture
Primary neuronal cultures were prepared from cerebral cortices of embryonic Day 14–16 CD1 mouse embryos. Cortices were dissected from embryonic brain after removal of the meninges. Cortices were dissociated by trituration and filtration and cells were resuspended in Neurobasal® (Gibco) medium supplemented with 10% foetal bovine serum, 2 mM GlutaMAX™, 100 U/ml penicillin, and 100 µg/ml streptomycin coated with 20 µg/ml poly-d-lysine (Sigma-Aldrich). After 4 h, medium was changed into Neurobasal®/B-27 [Neurobasal® medium containing 2% (v/v) B-27 supplement], 100 U/ml penicillin, 100 µg/ml streptomycin, and 2 mM GlutaMAX™. Cells were maintained at 37°C in 5% CO2 in a humidified incubator. Neurons were grown for 12–14 days in vitro before fixation for FLIM-FRET analysis.
Immunofluorescent staining for FLIM-FRET analysis
PSEN1 mouse embryonic fibroblast cells and primary neuronal cultures were fixed in 4% paraformaldehyde and then permeabilized in 0.01% Triton™. Cells were incubated in blocking solution consisting of 4% normal donkey serum and then double-immunostained for FLIM-FRET analysis with primary antibodies syn-1 (BD) and N-terminus (14–33 aa) of PSEN1 (Millipore), followed by Alexa Fluor® 488- and Cy3-conjugated secondary antibodies, respectively. Alexa Fluor®488 synuclein only immunostained cells were used as negative FRET control for the FLIM analysis. For immunohistochemical staining of human amygdala, the fixed tissue slices were washed of all cryoprotectant and then permeabilized in 0.1% Triton. Blocking solution consisted of 1% normal donkey serum and 0.1% Triton. The same primary and secondary antibody pairs as were used on cells were used for immunohistochemical staining of the tissue.
Fluorescent lifetime imaging microscopy
The interaction of α-synuclein with PSEN1 was monitored by previously established FLIM-FRET techniques, as described (Berezovska et al., 2003; Lleo et al., 2004). Briefly, the fluorescent lifetime of the Alexa Fluor® 488 donor fluorophore was established in the absence (donor only lifetime = t1) and presence (donor and acceptor lifetime = t2) of the Cy3 acceptor fluorophore. Upon excitation of the donor fluorophore (Alexa Fluor® 488) in the presence of an acceptor fluorophore (Cy3), some of the donor emission energy is non-radiatively transferred to the acceptor if the donor and acceptor are <5–10 nm apart. This results in a characteristic shortening of the donor fluorophore lifetime, measured as t2. The degree of donor fluorophore lifetime shortening (FRET efficiency, measured as EFRET) correlates with the change in distance between donor and acceptor molecules: high EFRET (short lifetime) indicates close proximity between the molecules. The percent EFRET is calculated using the following equation (Uemura et al., 2010):
Microscopy and SPC image lifetime analysis
A Chameleon pulsed laser was used to excite Alexa Fluor® 488 donor fluorophore. Becker&Hickl FLIM hardware and software were used to acquire the donor lifetime. Data analysis was performed using SPC Image (Becker&Hickl), in which donor fluorophore lifetimes are determined by fitting the data to one- (negative control) or two- (experimental conditions) component exponential decay curves, using the whole cell, cell body, or processes as a region of interest. In the two-component exponential model of lifetime analysis, the longer (no FRET) donor-only lifetime is ‘fixed’ as a t1 value, and the second, shorter lifetime reflecting the presence of FRET is calculated by the system as a t2 value. The t2 lifetime is used for comparisons between different experimental conditions. Thus, the ‘non-FRETing' component (t1) is excluded from the lifetime comparisons. For human tissue analysis, the lifetime data was fit to a two-component and three-component model as described previously (Wahlster et al., 2013), where the donor-only longer lifetime (t2) was established in the presence of a short autofluorescent lifetime (t1). In the presence of the acceptor, the donor lifetime was measured as (t3) and FRET efficiency was calculated as EFRET = 100 × (t2 − t3) / t2. Donor t2 or t3 lifetime pseudocolour images were produced using the SPC Image analysis software.
Cellular fractionation: cytosolic and membrane-enriched fractions
After collecting and washing cells with ice-cold PBS, cells were lysed in hypotonic buffer (10 mM Tris pH 7.4, 1 mM EDTA, 1 mM EGTA, EDTA-free protease inhibitor). Cells were passed through a 27-gauge needle 10 times and then centrifuged at 1000g for 15 min at 4°C to pellet the nuclear fraction. The post-nuclear fraction (supernatant) was separated into a heavy endoplasmic reticulum-enriched membrane fraction (pellet) and a lighter membrane and cytosol fraction (supernatant) by centrifugation at 16000g for 45 min at 4°C. The pellet was resuspended in RIPA buffer. The cytosolic and membrane fractions were analysed for α-synuclein levels by standard ELISA (Invitrogen) techniques. Samples loaded were equalized for total protein.
Gaussia luciferase assay
Fusion constructs syn-luc1 (N-terminal half of Gaussia luciferase) and syn-luc2 (C-terminal half of Gaussia luciferase) and full-length luciferase fusion with full-length α-synuclein, syn-luciferase, were previously generated (Outeiro et al., 2008). syn-luc1 and syn-luc2 or syn-luciferase and enhanced GFP were co-transfected into PSEN1 mouse embryonic fibroblast plated cells in a 96-well plate format using Lipofectamine™ 2000 (Invitrogen) per the manufacturer’s protocol. Due to interference of high foetal bovine serum concentrations with detection of luciferase luminescence, cells were cultured in 1% foetal bovine serum in Opti MEM® (Gibco) for 24 h after transfection. Twenty-four hours after transfection, culture media was transferred to a new 96-well plate. Cells were washed with PBS and replaced with serum- and phenol-red-free media. Luciferase activity from protein complementation and full length luciferase constructs was measured from live cells in an automated plate reader following the injection of the cell permeable substrate, coelenterazine (20 µM) (Prolume Ltd.) with a signal integration time of 2 s. GFP expression was measured by a fluorescent plate reader for each sample and used as an indirect measure of transfection efficiency.
Inhibition of γ-secretase activity
Cells were treated for 24 h with the following concentrations of γ-secretase inhibitors: DAPT 500 nM, Compound E 3 nM, and LY-450139 15 nM.
Statistics
Statistical analyses and graphs were generated using GraphPad Prism, version 5.0a. Median and interquartile ranges were graphed for the corresponding scatter-plots of non-normal data and bar graphs were expressed as mean and standard error of the mean (SEM) for normal data with n = 3 independent experiments or greater. Significance for non-Gaussian data (i.e. FLIM-FRET) was analysed using a Mann-Whitney test (two-tailed) or Kruskal-Wallis with a Dunns Multiple Comparison post hoc test. Significance for normal data was calculated using a one-way ANOVA with a Dunnett’s multiple comparison post hoc test or two-tailed t-test.
Results
PSEN1 and α-synuclein interact
To identify novel interactors of PSEN1 we incubated cortical lysates from adult wild-type CD1 mice with different GST-tagged PSEN1 fragments and then co-immunoprecipitated interacting proteins using a GST-specific antibody. Mass-spectrometry analysis (Table 1) of the extracted and SDS-PAGE separated proteins identified four unique peptides that together make up 44% of the α-synuclein sequence. α-synuclein was efficiently precipitated by two PSEN1 fragments comprising PSEN1 loop regions between transmembrane domains 1–2 and 6–7, but not by an N-terminal fragment of PSEN1, nor with the GST-tag alone. To verify these findings, we immunoprecipitated endogenous α-synuclein from whole brain lysates of wild-type CD1 adult mice and immunodetected endogenous PSEN1 (Fig. 1A and Supplementary Fig. 1A–C), and confirmed the reverse co-immunoprecipitation of α-synuclein by immunoprecipitation of PSEN1 (Supplementary Fig. 1A and B). Together with α-synuclein, both full-length PSEN1 and C-terminal fragments of PSEN1 were co-immunoprecipitated, consistent with our findings from mass spectrometry, whereas IgG control protein did not pull-down α-synuclein or PSEN1 (Supplementary Fig. 1C). We confirmed these findings in PS70 cells (stably expressing PSEN1 and APP) transiently transfected with α-synuclein. Pull-down of α-synuclein in this cell-line co-immunoprecipitated PSEN1 C-terminal fragments, whereas IgG did not pull-down α-synuclein or PSEN1 (Supplementary Fig. 1D).
Table 1.
Detection of α-synuclein as a novel interactor of PSEN1
| PSEN1 amino acid sequence | Corresponding region | Number of unique synuclein peptides detected (α, β, γ) | Number of unique α-synuclein peptides | % of total α-synuclein sequence |
|---|---|---|---|---|
| 1–80 | N-terminus | 0 | 0 | 0 |
| 263–376 | Loop 6–7 | 1 | 1 | 12.9 |
| 100–134 | Loop 1–2 | 4 | 2 | 44.3 |
| Overall interaction summary | 4 | 2 | 44.3 | |
| Positive control: 17 unique/ 24 delta catenin peptides detected (immunoprecipitated with Loop 6–7) | ||||
Three different regions of PSEN1 tagged with GST were used as ‘bait’ to co-immunoprecipitate interactors of PSEN1 from CD1 mouse cortical lysates. Peptide sequences of interactors were identified by mass spectrometry. α-synuclein was co-immunoprecipitated by two loop regions of PSEN1 (1-2 and 6-7) while an N-terminal fragment of PSEN1 did not co-immunoprecipitate α-synuclein. Four different peptides were identified that collectively covered 43% of the alpha-synuclein protein sequence. Two of these sequences were unique to α-synuclein while two sequences were common to α-, β-, and γ- synucleins. Co-imunoprecipitation and detection of known PSEN1 interactor, delta-catenin, was confirmed and is shown here as a positive control, while GST alone did not pull-down α-synuclein.
Figure 1.

Endogenous PSEN1 and α-synuclein interact in wild-type mouse brain on mitochondria and synaptic vesicles. (A) Immunoprecipitation (IP) of α-synuclein from CD1 mouse brian tissue co-immunoprecipitated full-length (FL) PSEN1 and C-terminal fragments (CTF) of PSEN1. (B) Representative electron micrographs of wild-type CD1 cortical tissue confirmed co-localization of α-synuclein (6 nm gold particles) and PSEN1 (15 nm gold particles) on mitochondria, synaptic vesicles, and at the plasma membrane. Scale bar = 100 nm.
We used immunoelectron microscopy to identify the subcellular localization of the interaction of PSEN1 and α-synuclein in non-homogenized mouse brain tissue. Immunogold labelling of endogenous PSEN1 and α-synuclein in cortical tissue slices from wild-type adult mice revealed a close association of PSEN1 (labelled with 15 nm gold particles) and α-synuclein (6 nm gold particles) on presynaptic vesicles, mitochondrial surfaces and the plasma membrane (Fig. 1B). These subcellular localizations are consistent with known organelle interactions or functions of PSEN1 and α-synuclein, and suggest a specific membrane-dependent interaction of these two proteins (Efthimiopoulos et al., 1998; Murphy et al., 2000; Torp et al., 2003; Fortin et al., 2004; Hansson et al., 2004; Guardia-Laguarta et al., 2014). To further validate the interaction in intact cells, we performed multiphoton FLIM-FRET of immunofluorescently labelled α-synuclein and PSEN1 in cortical neuronal cultures from wild-type mice. This powerful microscopy-based technique increases the spatial resolution of standard microscopy from ∼5 µm to 10 nm and allows for indirect quantification of protein–protein interactions in intact cells (Berezovska et al., 2003; Lleo et al., 2004). Using FLIM-FRET, we confirmed that PSEN1 and α-synuclein are within close proximity, as measured by the shortened lifetime of the donor fluorophore labelling α-synuclein, and expressed as increased percent FRET efficiency (EFRET), both in cell bodies [median 4.31%, interquartile range (IQR) 1.65–10.38] and in punctate synapse-like structures (median 11.75%, IQR 5.96–19.71) along processes (Fig. 2A and B). The significantly higher EFRET in processes compared to that in cell bodies indicates an increased proximity of PSEN1 and α-synuclein in neuronal processes.
Figure 2.

The interaction of PSEN1 (PS1) and α-synuclein occurs in neuronal cell bodies and synaptic-like structures. (A) Pseudocolour lifetime image of CD1 primary neurons immunostained for endogenous α-synculein/Alexa Fluor® 488 (donor) and α-synuclein/Alexa Fluor® 488 and PSEN1/Cy3 (donor and acceptor pair) shows close proximity [shorter lifetime (ps), higher EFRET] of α-synuclein and PSEN1 in the cell body and processes. The first image shows colour-coded distribution of the donor (α-synuclein) fluorophore lifetime in each pixel of the image in the absence of the acceptor (PSEN1); therefore, the blue colour is representative of the baseline lifetime of a fluorophore. The ranges of green, yellow, and red pixels in the second image are representative of shortened lifetimes due to close proximity of the donor (on α-synuclein) and the acceptor (on PSEN1) fluorophores. The shortest lifetimes reflect the closest interactions and are pseudocoloured in red, whereas yellow and green are intermediate interactions between red and blue (no interaction). (B) Scatter plot of % EFRET (median and interquartile range) in the cell body and processes shows increased close proximity interactions of α-synuclein and PSEN1 in processes compared to the cell-body (n = 3, Mann Whitney, two-tailed ***P-value < 0.001).
Increased interaction of PSEN1 and α-synuclein in dementia with Lewy bodies and PSEN1 familial Alzheimer’s disease amygdala
Mounting genetic data supports the idea that PSEN1 dysfunction plays a significant role in the development of α-synuclein pathology in dementia with Lewy bodies and familial Alzheimer’s disease with Lewy body pathology (Ishikawa et al., 2005; Snider et al., 2005; Meeus et al., 2012). Based on this and our data showing an endogenous interaction of α-synuclein and PSEN1 in mouse tissue and neuronal cultures, we hypothesize that PSEN1 dysfunction leads to an accumulation of α-synuclein levels through a dysregulation of the interaction between PSEN1 and α-synuclein. To directly test the relevance of this interaction to disease, we evaluated the interaction of PSEN1 and α-synuclein in human post-mortem tissue from patients with dementia with Lewy bodies (no known genetic association) (n = 4, mean age ± SD, 62.5 ± 5.2 years), patients with familial Alzheimer’s disease with known PSEN1 mutations (n = 4, 52.3 ± 8.4 years), and control subjects (with no signs of Parkinson’s disease, dementia with Lewy bodies or Alzheimer’s disease-related pathology or symptoms; n = 4, 59.5 ± 4.7 years). We focused on the amygdala as recent studies highlighted the presence of Lewy body pathology in the amygdala of sporadic Alzheimer’s disease cases and familial Alzheimer’s disease caused by PSEN1 mutations, suggesting that the amygdala is particularly vulnerable to the development of α-synuclein pathology.
Comparing disease and control cases, the most striking difference in the interaction of PSEN1 and α-synuclein (measured by increased EFRET) occurred in processes rather than in cell bodies. In axons and synaptic puncta (Fig. 3A) the interaction of PSEN1 and α-synuclein (%EFRET) was significantly increased in both dementia with Lewy bodies (median for disease group 12.58%, IQR 4.27–8.12) and the PSEN1 familial Alzheimer’s disease tissue (4.56%, IQR 0.38–10.66) compared with controls (0.29%, IQR 0.00–3.86) (Fig. 3A and B). Consistent with the relative extent of α-synuclein pathology normally seen in the amygdala for these diseases, the dementia with Lewy bodies group had the highest average EFRET. Importantly, this result did not correlate with Alzheimer’s disease pathology (Braak staging and thioflavin S) (Table 2).
Figure 3.

PSEN1 and α-synuclein interactions are elevated in axons from the amygdala of patients with dementia with Lewy bodies (DLB) and PSEN1 familial Alzheimer’s disease (PS1 FAD). (A) Representative pseudocolour lifetime images from donor and acceptor immunostained amygdala tissue from dementia with Lewy bodies, PSEN1 familial Alzheimer’s disease, and control cases shows enhanced PSEN1–α-synuclein interactions (shorter lifetime) in tissue from dementia with Lewy bodies and PSEN1 familial Alzheimer’s disease cases compared to healthy controls. (B) Scatter-plot of % Efret (and median and interquartile range) of axon-based region of interest analysis for each case showed a significant increase in α-synuclein-PSEN1 interactions in the axons of cases with dementia with Lewy bodies and with PSEN1–familial Alzheimer’s disease compared with controls (n = 4 cases for each disease group, Kruskal Wallis, Dunns multiple comparisons, **P < 0.01, ***P < 0.001).
Table 2.
Human tissue summary table for FLIM-FRET studies
| Case ID | Pathology diagnosis | Sex | Age at death | Alzheimer’s diease Braak and Braak | Thioflavin S medial amygdala | Thioflavin S lateral amygdala | Median (% EFRET) | 25–75% percentiles (% EFRET) | |
|---|---|---|---|---|---|---|---|---|---|
| Control 1 | Normal | F | 56 | 0 | 0 | 0 | 0.00 | 0.00–0.00 | |
| Control 2 | Normal | F | 64 | 1 | 0 | 0 | 0.00 | 0.00–2.59 | |
| Control 3 | Normal | M | 63 | 3 | 0 | 0 | 5.56 | 3.20–8.05 | |
| Control 4 | Normal/VaD | F | 55 | 0 | NA | NA | 0.00 | 0.00–1.01 | |
| PS1 FAD 1 | AD (PSEN1 G378V) | F | 51 | 6 | >50 | >50 | 0.41 | 0.00–2.48 | |
| PS1 FAD 2 | AD (PSEN1 G206A)/DLBD/VaD | M | 64 | 6 | >50 | 20–25 | 11.19 | 4.59–14.08 | |
| PS1 FAD 3 | AD (PSEN1 L392V) | M | 50 | 6 | >50 | 40–45 | 6.01 | 2.07–7.64 | |
| PS1 FAD 4 | AD (PSEN1 N135S)/CAA/ALB | M | 44 | 6 | >50 | 40–45 | 3.59 | 0.45–8.23 | |
| DLB 1 | DLBD/AD/CAA | F | 69 | 6 | >50 | 8–10 | 12.93 | 7.23–17.79 | |
| DLB 2 | DLBD | M | 61 | 2 | 0 | 0 | 4.10 | 0.00–7.17 | |
| DLB 3 | DLBD | M | 60 | 0 | 0 | 0 | 9.96 | 0.0, 16.68 | |
| DLB 4 | DLBD/AD/CAA | M | 64 | 6 | >50 | 6–8 | 18.63 | 14.83–22.83 |
Four cases from the following categories were included in this study: controls, no cognitive impairment as a result of Parkinson’s disease, dementia with Lewy bodies, or Alzheimer’s disease; familial history of Alzheimer’s disease associated with an identified PSEN1 mutation, and diagnosis of dementia with Lewy Bodies. Each case was assessed for the postmortem presence of neurodegeneration-associated pathologies. Disease class, pathology-based diagnosis (including known PSEN1 mutations in parentheses), sex, age-at-death, Alzheimer’s disease Braak and Braak staging, and plaque burden (thioflavin S) quantification from the medial and lateral amygdala are reported. The interaction of PSEN1 and α-synuclein was measured by FLIM-FRET and presented as the median and interquartile ranges for each case (as %EFRET). AD = Alzheimer’s disease; FAD = familial Alzheimer’s disease; VaD = vascular dementia; CAA = cerebral amyloid angiopathy; DLBD = diffuse Lewy body disease, a pathology diagnosis that parallels clinical dementia with Lewy bodies; ALB = amygdala predominant Lewy body disease; NA = not available.
PSEN1 familial Alzheimer’s disease mutations increase PSEN1 interaction with α-synuclein and α-synuclein membrane association
Our data suggests that PSEN1 familial Alzheimer’s disease mutations or ‘sporadic changes’ of PSEN1 cause a dysregulation of PSEN1–synuclein interactions and that an increase in this interaction is associated with advanced Lewy body pathology. To understand the functional consequences of this interaction, we used an in vitro cell culture system to measure the interaction of α-synuclein with PSEN1 familial Alzheimer’s disease mutations, delta E9 (dE9) and L166P. The dE9 and L166P PSEN1 mutations cause rapidly progressive early onset Alzheimer’s disease with onset of symptoms in the fourth decade of life. We used a previously characterized set of mouse embryonic fibroblast cell lines derived from a double knockout mouse with a loss of PSEN1 and PSEN2, and stably transfected with wild-type PSEN1, L166P PSEN1, or dE9 PSEN1 in four different cell lines (Herreman et al., 2000). We transiently transfected α-synuclein in these cell lines and pulled-down with α-synuclein antibody to assess the amount of PSEN1 that interacts with α-synuclein. The co-immunoprecipitation of full-length PSEN1 mutants by α-synuclein was increased by an average of 60.7% for L166P PSEN1 compared to full-length wild-type PSEN1 (quantified by ratio of immunoprecipitated PSEN1 to immunoprecipitated α-synuclein), whereas co-immunoprecipitation of C-terminal fragments of L166P and wild-type PSEN1 were not significantly different (Fig. 4A and B). Of note, the dE9 PSEN1 mutant lacks the endoproteolytic cleavage site and therefore remains in the full-length form (Fig. 4A and B). The amount of co-immunoprecipitated full-length dE9 PSEN1 was significantly higher than both wild-type and L166P, although it is not directly comparable to full-length L166P and wild-type PSEN1, which produce PSEN1 N/C-terminal fragments.
Figure 4.
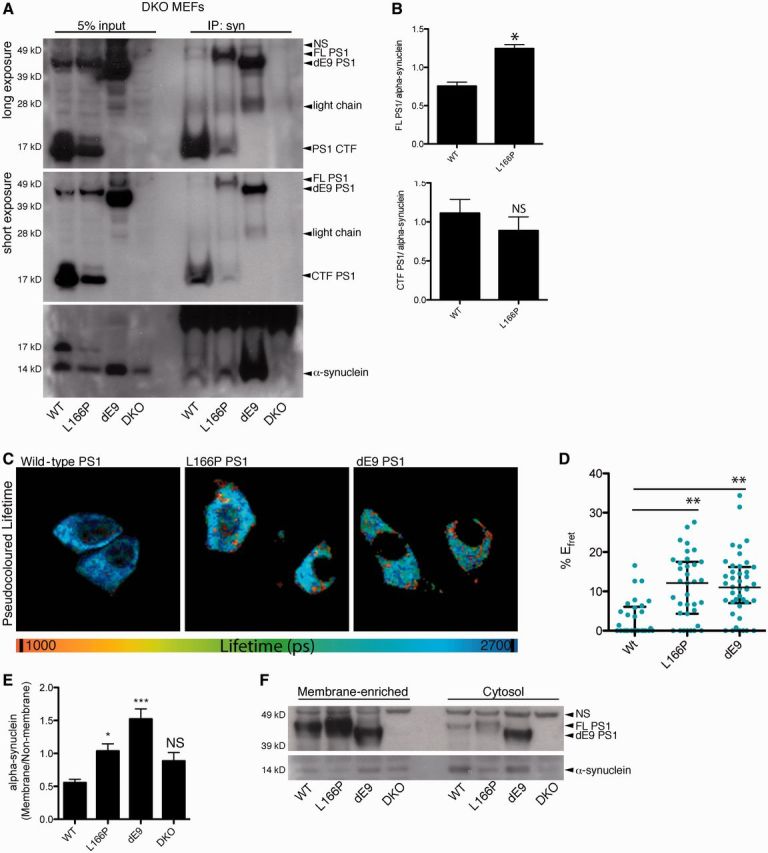
PSEN1 familial Alzheimer’s disease mutations, L166P and dE9, have increased interactions with α-synuclein in cell lines. (A) Representative image of SDS-PAGE showing immunoprecipitation (IP) of transiently expressed α-synuclein from cell lines expressing wild-type (WT), L166P, or dE9 PSEN1 α-synuclein co-immunoprecipitated more full-length (FL) L166P PSEN1 or full-length dE9 PSEN1 than full-length wild-type PSEN1. NS = non-specific band. (B) Quantification (mean and SEM) of co-immunoprecipitation of full-length PSEN1 or C terminal fragment PSEN1 (measured as the ratio of immunoprecipitateded α-synuclein to immunoprecipitated PSEN1) (n = 3, t-test, two-tailed, *P < 0.05, NS = not significant). (C) Representative pseudocolour lifetime image from donor and acceptor immunostained cells for wild-type (W), L166P, and dE9 cell lines. (D) Scatter plot (and median and IQR) for %EFRET showed increased interaction of α-synuclein and mutant PSEN1 compared with wild-type PSEN1 (n = 2, Kruskal Wallis, Dunns Multiple Comparison, ***P < 0.001). (E) Membrane fractionation of the PSEN1 mouse embryonic fibroblast cell lines showed increased α-synuclein association with membrane-enriched fractions (heavy membrane fraction) in the L166P and dE9 mutant PSEN1 lines compared to wild-type PSEN1. Quantification (mean and SEM) of α-synuclein was performed by ELISA from membrane (heavy membrane) and cytosol (cytosol and light membranes) fractions (n = 4, One-way ANOVA, Dunnett’s Multiple Comparison, *P < 0.05, ***P < 0.001, NS = not significant). (F) Representative image of SDS-PAGE for membrane separation. DKO = double knockout.
The increased co-immunoprecipitation of full-length PSEN1 by α-synuclein from cells that express dE9 and L166P PSEN1 indicate that both PSEN1 mutations stabilize the interaction of PSEN1 with α-synuclein. This observation was further confirmed by FLIM-FRET analysis (Fig. 4C and D), which is advantageous for detecting weak or transient interactions occurring at the time of fixation in intact cells.
The association of both α-synuclein and PSEN1 with multiple membrane compartments is highly characterized for both proteins. To determine if the increased interaction of PSEN1 mutations, with α-synuclein affected the subcellular localization or membrane association of α-synuclein we quantified α-synuclein levels by ELISA for membrane-enriched and cytosolic fractions in mutant, wild-type, and double knockout cell lines. Cell lines were transiently transfected with α-synuclein and then separated into cytosol and membrane-enriched fractions. α-synuclein was detected in both the cytosolic and membrane-enriched compartment, which was corroborated by our findings in mouse brain tissue with endogenous α-synuclein (data not shown) and previous publications (Lee et al., 2002). The mean ratio of membrane-associated α-synuclein to cytosolic α-synuclein was significantly higher for the L166P and dE9 cell lines compared to wild-type PSEN1 (Fig. 4E). Overall, the levels of α-synuclein were highest in the cytosolic (and light membrane) fraction for all cell lines, but there was a substantial increase in heavy membrane-association for the mutant lines, suggesting a possible role for PSEN1 in α-synuclein association or dissociation from membranes (Fig. 4E and F).
PSEN1 dysfunction increases α-synuclein accumulation independent of PSEN1 γ-secretase activity
To understand the functional consequences of increased α-synuclein–PSEN1 interactions we tested the effect of wild-type, L166P, dE9, and loss of PSEN1 on α-synuclein accumulation and oligomerization. Using two luciferase-based assays we can uniquely probe differences in both overall α-synuclein (full-length Gaussia luciferase-tagged α-synuclein, syn-luciferase) and oligomeric α-synuclein (α-synuclein tagged with N-terminal or C-terminal halves of G. luciferase, syn-luc1 and syn-luc2, respectively) (Remy and Michnick, 2004, 2006; Outeiro et al., 2008; Putcha et al., 2010). Syn-luciferase or syn-luc1 and syn-luc2, along with a transfection control enhanced GFP plasmid, were transfected into stable mouse embryonic fibroblast cell lines and levels of α-synuclein were measured relative to the transfection control (ratio of luciferase activity: GFP). The double knockout line and both mutant lines had significantly higher levels of total α-synuclein and oligomeric α-synuclein than the wild-type PSEN1 line (Fig. 5A and B). These data show that loss of PSEN1/PSEN2 and/or the presence of familial Alzheimer’s disease presenilin mutations result in increased α-synuclein accumulation.
Figure 5.

PSEN1 familial Alzheimer’s disease mutations cause increased α-synuclein accumulation. (A) PSEN1 mouse embryonic fibroblast cell lines were transfected with α-synuclein tagged with full-length lucifersase (syn-luc) and enhanced GFP as a transfection control. Luciferase and GFP levels for (A) and (B) were measured in each well for each condition and expressed as the mean and SEM of luciferase/GFP (n = 3, one-way ANOVA, Dunnett’s Multiple Comparison, ***P < 0.001). (B) PSEN1 mouse embryonic fibroblast cell lines were transfected with two α-synuclein constructs, one tagged with the N-terminus half of luciferase (syn-luc1) and one tagged with the C-terminus half of luciferase (syn-luc2), and enhanced GFP, to monitor complementation (oligomerization) of α-synuclein (n = 3, one-way ANOVA, Dunnett’s Multiple Comparison, **P < 0.01, ***P < 0.001). Mutant PSEN1 (L166P and dE9) or loss of PSEN1 increased the accumulation of total α-synuclein (A) and oligomeric α-synuclein (B) (as measured by luciferase activity/GFP levels). (C) Wild-type CHO cells were transfected with syn-luc1 and syn-luc2 and treated with γ-secretase inhibitors for 24 h. Inhibition of PSEN1 γ-secretase activity had no effect on the accumulation of oligomeric species (luciferase, mean and SEM) (n = 3, one-way ANOVA, Dunnett’s Multiple Comparison; NS = not significant). (D) CHO cells were transfected with either full-length, untagged wild-type human α-synuclein or empty vector pcDNA and amyloid-β40 and amyloid-β42 levels were measured by ELISA (mean and SD) from the conditioned media 24 h after tranfection. Overexpression of α-synuclein had no effect on the γ-secretase activity of PSEN1 measured indirectly by the amyloid-β42/ amyloid-β40 ratio [n = 2, t-test (on triplicate data sets), one-tailed, Dunnett’s Multiple Comparison, NS = not significant]. DKO = double knockout; DMSO = dimethylsulphoxide; DAPT = N-[(3,5-Difluorophenyl)acetyl]-L-alanyl-2-phenyl]glycine-1,1-dimethylethyl ester; CHO = Chinese hamster ovary.
Given the well-characterized γ-secretase activity of PSEN1, we tested if α-synuclein accumulation is affected by γ-secretase activity. Wild-type chinese hamster ovary cells were transfected with syn-luc1 and syn-luc2, and treated with known γ-secretase inhibitors: compound E, DAPT, and LY450139. Inhibition of γ-secretase activity had no significant effect on the accumulation of α-synuclein, as shown by comparable levels of luciferase activity in γ-secretase inhibitors and dimethyl sulphoxide vehicle-treated cells (Fig. 5C). Consistently, the presence of transiently transfected α-synuclein did not change the levels of amyloid-β42 or amyloid-β40 production in the media compared to cells transfected with empty vector, suggesting that the interaction of α-synuclein with PSEN1 does not affect γ-secretase activity (Fig. 5D).
Discussion
Overall, our data provide evidence for a novel interaction between PSEN1 and α-synuclein. This interaction occurs robustly in neuronal tissue at endogenous levels and is detectable in cell lines transfected with α-synuclein. Our immunoelectron microscopy data support this finding by visualization of PSEN1 and α-synuclein in close association in a number of different membrane compartments including synaptic vesicles, mitochondria, and plasma membrane. Quantitative FLIM-FRET studies in wild-type primary neuronal cultures show there is high a degree of PSEN1–α-synuclein interaction in both the cell body and along processes, with relatively higher levels of interaction in neuronal processes. Both PSEN1 and α-synuclein are known to associate with a number of membrane compartments including synaptic vesicles, endoplasmic reticulum membranes, Golgi, mitochondria and autophagosomes. PSEN1 is stably embedded in membranes, whereas α-synuclein is able to cycle on and off membranes by transitioning between a less ordered coil structure in aqueous environments to an α-helical structure upon membrane binding (Bodner et al., 2009; Vamvaca et al., 2009). It is believed that the ability of α-synuclein to transition between different conformations and different membrane states allows the protein to play an important role in the movement of membranes and the integration of different proteins into membranes. Future biophysical studies will be needed to further elucidate the nature of the PSEN1–synuclein interaction and determine how this interaction regulates each protein’s various functions and pathologically-relevant conformations, a source of continued debate (Lee et al., 2002).
As first steps towards understanding the function of this interaction, we used biochemical and genetic tool sets to determine if the interaction of PSEN1 and α-synuclein is relevant to human disease and to understand the early molecular consequences of this interaction intracellularly. From our FLIM-FRET studies in human brain tissue we discovered that the interaction of PSEN1 and α-synuclein is significantly increased in dementia with Lewy bodies and PSEN1 familial Alzheimer’s disease brain tissue compared with controls, with the highest overall interaction of PSEN1 and α-synuclein measured in dementia with Lewy bodies brain tissue. Our FLIM-FRET data suggest that an increase in the interaction of PSEN1 and α-synuclein generally increases with α-synuclein-related pathology with dementia with Lewy bodies having the most α-synuclein-related pathology and controls having the least. Alzheimer’s disease-relevant pathology, as measured by Braak staging and Thioflavin S plaque burden quantification in the amygdala, did not associate with the interaction of PSEN1 and α-synuclein. There was suggestive evidence of an association of Braak stage and PSEN1-synuclein interaction in controls, with less association in dementia with Lewy bodies tissue, but due to small sample size, this study is underpowered to robustly identify a true relationship. Overall, these data suggest an association between α-synuclein relevant pathology and the interaction of PSEN1 with α-synuclein, whereas Alzheimer’s disease-linked pathology has no conclusive influence on this interaction.
In addition to our human tissue studies, we show in vitro that the dE9 and L166P PSEN1 mutations known to cause familial Alzheimer’s disease have an increased interaction with α-synuclein. Through indirect luciferase-based measurements we show that these mutations significantly increase levels of both total and ‘oligomeric’ α-synuclein compared to wild-type PSEN1. Additionally, our findings suggest a possible role for PSEN1 in the targeting of α-synuclein to membranes. Given α-synuclein membrane association is increased in the presence PSEN1 mutations, we hypothesize that (i) through α-synuclein-PSEN1 interactions α-synuclein is sequestered into membrane compartments that have a lower rate of degradation due to accessibility of membrane-bound versus cytosolic α-synuclein; and/or (ii) direct and strong binding of mutant PSEN1 and α-synuclein inhibits effective release of α-synuclein to the proteasome, autophagosomes, or exocytosis. The interaction of PSEN1 and α-synuclein may also be indirectly modulated through an additional interacting partner or complex that has not yet been identified.
Although we have shown here that mutations in PSEN1 may alter interactions with α-synuclein, our data showing an increase in the strength of α-synuclein–PSEN1 interactions in the context of dementia with Lewy bodies is not explained by PSEN1 mutations. A plausible explanation for the parallel between PSEN1-linked familial Alzheimer’s disease and sporadic dementia with Lewy bodies is the sporadic occurrence of changes at the gene or protein level that phenocopy PSEN1 familial mutations. These changes could occur in PSEN1 or α-synuclein in response to decreases in quality controls systems occur in either PSEN1 or α-synuclein because of a decrease in protein-level quality control systems. These changes may include protein expression regulation, post-translational modifications, protein–protein interactions, protein folding, or membrane integration (Lionaki and Tavernarakis, 2013; Murshid et al., 2013; Tsakiri et al., 2013; Wang et al., 2013). Ageing-associated conformational alterations have been documented for PSEN1 (Wahlster et al., 2013). A number of familial mutations in PSEN1 are known to confer a pathogenic ‘closed’ conformation that alters γ-secretase activity (Berezovska et al., 2005); and we have recently shown that a subset of wild-type neurons in ageing and sporadic Alzheimer’s disease brain can also adopt such conformation (Wahlster et al., 2013). It is therefore possible, that structural alteration in PSEN1 is responsible for a stronger interaction with α-synuclein. We could speculate that this enhanced PSEN1-syn interaction can be normalized via allosteric modulation of PSEN1 conformation, a question for future studies.
In summary, our data provide the first evidence for the interaction of PSEN1 with α-synuclein suggesting a mechanistic convergence of Alzheimer’s disease-related dementias and Lewy body disease. Our findings are strongly supported by genetic data and the pathology seen in these diseases. Susceptibility and causal genes from both Alzheimer’s disease and Parkinson’s disease are linked to dementia with Lewy bodies whereas the associated pathologies are seen concomitantly in a number of diseases. By better understanding how PSEN1 affects α-synuclein accumulation we may develop future therapeutics that target this interaction specifically. This is important in light of recent studies showing that Lewy body density in cortical regions may be a stronger correlate of dementia in Parkinson’s disease and dementia with Lewy bodies than amyloid-β plaque load or neurofibrillary tangles (Horvath et al., 2013). This is supported by studies showing that even in the absence of Lewy body pathology soluble levels of α-synuclein increase in sporadic Alzheimer’s disease and that levels of soluble α-synuclein are a stronger correlate of cognitive impairment than soluble amyloid-β or tau protein levels (Larson et al., 2012). Future studies will need to address critical gaps in our understanding by explaining why these pathways and proteins converge in different ways in different diseases.
Supplementary Material
Acknowledgements
We thank Dr. Marian DiFiglia for use of the transmission electron microscope and Allyson Roe for assistance in tissue processing. We thank Anika Bronnhuber for mouse brain lysate preparation for crosslinking experiments.
Glossary
Abbreviations
- FLIM-FRET
fluorescence lifetime image microscopy based on Forster resonance energy transfer
- GST
glutathione S-transferase
Funding
This work was supported by the National Institute of Health (NIH) through grants 5T32AG023480-07 (through Beth Israel Deaconess) to A.R.W., The Mayo Clinic Udall Centre for Excellence in Parkinson’s disease research P50 NS072187 to D.W.D., NIH AG16574-15 to P.J.M., and NIH AG15379 to O.B.
Supplementary material
Supplementary material is available at Brain online.
References
- Berezovska O, Ramdya P, Skoch J, Wolfe MS, Bacskai BJ, et al. Amyloid precursor protein associates with a nicastrin-dependent docking site on the presenilin 1-gamma-secretase complex in cells demonstrated by fluorescence lifetime imaging. J Neurosci. 2003;23:4560–6. doi: 10.1523/JNEUROSCI.23-11-04560.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezovska O, Lleo A, Herl LD, Frosch MP, Stern EA, Bacskai BJ, et al. Familial Alzheimer's disease presenilin 1 mutations cause alterations in the conformation of presenilin and interactions with amyloid precursor protein. J Neurosci. 2005;25:3009–17. doi: 10.1523/JNEUROSCI.0364-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodner CR, Dobson CM, Bax A. Multiple tight phospholipid-binding modes of alpha-synuclein revealed by solution NMR spectroscopy. J Mol Biol. 2009;390:775–90. doi: 10.1016/j.jmb.2009.05.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner LT, Tsuang DW, Cherrier MM, Eugenio CJ, Du Jennifer Q, Steinbart EJ, et al. Familial dementia with Lewy bodies with an atypical clinical presentation. J Geriatr Psychiatry Neurol. 2003;16:59–64. doi: 10.1177/0891988702250585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brett FM, Henson C, Staunton H. Familial diffuse Lewy body disease, eye movement abnormalities, and distribution of pathology. Arch Neurol. 2002;59:464–7. doi: 10.1001/archneur.59.3.464. [DOI] [PubMed] [Google Scholar]
- Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–7. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–96. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Chen RH, Wislet-Gendebien S, Samuel F, Visanji NP, Zhang G, Marsilio D, et al. alpha-Synuclein membrane association is regulated by the Rab3a recycling machinery and presynaptic activity. J Biol Chem. 2013;288:7438–49. doi: 10.1074/jbc.M112.439497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BK, Choi MG, Kim JY, Yang Y, Lai Y, Kweon DH, et al. Large alpha-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc Natl Acad Sci USA. 2013;110:4087–92. doi: 10.1073/pnas.1218424110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 2006;313:324–8. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denson MA, Wszolek ZK, Pfeiffer RF, Wszolek EK, Paschall TM, McComb RD. Familial parkinsonism, dementia, and Lewy body disease: study of family G. Ann Neurol. 1997;42:638–43. doi: 10.1002/ana.410420415. [DOI] [PubMed] [Google Scholar]
- Efthimiopoulos S, Floor E, Georgakopoulos A, Shioi J, Cui W, Yasothornsrikul S, et al. Enrichment of presenilin 1 peptides in neuronal large dense-core and somatodendritic clathrin-coated vesicles. J Neurochem. 1998;71:2365–72. doi: 10.1046/j.1471-4159.1998.71062365.x. [DOI] [PubMed] [Google Scholar]
- Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH. Lipid rafts mediate the synaptic localization of alpha-synuclein. J Neurosci. 2004;24:6715–23. doi: 10.1523/JNEUROSCI.1594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler AD, Bevis BJ, Shorter J, Strathearn KE, Hamamichi S, Su LJ, et al. The Parkinson's disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc Natl Acad Sci USA. 2008;105:145–50. doi: 10.1073/pnas.0710685105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KN, Demuro A, Akbari Y, Hitt BD, Smith IF, Parker I, et al. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J Gen Physiol. 2008;132:i1. doi: 10.1085/JGP1322OIA1. [DOI] [PubMed] [Google Scholar]
- Guardia-Laguarta C, Area-Gomez E, Rub C, Liu Y, Magrane J, Becker D, et al. alpha-Synuclein is localized to mitochondria-associated ER membranes. J Neurosci. 2014;34:249–59. doi: 10.1523/JNEUROSCI.2507-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson CA, Frykman S, Farmery MR, Tjernberg LO, Nilsberth C, Pursglove SE, et al. Nicastrin, presenilin, APH-1, and PEN-2 form active gamma-secretase complexes in mitochondria. J. Biol Chem. 2004;279:51654–60. doi: 10.1074/jbc.M404500200. [DOI] [PubMed] [Google Scholar]
- Herreman A, Serneels L, Annaert W, Collen D, Schoonjans L, De Strooper B. Total inactivation of gamma-secretase activity in presenilin-deficient embryonic stem cells. Nat Cell Biol. 2000;2:461–2. doi: 10.1038/35017105. [DOI] [PubMed] [Google Scholar]
- Horvath J, Herrmann FR, Burkhard PR, Bouras C, Kovari E. Neuropathology of dementia in a large cohort of patients with Parkinson's disease. Parkinsonism Relat Disord. 2013;19:864–8. doi: 10.1016/j.parkreldis.2013.05.010. [DOI] [PubMed] [Google Scholar]
- Ishikawa A, Piao YS, Miyashita A, Kuwano R, Onodera O, Ohtake H, et al. A mutant PSEN1 causes dementia with Lewy bodies and variant Alzheimer's disease. Ann Neurol. 2005;57:429–34. doi: 10.1002/ana.20393. [DOI] [PubMed] [Google Scholar]
- Larson ME, Sherman MA, Greimel S, Kuskowski M, Schneider JA, Bennett DA, et al. Soluble alpha-synuclein is a novel modulator of Alzheimer's disease pathophysiology. J Neurosci. 2012;32:10253–66. doi: 10.1523/JNEUROSCI.0581-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lee HJ, Choi C, Lee SJ. Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J Biol Chem. 2002;277:671–8. doi: 10.1074/jbc.M107045200. [DOI] [PubMed] [Google Scholar]
- Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–58. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leftin A, Job C, Beyer K, Brown MF. Solid-State C NMR reveals annealing of raft-like membranes containing cholesterol by the intrinsically disordered protein alpha-synuclein. J Mol Biol. 2013;425:2973–87. doi: 10.1016/j.jmb.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leverenz JB, Fishel MA, Peskind ER, Montine TJ, Nochlin D, Steinbart E, et al. Lewy body pathology in familial Alzheimer disease: evidence for disease- and mutation-specific pathologic phenotype. Arch Neurol. 2006;63:370–6. doi: 10.1001/archneur.63.3.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionaki E, Tavernarakis N. Oxidative stress and mitochondrial protein quality control in aging. J Proteomics. 2013;92:181–94. doi: 10.1016/j.jprot.2013.03.022. [DOI] [PubMed] [Google Scholar]
- Lleo A, Berezovska O, Herl L, Raju S, Deng A, Bacskai BJ, et al. Nonsteroidal anti-inflammatory drugs lower Abeta42 and change presenilin 1 conformation. Nat Med. 2004;10:1065–6. doi: 10.1038/nm1112. [DOI] [PubMed] [Google Scholar]
- McKeith I, Mintzer J, Aarsland D, Burn D, Chiu H, Cohen-Mansfield J, et al. Dementia with Lewy bodies. Lancet Neurol. 2004;3:19–28. doi: 10.1016/s1474-4422(03)00619-7. [DOI] [PubMed] [Google Scholar]
- Meeus B, Verstraeten A, Crosiers D, Engelborghs S, Van den Broeck M, Mattheijssens M, et al. DLB and PDD: a role for mutations in dementia and Parkinson disease genes? Neurobiol Aging. 2012;33:629 e5–629 e18. doi: 10.1016/j.neurobiolaging.2011.10.014. [DOI] [PubMed] [Google Scholar]
- Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–20. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshid A, Eguchi T, Calderwood SK. Stress proteins in aging and life span. Int J Hyperthermia. 2013;29:442–7. doi: 10.3109/02656736.2013.798873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nervi A, Reitz C, Tang MX, Santana V, Piriz A, Reyes D, et al. Familial aggregation of dementia with Lewy bodies. Arch Neurol. 2011;68:90–3. doi: 10.1001/archneurol.2010.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohara K, Takauchi S, Kokai M, Morimura Y, Nakajima T, Morita Y. Familial dementia with Lewy bodies (DLB) Clin Neuropathol. 1999;18:232–9. [PubMed] [Google Scholar]
- Ohta K, Mizuno A, Ueda M, Li S, Suzuki Y, Hida Y, et al. Autophagy impairment stimulates PS1 expression and gamma-secretase activity. Autophagy. 2010;6:345–52. doi: 10.4161/auto.6.3.11228. [DOI] [PubMed] [Google Scholar]
- Outeiro TF, Putcha P, Tetzlaff JE, Spoelgen R, Koker M, Carvalho F, et al. Formation of toxic oligomeric alpha-synuclein species in living cells. PloS One. 2008;3:e1867. doi: 10.1371/journal.pone.0001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pranke IM, Morello V, Bigay J, Gibson K, Verbavatz JM, Antonny B, et al. alpha-Synuclein and ALPS motifs are membrane curvature sensors whose contrasting chemistry mediates selective vesicle binding. J Cell Biol. 2011;194:89–103. doi: 10.1083/jcb.201011118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha P, Danzer KM, Kranich LR, Scott A, Silinski M, Mabbett S, et al. Brain-permeable small-molecule inhibitors of Hsp90 prevent alpha-synuclein oligomer formation and rescue alpha-synuclein-induced toxicity. J Pharmacol Exper Therapeutics. 2010;332:849–57. doi: 10.1124/jpet.109.158436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy I, Michnick SW. A cDNA library functional screening strategy based on fluorescent protein complementation assays to identify novel components of signaling pathways. Methods. 2004;32:381–8. doi: 10.1016/j.ymeth.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Remy I, Michnick SW. A highly sensitive protein-protein interaction assay based on Gaussia luciferase. Nat Methods. 2006;3:977–9. doi: 10.1038/nmeth979. [DOI] [PubMed] [Google Scholar]
- Scott D, Roy S. alpha-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J Neurosci. 2012;32:10129–35. doi: 10.1523/JNEUROSCI.0535-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AL, Friedman DB, Yu H, Carnahan RH, Reynolds AB. ReCLIP (reversible cross-link immuno-precipitation): an efficient method for interrogation of labile protein complexes. PloS One. 2011;6:e16206. doi: 10.1371/journal.pone.0016206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider BJ, Norton J, Coats MA, Chakraverty S, Hou CE, Jervis R, et al. Novel presenilin 1 mutation (S170F) causing Alzheimer disease with Lewy bodies in the third decade of life. Arch Neurol. 2005;62:1821–30. doi: 10.1001/archneur.62.12.1821. [DOI] [PubMed] [Google Scholar]
- Torp R, Ottersen OP, Cotman CW, Head E. Identification of neuronal plasma membrane microdomains that colocalize beta-amyloid and presenilin: implications for beta-amyloid precursor protein processing. Neuroscience. 2003;120:291–300. doi: 10.1016/s0306-4522(03)00320-8. [DOI] [PubMed] [Google Scholar]
- Tsakiri EN, Sykiotis GP, Papassideri IS, Gorgoulis VG, Bohmann D, Trougakos IP. Differential regulation of proteasome functionality in reproductive vs. somatic tissues of Drosophila during aging or oxidative stress. FASEB J. 2013;27:2407–20. doi: 10.1096/fj.12-221408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuang DW, DiGiacomo L, Bird TD. Familial occurrence of dementia with Lewy bodies. Am J Geriatr Psychiatry. 2004;12:179–88. [PMC free article] [PubMed] [Google Scholar]
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, et al. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell. 2006;126:981–93. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vamvaca K, Volles MJ, Lansbury PT., Jr The first N-terminal amino acids of alpha-synuclein are essential for alpha-helical structure formation in vitro and membrane binding in yeast. J Mol Biol. 2009;389:413–24. doi: 10.1016/j.jmb.2009.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlster L, Arimon M, Nasser-Ghodsi N, Post KL, Serrano-Pozo A, Uemura K, et al. Presenilin-1 adopts pathogenic conformation in normal aging and in sporadic Alzheimer's disease. Acta Neuropathol. 2013;125:187–99. doi: 10.1007/s00401-012-1065-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CH, Wu SB, Wu YT, Wei YH. Oxidative stress response elicited by mitochondrial dysfunction: Implication in the pathophysiology of aging. Exp Biol Med. 2013;238:450–60. doi: 10.1177/1535370213493069. [DOI] [PubMed] [Google Scholar]
- Waters CH, Miller CA. Autosomal dominant Lewy body parkinsonism in a four-generation family. Ann Neurol. 1994;35:59–64. doi: 10.1002/ana.410350110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.