

Abstract
Bovine viral diarrhea virus (BVDV) is the prototype Pestivirus. BVDV infection is distributed worldwide and causes serious problems for the livestock industry. The thiosemicarbazone of 5,6-dimethoxy-1-indanone (TSC) is a non-nucleoside polymerase inhibitor (NNI) of BVDV. All TSC-resistant BVDV variants (BVDV-TSCr T1–5) present an N264D mutation in the NS5B gene (RdRp) whereas the variant BVDV-TSCr T1 also presents an NS5B A392E mutation. In the present study, we carried out twenty passages of BVDV-TSCr T1–5 in MDBK cells in the absence of TSC to evaluate the stability of the resistance. The viral populations obtained (BVDV R1–5) remained resistant to the antiviral compound and conserved the mutations in NS5B associated with this phenotype. Along the passages, BVDV R2, R3 and R5 presented a delay in the production of cytopathic effect that correlated with a decrease in cell apoptosis and intracellular accumulation of viral RNA. The complete genome sequences that encode for NS2 to NS5B, Npro and Erns were analyzed. Additional mutations were detected in the NS5B of BVDV R1, R3 and R4. In both BVDV R2 and R3, most of the mutations found were localized in NS5A, whereas in BVDV R5, the only mutation fixed was NS5A V177A. These results suggest that mutations in NS5A could alter BVDV cytopathogenicity. In conclusion, the stability of the resistance to TSC may be due to the fixation of different compensatory mutations in each BVDV-TSCr. During their replication in a TSC-free medium, some virus populations presented a kind of interaction with the host cell that resembled a persistent infection: decreased cytopathogenicity and viral genome synthesis. This is the first report on the stability of antiviral resistance and on the evolution of NNI-resistant BVDV variants. The results obtained for BVDV-TSCr could also be applied for other NNIs.
Introduction
Bovine viral diarrhea virus (BVDV), along with classical swine fever virus and border disease virus, is a member of the genus Pestivirus. This genus belongs to the family Flaviviridae, which also includes the genera Hepacivirus (hepatitis C virus) and Flavivirus (yellow fever virus, Dengue fever virus and West Nile virus).
BVDV infection is distributed worldwide and tends to be endemic in many countries [1], [2]. In Argentina, the prevalence of BVDV antibodies in adult cattle is around 70% [3], [4]. BVDV infections lead to a reduction in milk production, lower conception rates, respiratory disorders and death of the animals that acquire the acute infection, thus causing serious economic losses for the livestock industry. Moreover, fetal infection leads to persistently infected cattle, which are generally smaller, are more susceptible to other infections and eventually develop a lethal mucosal disease.
BVDV is an enveloped single-stranded (+) RNA virus. Its genomic RNA is about 12.5 kb long and consists of a single open reading frame (ORF) flanked by 5′ and 3′ non-translated regions (NTRs). The ORF encodes a polyprotein of approximately 3,900 amino acids that is co- and post-translationally processed to mature viral proteins by cellular and viral proteases. The viral proteins are sequentially designated Npro, C, Erns, E1, E2, p7, NS2, NS3, NS4A, NS4B, NS5A and NS5B. The autoprotease Npro is a non-structural (NS) protein that plays a role in blocking IFN-α/β induction [5]–[8]. The capsid protein (C) is followed by three virion glycoproteins: Erns, E1, and E2. Erns encodes an RNase that is secreted in nonvirion forms [9] and targets extracellular RNA, a major viral signal that triggers IFN synthesis [10], [11]. The rest of the polyprotein is processed to NS proteins, of which only NS3 through NS5B are required for RNA replication [12], [13]. NS2, together with the amino terminus of NS3, functions as an autoprotease that cleaves the NS2-NS3 junction of the polyprotein. This cleavage is required for RNA replication and is linked to BVDV cytopathogenicity and pathogenesis [14], [15]. NS3 is a multifunctional protein with a helicase/nucleoside triphosphatase and serine protease activity responsible for all downstream polyprotein cleavages [16]–[19]. NS4A is tightly associated with NS3 and serves both as a cofactor for NS3 protease activity and as a tether that localizes NS3 to membranes [17], [20]. NS4B is believed to function as an integral membrane scaffold upon which the replicase complex (RC) assembles [21]. NS5A is a phosphoprotein that plays an essential role in BVDV replication and pathogenesis [22]–[24]. Finally, NS5B is the viral RNA-dependent RNA polymerase that catalyzes viral RNA synthesis [25]–[29].
BVDV isolates are categorized into two biotypes according to their effect on cultured cells: noncytopathic (ncp) isolates, which infect permissive host cells without causing cell death, and cytopathic (cp) isolates, which produce rapid cytopathic effects (CPE) and kill cells [30]. Only ncp isolates are able to establish the persistent infection. This difference is associated with distinct interactions between each biotype and the host innate immune response against the viral infection, which is active early during gestation. Cp biotypes emerge from ncp biotypes exclusively in persistently infected animals and are invariably associated with the mucosal disease [12], [31]. The induction of apoptosis plays a significant role in the pathology of the cp isolates both in infected cell cultures and in the clinical manifestations of the mucosal disease [32]–[35].
The thiosemicarbazone of 5,6-dimethoxy-1-indanone (TSC) is a non-nucleoside polymerase inhibitor of BVDV (EC50 1.8±0.6.µM) [36]–[38]. In a previous study, we selected five independent populations of TSC-resistant BVDV (BVDV-TSCr; T1–5) (TSC EC50>80.0 µM). All of them carry an NS5B N264D mutation, whereas BVDV-TSCr T1 also shows an NS5B A392E mutation [38].
Studies on the resistance to an antiviral agent are highly important for the development and therapeutic application of such antiviral agent. Given that the impact of resistance is difficult to predict, it is important to evaluate not only the emergence of resistance but also its stability and the effect of the associated mutations on the viral replicative fitness in an antiviral-free environment. Therefore, the aim of the present work was to evaluate the stability of the resistance to TSC. To this end, we carried out 20 passages of BVDV-TSCr T1–5 in the absence of TSC. We also describe the molecular and biological characterization of the viral populations obtained, in terms of infectious virus production and cytopathogenesis.
Materials and Methods
Cells and virus
Madin Darby bovine kidney cells (MDBK NBL-1; ATCC CCL-22) were grown in Eagle's Minimal Essential Medium (EMEM), supplemented with 10% γ irradiated fetal bovine serum (FBS, PAA Laboratories, Pasching, Austria) (growing medium). BVDV type 1 NADL strain, cytopathic biotype (BVDV-1, ATCC VR 534) was provided by Dr Laura Weber, INTA Castelar, Argentina. Wild type (wt) BVDV p0 was obtained after three successive steps of biological cloning of BVDV NADL in MDBK cells. TSC-resistant BVDV (BVDV-TSCr T1–5) were obtained from wt BVDV p0 after ten passages in MDBK cells with increasing concentrations of TSC [38].
Passages of BVDV-TSCr in the absence of TSC
MDBK cells were infected with BVDV-TSCr T1–5 at a multiplicity of infection (MOI) of 0.01. Cell cultures were incubated in infection medium (infection medium: EMEM supplemented with 2.5% Donor Horse Serum -DHS- Gibco) in a 5% CO2 incubator at 37°C until cell monolayers presented 80–90% of viral CPE. The supernatant was collected and clarified by centrifugation for 10 min at 1,000×g at 4°C. After titration of the virus in MDBK cells, the virus was used to infect new cell monolayers to generate the next passage. This procedure was repeated twenty times, and the virus obtained from each BVDV-TSCr T1–5 was named BVDV R1–5.
Plaque reduction assay for BVDV
To evaluate the antiviral activity of TSC against BVDV R1–5, MDBK cells were seeded in growing medium at a density of 1.3×105 cells per well in a 24-well plate and incubated for 24 h at 37°C in a 5% CO2 incubator. Then, cells were infected with approximately 100 plaque-forming units (PFUs) of BVDV R1–5 or wt BVDV p0. Following 1 h of adsorption at 37°C, the inoculum was removed, cell monolayers were washed twice with phosphate-buffered saline (PBS), and 1 ml of overlay medium (2.5% DHS and 0.8% methylcellulose) containing 80 µM of TSC was added in each well. Mock-infected cells with and without TSC, and infected cells without TSC were included as controls. After 72 h of incubation at 37°C with 5% CO2, cells were fixed with formalin 10%, stained with 0.75% crystal violet, and the viral plaques counted.
Apoptosis measurements
MDBK cells were seeded in a 24-well plate as described above. After 24 h in a 5% CO2 incubator at 37°C, cells were infected with wt BVDV p0 or R1–5 (MOI 0.01). Following 1 h of adsorption at 37°C, the inoculum was removed, cell monolayers were washed twice with PBS and 1 ml of infection medium was added in each well. Mock-infected cells were included as controls. After 48 h of incubation at 37°C with 5% CO2, apoptosis was measured in infected and mock-infected cell cultures.
For apoptosis measurements, PE Annexin V Apoptosis Detection Kit I (BD Pharmigen, NJ, USA) was used following the manufacturer's instructions. Briefly, supernatants from infected and mock-infected cells were harvested and cells in the monolayer were collected by trypsinization and resuspended in 0.5 ml of infection medium. Adherent and floating cells were pooled and pelleted by centrifugation at 500×g for 10 min. Pelleted cells were washed twice with 0.5 ml of cold PBS. Before the second centrifugation (500×g, 10 min), cells were counted in a Neubauer chamber. To obtain the dead-cell control for apoptosis measurements, one mock-infected cell suspension was heated at 65°C for 30 min. Then, cells were resuspended in binding buffer (4×106 cells/ml) and 100 µl of each cell suspension was stained with 5 µl Annexin V (conjugated with Phycoerythrin- PE-) and 5 µl 7-Amino-Actinomycin (7AAD) and incubated for 15 min at room temperature in the dark. Finally, 400 µl of binding buffer was added and the cells were analyzed by flow cytometry (PARTEC, PAS-III). PE Annexin V and 7AAD fluorescence was collected at FL 2 (550–600 nm) and FL4 (640–700 nm) channels, respectively. Histograms and density plots of 20,000 events were obtained and analyzed with WinMDI 2.9 software [39].
At 48 h p.i. some infected cultures already presented viral CPE, which is caused by cell apoptosis. The number of cells that would have died by this mechanism and could no longer be detected was estimated by the difference between the number of cells counted in a Neubauer chamber in the mock-infected cultures (MIC) and those counted in the infected ones (IC) (A: N ° undetected dead cells = N° cells MIC − N° cells IC). Then, the number of apoptotic cells in each infection (i.e. in the remaining adherent and floating cells) was quantified by AnV binding and flow cytometry as stated above (B: N° apoptotic cells = % AnVpos × N° cells IC/100). Finally, the total cells that would die by each viral infection was estimated as % dead cells (normalized to uninfected) = (A+B) × 100/N° cells MIC.
Multiple-step growth curve
To quantify the infectious virus produced by BVDV R1–5 in the absence of TSC, MDBK cells were seeded in a 24-well plate as stated above and infected with BVDV R1–5 or wt BVDV p0 (MOI 0.01). Following 1 h of adsorption at 37°C, the inoculum was removed, cell monolayers were washed twice with PBS, and 1 ml of infection medium was added to each well. The culture supernatant was collected every 24 h for 7 days and clarified by centrifugation for 10 min at 1,000×g at 4°C. The infectious virus in each sample was quantified by the PFU method in MDBK cells.
Given that BVDV R4 and R5 showed CPE and levels of cell apoptosis similar to those of BVDV R1 and R2, respectively (see results section), they were not included in the following assays (unless otherwise indicated).
Fluorescent microscopy
MDBK cells were seeded in a 24-well plate at a density of 1.3 ×105 cells per well containing a circular slide cover of 10 mm in diameter. After 24 h in a 5% CO2 incubator at 37°C, cells were infected with BVDV R1, R2, R3 or wt BVDV p0 (MOI 0.01). Following 1 h at 37°C, the inoculum was removed, cell monolayers were washed twice with PBS, and 1 ml of infection medium was added to each well. At 24, 48 and 72 h p.i., the cell culture medium was removed and cell monolayers were fixed with 4% formaldehyde/PBS for 15 min at room temperature. After washing three times with PBS, cultures were incubated with 0.5 ml of blocking buffer (10% Bovine seralbumin -BSA-; 0.3% Triton X-100 in PBS) for 30 min. Then, slide covers were removed and incubated overnight at 4°C with 30 µl of anti-BVDV NS3 Monoclonal Antibody (diluted 1∶1000 in PBS 0.1% Triton X-100; 1% BSA), kindly provided by Dr Alejandra Capozzo (Instituto de Virología, INTA, Castelar, Buenos Aires, Argentina). Slide covers were washed three times with PBS and incubated protected from light for 2 h at room temperature with Alexa Fluor 488 Goat Anti-Mouse antibody (Invitrogen) (diluted 1∶1000 in PBS 0.1% Triton X-100; 1% BSA). Afterwards, cells were washed three times with PBS and the nucleus stained with 300 µl of DAPI (Invitrogen) (dilution 1∶100) for 5 min. The slides were mounted with FluorSave (Calbiochem) and the cells were observed with a Nikon Eclipse 80i microscope with a 20× objective (numerical aperture 0.40). The images were captured with a digital camera (Nikon Digital Sight DS-Qi1Mc) and processed using ImageJ software.
Viral RNA production
MDBK cells were seeded in a 24-well plate as stated above. After 24 h in a 5% CO2 incubator at 37°C, cells were infected with BVDV R1, R2, R3 or wt BVDV p0 (MOI 1). Following 1 h of adsorption on ice, the inoculum was removed, cell monolayers were washed twice with PBS, and 1 ml of infection medium was added to each well. After 24 h of incubation at 37°C, intracellular RNA was extracted with TRIzol reagent (Gibco-BRL, CA, USA). RNA was retrotranscribed with random hexamers (Biodynamics) at 37°C for 90 min, using 0.5 U/μl M-MLV reverse transcriptase (Promega, Madison, WI, USA), and quantified by Real-time PCR.
Real-time PCR
To measure the intracellular viral RNA (ivRNA), a 108-bp fragment from the 5′NTR was amplified with the following oligonucleotides: 5′NTR forward 5′GAGGCTAGCCATGCCCTTAGT3′ and 5′NTR reverse 5′TCGAACCACTGACGACTACCCT3′. The reaction was carried out in 2X Power SYBR Green PCR Master Mix (Applied Biosystems, UK) in an ABI 7500 apparatus (Applied Biosystems), using the following experimental run protocol: 10 min of activation at 95°C, followed by 45 cycles of amplification and quantification (15 sec at 95.0°C, 1 min at 60.0°C and 35 sec at 78.5°C) during which the SYBR GREEN signal was measured. To normalize target amplification data, a 91-bp fragment of bovine β-actin mRNA was amplified simultaneously in each sample and used as endogenous control (forward oligonucleotide: 5′CCCACACGGTGCCCATCTAT3′ and reverse oligonucleotide: 5′ CCACGCTCCGTGAGGATCTTC3′). Each sample was tested in triplicate and both positive and negative controls were included.
Data were analyzed by means of the relative standard curve method [40] to quantify the ivRNA at 24 h p.i. in the cell cultures infected with BVDV R1, R2 and R3 in relation with those infected with wt BVDV p0.
Sequencing
Total RNA was extracted with TRIzol reagent (Gibco-BRL) from wt BVDV p0, BVDV-TSCr T1–5 and BVDV R1–5 virus suspensions obtained from clarified supernatants of infected cells. The RNA obtained was denatured at 70°C for 5 min and primed with the designed specific oligonucleotides (Table 1). The reverse transcription reaction was performed at 42°C for 90 min, using 10 U/μl of ArrayScript™ Reverse Transcriptase (Ambion, Inc.). The PCR fragments that cover the complete coding region of all the non-structural proteins (Npro, NS2-NS5B) and the glycoprotein Erns were obtained using specific oligonucleotides (Table 1) by PCR amplification with 0.025 U/μl of PFU DNA polymerase (Promega). Both DNA strands were sequenced using an ABI ABI3130XL sequencer 131 (Applied Biosystems; Unidad de Genómica, Instituto de Biotecnología, INTA, Castelar, Buenos Aires, Argentina). The sequences were aligned using ClustalX 1.83 [41], edited with BioEdit v 7.0.9.0 [42] and compared with the BVDV strain NADL (p0 and GenBank accession no. M31182.1; AJI33738.1; NP_776270.1; NP_776271.1; NC_001461.1).
Table 1. Oligonucleotides used for retrotranscription (RT) reactions and PCR amplifications.
| Primer | Sequence (5′-3′) | Position in the genome |
| Npro region | ||
| PR S1 | GCCCACTGTATTGCTAC | 344–360 |
| PR AS1 | CCCCTTTCTAGTCTGTCG | 937–954 |
| Erns region | ||
| PR S2 | AGGAATCACGCAAGAAACTG | 1,116–1,135 |
| PR AS2 | GTCACTAGCCCTATCATACC | 1,539–1,558 |
| PR S3 | TTGAACCCTGGATTCTAGTC | 1,455–1,474 |
| PR AS3 | AATTTCCCAGGGCCGAC | 1,970–1,986 |
| NS2-NS3 region | ||
| RT1 | CTTCCCCTTCCTTATAGAGAG | 8,261–8,281 |
| PR S4 | AATACTGGTTTGACCTGGAG | 3,435–3,454 |
| PR AS4 | CGCCAGTTCTTTCAGTTCAGT | 7,646–7,666 |
| SR S5 | CGCTGAGTCCATATTAGTGGT | 3,481–3,501 |
| SR AS5 | ACTGAACTTATCCCGCCT | 5,605–5,622 |
| SR S6 | CTGCCGTGTGTAAGAAGAT | 5,427–5,445 |
| SR AS6 | GTCTTCTAGTCTCTGGTCCT | 7,575–7,594 |
| NS4A-NS4B region | ||
| RT2 | GTAGTGTTAGCTTGAGGTAGATA | 12,477–12,498 |
| PR S7 | TAGACTGGCCTGATCCTG | 7,401–7,418 |
| PR AS7 | GTTGGCAATCTGATCCTCT | 8,826–8,844 |
| NS5A-NS5B region | ||
| RT2 | GTAGTGTTAGCTTGAGGTAGATA | 12,477–12,498 |
| PR S8 | CTGTATGGGGTTTACTACAAAGG | 8,567–8,589 |
| PR AS8 | TCTTCTTGAGGTGGACGG | 12,417–12,434 |
| SR S9 | CTGTATGGGGTTTACTACAAAGG | 8,567–8,589 |
| SR AS9 | TTGGTAAGCTGATGCCATGTG | 10,295–10,315 |
| SR S10 | AGGAGATGTTGGAGAGGTAA | 10,090–10,109 |
| SR AS10 | TCTTCTTGAGGTGGACGG | 12,417–12,434 |
Sequences and positions in the genome of BVDV NADL of the oligonucleotides designed and used for RT reactions and PCR amplifications of Npro, glycoprotein Erns and non-structural proteins coding region. Positions in the BVDV genome are according to BVDV NADL genome sequence GenBank accession no AJ133738.1.
Statistical analysis
The results were analyzed by Student's t test using InfoStat version 2011 [43]. A statistically significant difference was considered if p values were less than 0.050.
Results
The resistance to TSC is conserved after 20 passages
BVDV R1–5 were obtained after 20 passages of BVDV-TSCr T1–5 in the absence of TSC. BVDV R1–4 were not significantly inhibited by TSC, whereas wt BVDV p0 was completely inhibited (TSC vs untreated: R1 p = 0.560; R2 p = 0.054; R3 p = 0.449; R4 p = 0.382; p0 p = 0.003) (Figure 1). However, with the number of PFUs analyzed, BVDV R5 showed a reduction in the number of viral plaques of approximately 44%, suggesting a slight decrease in drug resistance (TSC vs untreated: R5 p = 0.002).
Figure 1. Antiviral activity of TSC against BVDV R1–5.
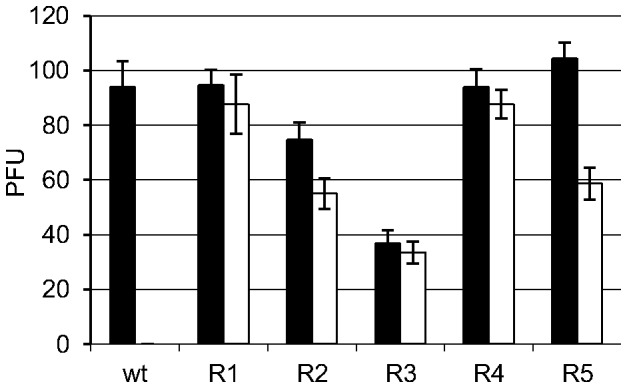
BVDV-TSCr T1–5 were propagated in MDBK cells in the absence of TSC during 20 passages. The antiviral activity of 80 µM of TSC against the viral populations obtained (BVDV R1–R5) and wt BVDV p0 was evaluated by plaque reduction assays. White and black bars indicate the number of viral plaque forming units (PFUs) formed in the presence or in the absence of TSC, respectively. (TSC vs untreated: R1 p = 0.560; R2 p = 0.054; R3 p = 0.449; R4 p = 0.382; R5 p = 0.002; p0 p = 0.003).
A delay in CPE production is observed in some viral populations during passages
The infection of MDBK cells with wt BVDV NADL cp at a MOI of 0.01 required 48 h of incubation at 37°C to reach 80–90% of CPE in the cell monolayer. However, along the passages of BVDV-TSCr T1–5 in the same conditions, some viral populations showed a delay in producing CPE. To reach 80–90% of CPE, the infections with BVDV-TSCr T2 and T5 required 72 h of incubation from the 1st to the 3rd passage and then 120 h of incubation from the 4th to the 20th passage. Similarly, BVDV-TSCr T3 required 120 h of incubation from the 13th to the 20th passage. This delay in producing CPE was also observed in the growth of viral plaques when the infected cultures were incubated in semi-solid medium. BVDV R2, R3, and R5 needed 72–96 additional hours of incubation to properly exhibit the viral plaques.
The delay in producing CPE is correlated with a reduced induction of apoptosis in MDBK cells
BVDV cp isolates kill cells by triggering apoptosis [30], [32], [44]. In accordance with the microscopic observations of the viral CPE, the infection of MDBK cells with BVDV R1 or R4 produced levels of apoptosis similar to those produced by wt BVDV p0 (13.1%, 10.8%, and 11.0% of PE Annexin-positive cells, respectively), whereas the infections with BVDV R2 and R5 produced lower level of apoptosis (5.7% and 5.5% of PE Annexin-positive cells, respectively). BVDV R3 showed an intermediate value (9.1% of PE Annexin-positive cells) (Figure 2).
Figure 2. Apoptosis measurements.

MDBK cells were infected with wt BVDV p0 or BVDV R1–5 (MOI 0.01) and at 48 h p.i. apoptotic cells were measured by incorporation of PE Annexin V and 7AAD dyes and analyzed by flow cytometry. Mock-infected cultures were added as control. Representative density plots (20,000 events) obtained with WinMDI software are displayed. Quadrants delimitating positive (pos) and negative (neg) events are shown and the percentage of events in each quadrant is indicated. The populations of PE Annexin V−/7AAD−, PE Annexin V+/7AAD− and PE Annexin V+/7AAD+ correspond to viable cells, early apoptotic cells, and late apoptotic cells.
Due to viral CPE, the number of remaining cells at 48 h p.i. in the cultures infected with wt BVDV p0, R1 or R4 was lower than that in the cultures infected with BVDV R2, R3 or R5 (2.4–3.0×105 and 4.8–5.5×105, respectively) and that in mock-infected cultures (6.4×105). Taking into account that apoptotic cells could be quantified only in the remaining adherent and floating cells, the total number of cells that would die by viral CPE in each infection was estimated (see Materials and Methods Section). Having considered the differences in CPE between each viral infection, the results showed that the number of cells that would die by BVDV R2, R3 and R5 infections was markedly lower than that in wt BVDV p0 infection (15.6%, 27.5%, 15.5% and 48.6% of estimated dead cells, respectively) (Figure 3).
Figure 3. Infection with BVDV R2, R3 and R5 leads to a decrease in dead cells.
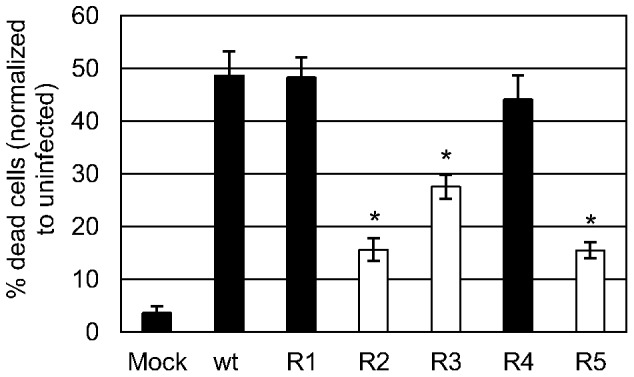
MDBK cells were infected with wt BVDV p0 or BVDV R1–5 (MOI 0.01) and at 48 h p.i. apoptotic cells were measured as stated in Figure 2. Considering the differences in CPE between each viral infection, the total cells that would die by viral CPE was estimated (see Materials and Methods Section). The results are summarized in a bar graph as mean ± SD of % dead cells in relation to the mock-infected culture. *p<0.050; Student's t test vs wt BVDV p0.
The delay in CPE is not caused by a decreased infectious virus production
Although some viral strains produced no CPE, all viral populations showed the highest titers of infectious virus at 48 h p.i. (Figure 4). At that time, most BVDV R (R1 and R3–5) showed slightly higher virus titers (approximately three times higher) than wt BVDV p0, but, notably, BVDV R2 showed viral titers approximately 30 times higher.
Figure 4. Multiple-step growth curve.
MDBK cells were infected with wt BVDV p0 or BVDV R1–5 (MOI 0.01). The viral progeny in the culture supernatant was collected every 24 h for 7 days and titrated by PFU. Viruses with altered CPE (BVDV R2, R3, and R5) are displayed in dashed lines.
On the other hand, wt BVDV p0, R1 and R4 showed a marked decrease in viral titers at 72 h p.i., consistent with the lack of viable host cells as a result of the viral CPE. However, MDBK cells infected with R2, R3 and R5 continued producing infectious virus for at least four additional days.
In addition, expression of the NS3 antigen was monitored by indirect immunofluorescence. In accordance with the results described above, cell monolayers infected with BVDV R2 or R3 were still confluent until 72 h p.i and maintained a continuous production of NS3 (Figure 5).
Figure 5. MDBK cells infected with wt BVDV and BVDV R viruses.

MDBK cells were infected with wt BVDV p0, BVDV R1, R2, or R3 (MOI: 0.1) and IF-stained using a monoclonal antibody against the NS3 protein at 24, 48 and 72 h p.i. The nucleus was stained with DAPI. Images were taken with a 20× objective (numerical aperture 0.40) and processed using ImageJ software (the nuclei were pseudocolored with red for better illustration).
Viruses that have a delayed CPE exhibit less accumulation of viral RNA
One of the factors that contribute to cell apoptosis and viral cytopathogenicity is the accumulation of viral RNA in the cells infected with cp BVDV [45]. In line with this, our results showed that the ivRNA levels in BVDV R2 and R3 infections were lower than those of wt BVDV p0 and R1. BVDV R2, which together with BVDV R5 exhibited the lowest level of cell apoptosis in the infected cultures, also presented the lowest level of ivRNA accumulation (approximately three times lower than wt BVDV p0). On the other hand, BVDV R3, which showed an intermediate level of decrease in cell apoptosis, also presented intermediate levels of ivRNA (Figure 6).
Figure 6. Viral RNA production.
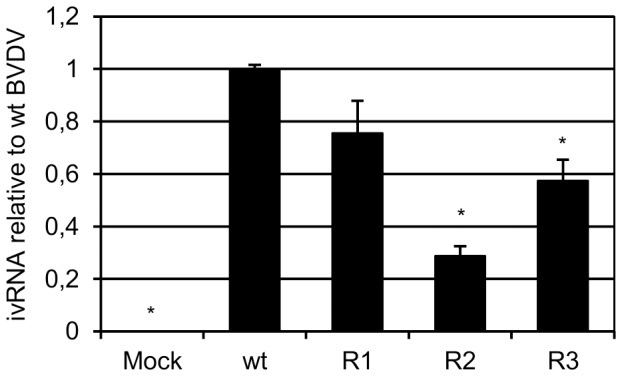
MDBK cells were infected with wt BVDV p0, or BVDV R1, R2 or R3 (MOI 1) and the intracellular viral RNA (ivRNA) was quantified at 24 h p.i. by real-time PCR. For each infection with BVDV R viruses, the results are expressed as ivRNA relative to wt BVDV p0. Mock-infected cells were added as control in each experiment. The results are shown as mean ± SD from two independent experiments in triplicate. *p<0.050; Student's t test vs wt BVDV p0.
Resistance mutations are conserved and additional substitutions were fixed in BVDV R1–5
The nucleotide sequence of Npro, Erns and the complete NS region of wt BVDV p0, BVDV-TSCr T1–5 and BVDV R1–5 genomes were obtained by direct sequencing (GenBank accession numbers KJ608467-KJ608499).
After 20 passages without TSC, the nucleotide sequences of the genomic region that codifies the NS5B protein showed that BVDV R1–5 conserved the mutations previously associated with the resistance to TSC [38]: A10,982G (N264D) in BVDV R1–5 and C11,367A (A392E) in BVDV R1 (which showed mixed populations of nucleotides A/G and C/A in positions 10,982 and 11,367, respectively) (Figure 7).
Figure 7. Molecular characterization of BVDV R1–5.

The complete nucleotide sequence of the Erns, Npro and NS region from wt BVDV p0, BVDV-TSCr T1–5 and BVDV R1–5 genomes was obtained by direct sequencing. The nucleotide sequences are arranged in three or four codons and the position in the genome of the first nucleotide of each group of sequences is indicated above according to the complete genome of BVDV (GenBank accession no. NC_001461.1). Only the genome regions which showed mutations are displayed. Each BVDV-TSCr T-R pair is shown in gray or white rows and, for non-synonymous mutations, amino acid changes are indicated below the corresponding nucleotide sequence, indicating the number of amino acids for each protein. Viruses that showed altered CPE (BVDV R2, R3 and R5) are indicated in boldface.
Secondly, several mutations were fixed along the sequences studied: in BVDV R1: NS2 G5,207A (G472R); in BVDV R2: Erns T1,315Y (Y: mix of T and C), NS5A A8,731G (I9M), NS5A G8,860A, NS5A A9,617T (N305Y), and NS5B C11,987T; in BVDV R3: Erns T1,706G (C171G), NS4B A7,745R (R: mix of A and G) (E28G), NS5A A9,287R (K195E), NS5A G9,812R (E370K), NS5B A10,592G (I134V) and A10,975G (I261M); in BVDV R4: NS3 T6,912C, NS5A T9,285Y (M94T), NS5B C10,768T and NS5B G11,300C (K370N). Some other mutations (not associated with antiviral resistance) were conserved between BVDV-TSCr T1–5 and their respective BVDV R1–5: NS5B A10,723G in T1-R1 and NS5A T9,234C in T2-R2 (V177A). Notably, no new mutation was detected in BVDV R5 after 20 passages. However, whereas BVDV-TSCr T5 presented mixed population of T and C in nucleotide T9,234 (NS5A V177), BVDV R5 presented only C (A177) (Figure 7). This suggests that this mutation would have become fixed in the viral population during the passages in the absence of TSC.
Discussion
We have previously reported the selection of TSC-resistant BVDV mutants (BVDV-TSCr T1–5) with mutations in NS5B (T1–5: N264D; T1: A392E) [38]. In the present study, we performed 20 passages of BVDV-TSCr T1–5 in the absence of TSC to analyze their genetic stability.
The resistance to TSC remained stable and mutations in NS5B associated with resistance were conserved in all the viral populations obtained (BVDV R1–5). However, BVDV R1–5 produced higher titers of infectious virus than wt BVDV p0 and, along the passages in the antiviral-free environment, three out of the five viral populations (BVDV R2, R3 and R5) showed a delay in producing CPE.
It is known that drug-resistant mutants may have fitness costs in the absence of the selecting drug, but the occurrence of compensatory mutations may allow them to partially or even completely regain fitness [46]–[48]. A previous work has shown that BVDV variants that carry the NS5B N264D mutation, which confers resistance to TSC and to other non-nucleoside polymerase inhibitors (NNI) [49], [50], present a replication disadvantage when competing with the wild-type (wt) virus in an antiviral-free environment [51]. In the present work, the replication of BVDV-TSCr in the absence of TSC led to the fixation of different mutations along the NS region that could compensate for the reduced fitness caused by the N264D mutation and confer some stability to the TSC-resistant phenotype. Specifically, mutations were detected in NS2 (BVDV R1), NS4B (BVDV R3), NS5A (BVDV R2–5) and NS5B (BVDV R3 and R4). Particularly, mutation NS5B I261M (BVDV R3) has been reported to compensate for the reduced fitness caused by the N264D mutation and to confer resistance to other BVDV NNI [49]–[51]. Additional mutations suggest that the effect of the NS5B N264D mutation on viral fitness could be also counterbalanced by a change in other NS proteins. Because BVDV-TSCr were not replicated under a selective pressure, these additional mutations may have resulted from the genetic drift during the passages and could have also been detected in the wild type virus if it had been passaged in conditions similar to those of BVDV-TSCr.
As mentioned above, BVDV R2, R3, and R5 showed a delay in producing CPE. The cp biotypes of BVDV induce cell death by apoptosis [44], [52], triggered by the accumulation of viral double-strand RNA [45], [53] and the continuous expression of NS3, the cleaved form of the nonstructural protein NS2-3 [14], [15]. Although viral RNA production in cp BVDV infections is higher than that in ncp ones, both show similar production of infectious viral particles [18], [53]–[56]. Accordingly, cell cultures infected with BVDV R2, R3 and R5 showed a decrease in the induction of apoptosis and in the accumulation of viral RNA. However, BVDV R1–5 produced higher infectious virus titers than wt BVDV p0, and due to host cell survival, BVDV R2, R3 and R5 produced viral antigens and infectious viral progeny for at least four additional days.
BVDV NADL cytopathogenicity is determined by an in-frame insertion in the NS2 coding region of a 270-nucleotide cellular mRNA sequence (called cIns), which modulates NS3 production and upregulates RNA replication [18]. As for other cp BVDV strains, the deletion of the inserted host-derived sequences could lead to reversion to the ncp phenotype [54], [57], [58]. In this work, BVDV R1–5 showed no deletion (or insertion) in the cIns or in any of the viral sequences. In addition, BVDV R2, R3 and R5 showed no amino acid changes in NS2 or NS3, whose bond cleavage is required for RNA replication and is linked to BVDV cytopathogenicity [14], [15]. No amino acid changes were detected in NS3-cofactor NS4A or in Npro and Erns, which may play a role in blocking IFN induction and therefore could also affect the viral CPE [5]–[8], [10], [11]. This suggests that the delay of BVDV R2, R3 and R5 in producing CPE may not result from an alteration in the production or function of these viral proteins. On the other hand, BVDV R3 showed a mutation in NS4B (E28G). This protein would also play a role in BVDV cytopathogenicity [21], and substitutions for residue NS4B Y15 have been shown to attenuate viral CPE despite NS3 production [55].
Interestingly, most of the additional amino acid changes found in BVDV R2 and R3 and the only one detected in BVDV R5 were in NS5A. BVDV NS5A is a zinc metalloprotein essential for the formation of a functional replicase complex [22], [59], [60] that seems to inhibit TNF-α and double-strand RNA-induced NF-kB activation, which is involved in inflammation, innate immune response, cell survival, and pathogenesis or persistence of BVDV infections [24], [61], [62]. BVDV NS5A is composed of three domains. Mutations were found in domain I (I9M in BVDV R2, V177A in BVDV R5 and K195E in BVDV R3) and domain II (N305Y in BVDV R2 and E370K in BVDV R3), both critical for RNA replication [22], [63]. Thus, the additional mutations in NS5A could alter its function during viral RNA replication and/or interfere with its interaction with cellular proteins involved in the induction of apoptosis, thus leading to the observed reduction in viral cytopathogenesis.
To our knowledge, this is the first report on the stability and evolution of non-nucleoside polymerase inhibitor-resistant BVDV. The study of the TSC-resistant viruses that showed reduced CPE and prolonged virus production may be an interesting tool to learn more about the virus-cell interaction, viral cytopathogenesis, attenuation and persistence. Further studies and reverse genetic experiments would be needed to identify the specific phenotype of each of the additional mutations observed.
Acknowledgments
We are grateful to D. Alvarez (Instituto de Investigaciones Biotecnológicas, Universidad de San Martin-CONICET, Buenos Aires, Argentina) for providing the fluorescence microscope and for the advice and discussion of our results. We thank A. Capozzo (Instituto de Virología, CNIA- INTA Castelar, Buenos Aires, Argentina) for providing the monoclonal antibody directed against BVDV NS3.
Funding Statement
This research was supported by grants from Agencia Nacional para la Promoción Científica y Tecnológica (PICT 2012-2867) (http://www.agencia.mincyt.gob.ar/) and Universidad de Buenos Aires (UBACYT 2011-2014, N° 20020100100612) (http://www.uba.ar/secyt/) from Argentina. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Baker JC (1995) The clinical manifestations of bovine viral diarrhea infection. Vet. Clin. North Am 11: 425–445. [DOI] [PubMed] [Google Scholar]
- 2. Chimeno Zoth S, Taboga O (2006) Multiple recombinants ELISA for the detection of bovine viral diarrhea virus antibodies in cattle sera. J Virol Methods 138: 99–108. [DOI] [PubMed] [Google Scholar]
- 3. Pacheco J, Lager I (2003) Indirect method ELISA for the detection of antibodies against bovine diarrhea virus in bovine serum. Rev. Argent. Microbiol 35: 19–23. [PubMed] [Google Scholar]
- 4. Rweyemamu MM, Fernandez AA, Espinosa AM, Schudel AA, Lager IA, et al. (1990) Incidence, epidemiology and control of bovine viral diarrhea virus in South America. Rev. Sci. Tech. (Off. Int. Epizoot.) 9: 207–214. [DOI] [PubMed] [Google Scholar]
- 5. Chen Z, Rijnbrand R, Jangra RK, Devaraj SG, Qu L, et al. (2007) Ubiquitination and proteasomal degradation of interferon regulatory factor-3 induced by Npro from a cytopathic bovine viral diarrhea virus. Virology 366: 277–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gil LH, Ansari IH, Vassilev V, Liang D, Lai VC, et al. (2006) The amino-terminal domain of bovine viral diarrhea virus Npro protein is necessary for alpha/beta interferon antagonism. J Virol 80: 900–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hilton L, Moganeradj K, Zhang G, Chen YH, Randall RE, et al. (2006) The NPro product of bovine viral diarrhea virus inhibits DNA binding by interferon regulatory factor 3 and targets it for proteasomal degradation. J Virol 80: 11723–11732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meyers G, Ege A, Fetzer C, von Freyburg M, Elbers K, et al. (2007) Bovine viral diarrhea virus: prevention of persistent fetal infection by a combination of two mutations affecting Erns RNase and Npro protease. J Virol 81: 3327–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rümenapf T, Unger G, Strauss JH, Thiel HJ (1993) Processing of the envelope glycoproteins of pestiviruses. J Virol 67: 3288–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Magkouras I, Mätzener P, Rümenapf T, Peterhans E, Schweizer MJ (2008) RNase-dependent inhibition of extracellular, but not intracellular, dsRNA-induced interferon synthesis by Erns of pestiviruses. Gen Virol 89: 2501–2506. [DOI] [PubMed] [Google Scholar]
- 11. Mätzener P, Magkouras I, Rümenapf T, Peterhans E, Schweizer M (2009) The viral RNase E(rns) prevents IFN type-I triggering by pestiviral single- and double-stranded RNAs. Virus Res 140: 15–23. [DOI] [PubMed] [Google Scholar]
- 12.Lindenbach BD, Heinz-Jürgen T, Rice CM (2007) Flaviviridae: The Viruses and Their Replication. In Knipe DM, Howley PM, editors. Fields of Virology, 5th ed., vol 1 . Lippincott, Williams & Wilkins pp. 1126–1131. [Google Scholar]
- 13. Tautz N, Harada T, Kaiser A, Rinck G, Behrens S, et al. (1999) Establishment and characterization of cytopathogenic and noncytopathogenic pestivirus replicons. J Virol. 73: 9422–9432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kümmerer BM, Meyers G (2000) Correlation between point mutations in NS2 and the viability and cytopathogenicity of Bovine viral diarrhea virus strain Oregon analyzed with an infectious cDNA clone. J Virol. 74: 390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lackner T, Muller A, Pankraz A, Becher P, Thiel HJ, et al. (2004) Temporal modulation of an autoprotease is crucial for replication and pathogenicity of an RNA virus. J Virol. 78: 10765–10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Warrener P, Collett MS (1995) Pestivirus NS3 (p80) protein possesses RNA helicase activity. J Virol. 69: 1720–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu J, Mendez E, Caron PR, Lin C, Murcko MA, et al. (1997) Bovine viral diarrhea virus NS3 serine proteinase: polyprotein cleavage sites, cofactor requirements, and molecular model of an enzyme essential for pestivirus replication. J Virol 71: 5312–5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mendez E, Ruggli N, Collett MS, Rice CM (1998) Infectious bovine viral diarrhea virus (strain NADL) RNA from stable cDNA clones: a cellular insert determines NS3 production and viral cytopathogenicity. J Virol 72: 4737–4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tautz N, Kaiser A, Thiel HJ (2000) NS3 serine protease of bovine viral diarrhea virus: characterization of active site residues, NS4A cofactor domain, and protease-cofactor interactions. Virology 273: 351–363. [DOI] [PubMed] [Google Scholar]
- 20. Tautz N, Elbers K, Stoll D, Meyers G, Thiel HJ (1997) Serine protease of pestiviruses: determination of cleavage sites. J Virol 71: 5415–5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weiskircher E, Aligo J, Ning G, Konan KV (2009) Bovine viral diarrhea virus NS4B protein is an integral membrane protein associated with Golgi markers and rearranged host membranes. Virol J 6: 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tellinghuisen TL, Paulson MS, Rice CM (2006) The NS5A protein of bovine viral diarrhea virus contains an essential zinc binding site similar to that of the hepatitis C virus NS5A protein. J Virol 80: 7450–7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Johnson CM, Perez DR, French R, Merrick WC, Donis RO (2001) The NS5A protein of bovine viral diarrhoea virus interacts with the alpha subunit of translation elongation factor-1. J Gen Virol 82: 2935–2943. [DOI] [PubMed] [Google Scholar]
- 24. Zahoor MA, Yamane D, Mohamed YM, Suda Y, Kobayashi K, et al. (2010) Bovine viral diarrhea virus non-structural protein 5A interacts with NIK- and IKKbeta-binding protein. J Gen Virol 91: 1939–1948. [DOI] [PubMed] [Google Scholar]
- 25. Choi KH, Groarke JM, Young DC, Kuhn RJ, Smith JL, et al. (2004) The structure of the RNA-dependent RNA polymerase from bovine viral diarrhea virus establishes the role of GTP in de novo initiation. Proc Natl Acad Sci U S A 101: 4425–4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Collett MS, Anderson DK, Retzel E (1988) Comparisons of the pestivirus bovine viral diarrhoea virus with members of the Flaviviridae. J Gen Virol 69: 2637–2643. [DOI] [PubMed] [Google Scholar]
- 27. Zhong W, Gutshall LL, Del Vecchio AM (1998) Identification and characterization of an RNA-dependent RNA polymerase activity within the nonstructural protein 5B region of bovine viral diarrhea virus. J Virol 72: 9365–9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lai VC, Kao CC, Ferrari E, Park J, Uss AS, et al. (1999) Mutational analysis of bovine viral diarrhea virus RNA-dependent RNA polymerase. J Virol 73: 10129–10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Steffens S, Thiel HJ, Behrens SE (1999) The RNA-dependent RNA polymerases of different members of the family Flaviviridae exhibit similar properties in vitro. J Gen Virol 80: 2583–2590. [DOI] [PubMed] [Google Scholar]
- 30. Moennig V, Plagemann PG (1992) The pestiviruses. Adv Virus Res 41: 53–98. [DOI] [PubMed] [Google Scholar]
- 31. Peterhans E, Bachofen C, Stalder H, Schweizer M (2010) Cytopathic bovine viral diarrhea viruses (BVDV): emerging pestiviruses doomed to extinction. Vet Res 41: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schweizer M, Peterhans E (1999) Oxidative stress in cells infected with bovine viral diarrhoea virus: a crucial step in the induction of apoptosis. J Gen Virol 80: 1147–1155. [DOI] [PubMed] [Google Scholar]
- 33. Grummer B, Bendfeldt S, Wagner B, Greiser-Wilke I (2002) Induction of the intrinsic apoptotic pathway in cells infected with cytopathic bovine virus diarrhoea virus. Virus Res 90: 143–153. [DOI] [PubMed] [Google Scholar]
- 34. Jordan R, Wang L, Graczyk TM, Block TM, Romano PR (2002) Replication of a cytopathic strain of bovine viral diarrhea virus activates PERK and induces endoplasmic reticulum stress-mediated apoptosis of MDBK cells. J Virol 76: 9588–9599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maeda K, Fujihara M, Harasawa R (2009) Bovine viral diarrhea virus 2 infection activates the unfolded protein response in MDBK cells, leading to apoptosis. J Vet Med Sci 71: 801–805. [DOI] [PubMed] [Google Scholar]
- 36. Finkielsztein LM, Castro EF, Fabián LE, Moltrasio GY, Campos RH, et al. (2008) New 1-indanone thiosemicarbazone derivatives active against BVDV. Eur J Med Chem 43: 1767–1773. [DOI] [PubMed] [Google Scholar]
- 37. Finkielsztein LM, Moltrasio GY, Caputto ME, Castro EF, Cavallaro LV, et al. (2010) What is known about the antiviral agents active against bovine viral diarrhea virus (BVDV)? Curr Med Chem 17: 2933–2955. [DOI] [PubMed] [Google Scholar]
- 38. Castro EF, Fabian LE, Caputto ME, Gagey D, Finkielsztein LM, et al. (2011) Bovine viral diarrhea virus RNA synthesis inhibition by the thiosemicarbazone derived from 5,6-dimethoxy-1-indanone. J Virol 85: 5436–5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trotter J (2011) WinMDI 2.9 (Windows Multiple Document Interface for Flow Cytometry). Available: http://www.mybiosoftware.com/cytometry/1677.
- 40.Applied Biosystems (2004) 7900HT Fast Real-Time PCR System and 7300/7500/7500 Fast Real-Time PCR Systems. Chemistry Guide. Chapter 3.
- 41. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The clustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucelic Acids Res 25: 7876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hall TA (1999) Bioedit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/nt. Nucleic Acids Symp Ser 41: 95–98. [Google Scholar]
- 43.Di Rienzo JA, Casanoves F, Balzarini MG, Gonzalez L, Tablada M, et al. (2011) InfoStat versión 2011. Grupo InfoStat, FCA, Universidad Nacional de Córdoba, Argentina. Available: http://www.infostat.com.ar.
- 44. Hoff HS, Donis RO (1997) Induction of apoptosis and cleavage of poly(ADP-ribose) polymerase by cytopathic bovine viral diarrhea virus infection. Virus Res 49: 101–113. [DOI] [PubMed] [Google Scholar]
- 45. Vassilev VB, Donis RO (2000) Bovine viral diarrhea virus induced apoptosis correlates with increased intracellular viral RNA accumulation. Virus Res 69: 95–107. [DOI] [PubMed] [Google Scholar]
- 46. Govorkova EA, Ilyushina NA, Marathe BM, McClaren JL, Webster RG (2010) Competitive fitness of oseltamivir-sensitive and -resistant highly pathogenic H5N1 influenza viruses in a ferret model. J Virol 84: 8042–8050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hu Z, Kuritzkes DR (2010) Effect of raltegravir resistance mutations in HIV-1 integrase on viral fitness. J Acquir Immune Defic Syndr 55: 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wu W, Blythe DC, Loyd H, Mealey RH, Tallmadge RL, et al. (2011) Decreased infectivity of a neutralization-resistant equine infectious anemia virus variant can be overcome by efficient cell-to-cell spread. J Virol 85: 10421–10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Giliberti G, Ibba C, Marongiu E, Loddo R, Tonelli M, et al. (2010) Synergistic experimental/computational studies on arylazoenamine derivatives that target the bovine viral diarrhea virus RNA-dependent RNA polymerase. Bioorg Med Chem 18: 6055–6068. [DOI] [PubMed] [Google Scholar]
- 50. Tonelli M, Boido V, La Colla P, Loddo R, Posocco P, et al. (2010) Pharmacophore modeling, resistant mutant isolation, docking, and MM-PBSA analysis: Combined experimental/computer-assisted approaches to identify new inhibitors of the bovine viral diarrhea virus (BVDV) Bioorg Med Chem. 18: 2304–2316.50. [DOI] [PubMed] [Google Scholar]
- 51. Newcomer BW, Neill JD, Marley MS, Ridpath JF, Givens MD (2013) Mutations induced in the NS5B gene of bovine viral diarrhea virus by antiviral treatment convey resistance to the compound. Virus Res 174: 95–100. [DOI] [PubMed] [Google Scholar]
- 52. Zhang G, Aldridge S, Clarke MC, McCauley JW (1996) Cell death induced by cytopathic bovine viral diarrhoea virus is mediated by apoptosis. J Gen Virol 77: 1677–1681. [DOI] [PubMed] [Google Scholar]
- 53. Yamane D, Kato K, Tohya Y, Akashi H (2006) The double-stranded RNA-induced apoptosis pathway is involved in the cytopathogenicity of cytopathogenic Bovine viral diarrhea virus. J Gen Virol 87: 2961–2970. [DOI] [PubMed] [Google Scholar]
- 54. Becher P, Orlich M, Thiel HJ (2001) RNA recombination between persisting pestivirus and a vaccine strain: generation of cytopathogenic virus and induction of lethal disease. J Virol 75: 6256–6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Qu L, McMullan LK, Rice CM (2001) Isolation and characterization of noncytopathic pestivirus mutants reveals a role for nonstructural protein NS4B in viral cytopathogenicity. J Virol 75: 10651–10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pankraz A, Preis S, Thiel HJ, Gallei A, Becher P (2009) A single point mutation in nonstructural protein NS2 of bovine viral diarrhea virus results in temperature-sensitive attenuation of viral cytopathogenicity. J Virol 83: 12415–12423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gallei A, Orlich M, Thiel HJ, Becher P (2005) Noncytopathogenic pestivirus strains generated by nonhomologous RNA recombination: alterations in the NS4A/NS4B coding region. J Virol 79: 14261–14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Baroth M, Orlich M, Thiel HJ, Becher P (2000) Insertion of cellular NEDD8 coding sequences in a pestivirus. Virology 278: 456–466. [DOI] [PubMed] [Google Scholar]
- 59. Zahoor MA, Yamane D, Mohamed YM, Kobayashi K, Kato K (2009) Characterization and application of monoclonal antibodies to bovine viral diarrhea virus nonstructural protein 5A. Arch Virol 154: 1745–1754. [DOI] [PubMed] [Google Scholar]
- 60. Sapay N, Montserret R, Chipot C, Brass V, Moradpour D, et al. (2006) NMR structure and molecular dynamics of the in-plane membrane anchor of nonstructural protein 5A from bovine viral diarrhea virus. Biochemistry 45: 2221–2233. [DOI] [PubMed] [Google Scholar]
- 61. Häcker H, Karin M (2006) Regulation and function of IKK and IKK-related kinases. Sci STKE 2006: re13. [DOI] [PubMed] [Google Scholar]
- 62. Hatada EN, Krappmann D, Scheidereit C (2000) NF-kappaB and the innate immune response. Curr Opin Immunol 12: 52–58. [DOI] [PubMed] [Google Scholar]
- 63. Isken O, Langerwisch U, Schönherr R, Lamp B, Schröder K, et al. (2014) Functional Characterization of Bovine Viral Diarrhea Virus 1 nonstructural protein 5A by reverse genetic analysis and live cell imaging. J Virol 88: 82–98. [DOI] [PMC free article] [PubMed] [Google Scholar]