

Abstract
Background
Antibody fragments selected from large combinatorial libraries have numerous applications in diagnosis and therapy. Most existing antibody repertoires are derived from human immunoglobulin genes. Genes from other species can, however, also be used. Because of the way in which gene conversion introduces diversity, the naïve antibody repertoire of the chicken can easily be accessed using only two sets of primers.
Results
With in vitro diagnostic applications in mind, we have constructed a large library of recombinant filamentous bacteriophages displaying single chain antibody fragments derived from combinatorial pairings of chicken variable heavy and light chains. Synthetically randomised complementarity determining regions are included in some of the heavy chains. Single chain antibody fragments that recognise haptens, proteins and virus particles were selected from this repertoire. Affinities of three different antibody fragments were determined using surface plasmon resonance. Two were in the low nanomolar and one in the subnanomolar range. To illustrate the practical value of antibodies from the library, phage displayed single chain fragments were incorporated into ELISAs aimed at detecting African horsesickness and bluetongue virus particles. Virus antibodies were detected in a competitive ELISA.
Conclusion
The chicken-derived phage library described here is expected to be a versatile source of recombinant antibody fragments directed against a wide variety of antigens. It has the potential to provide monoclonal reagents with applications in research and diagnostics. For in vitro applications, naïve phage libraries based on avian donors may prove to be useful adjuncts to the selectable antibody repertoires that already exist.
Background
Recombinant antibodies derived from phage display libraries [1-10] are by now well established in medicine and biotechnology. A major objective of recombinant antibody research so far has been to develop new therapeutic agents for use in human patients. Accordingly, most of the existing large combinatorial libraries are based upon human immunoglobulin genes. Phage antibodies can, however, be derived from a number of alternative donors such as rabbits [11], camels [12], cattle [13], sheep [14] and chickens [15]. From a practical point of view, the chicken is a particularly attractive source of immunoglobulin genes. To amplify each of the V gene segments in the human or mouse requires a number of PCR primer sets. In contrast, the chicken antibody repertoire can be accessed relatively easily because of the way in which its diversity is generated. In birds, single immunoglobulin variable and joining gene segments at each heavy (H) and light (L) chain locus are subjected to VJ rearrangement in the case of the L chain and VDJ for the H chain. Variability then arises by gene conversion resulting from the incorporation of pseudo V region genes [16-19]. As a result, essentially all chicken V regions can be expected to have virtually identical amino acid sequences at both termini. The naïve antibody repertoire can thus be conveniently amplified by using only a single set of PCR primers for the H chain and another for the L chain. This property was first exploited by Davies and co-workers [15] who constructed and expressed an scFv library derived from chicken bursal lymphocyte RNA. They demonstrated the potential of the chicken as a source of recombinant antibody fragments by constructing a relatively small (2.7 × 107 clones), but nonetheless effective, naïve library which yielded scFv phage antibodies against three different proteins. Antibody fragments that recognise proteins are not always readily obtained from small libraries of human origin [3].
The accessibility by PCR of chicken antibody gene repertoires also facilitates the construction of immune phage libraries [20-22]. If, however, the aim is to bypass immunisation entirely, it becomes important to increase the probability of isolating high affinity binders by having as large and diverse a repertoire as possible [5-7,23,24]. While the construction of a large antibody library is not a trivial undertaking, the technology ultimately offers several advantages when compared to conventional sources of antibodies, particularly the way in which the need for animals or cell culture facilities is circumvented. To be able to understand and control new or re-emerging diseases, new reagents are constantly required for use in veterinary and agricultural diagnostic tests and as research tools. A rapid and economical way of obtaining pathogen-specific antibodies is therefore likely to be especially useful in developing countries where budgets and facilities are often severely limited. Because of the potential of recombinant antibody technology to provide reagents that can be used in a variety of standard immunodiagnostic tests [4,25-29] we have constructed a large (approximately 2 × 109 clones) and diverse scFv library, primarily with in vitro diagnostics in mind. This library combines the naïve chicken immunoglobulin repertoire with a sublibrary in which amino acid residues comprising the third H chain complementarity determining region (CDR3) have been synthetically randomised. The overall repertoire was found to be diverse enough to yield antibody fragments that recognise haptens, proteins and viruses. Its potential as a source of in vitro veterinary diagnostic reagents was illustrated by finding antibodies which could be used in ELISA to detect African horsesickness virus (AHSV), bluetongue virus (BTV) and BTV antibodies.
Results
Antibody fragment library construction
With the goal of creating a widely diverse antibody repertoire, two separate phage display sublibraries were constructed. The first was aimed at representing the naïve immunoglobulin repertoire of the chicken. For this, pooled RNA extracted from five chicken bursae was used. A strategy was employed in which the 5' PCR primer used to amplify the L chain and the 3' of the H chain included complementary sequences, which when extended, would serve to link the H and L chains via a (Gly4 Ser)3 linker [30]. This naïve sublibrary comprised a total of 3.75 × 108 phage clones. DNA from a number of these clones was sequenced to verify that joining had taken place correctly and that reading frames had been preserved. This analysis also revealed that the CDR3 of the H chain of the sequenced clones varied in size from 6 to 19 residues (Figure 1A). Attempts to use this same joining strategy to introduce artificially randomised CDR3s of defined size were not consistently successful. Accordingly, a separate linker sequence was amplified from a DNA template obtained from a clone in the naïve library and splicing by overlap extension (SOE) PCR [31] was used to join separate H, L and linker sequences [32]. The H chains included artificially randomised CDR3s ranging in size from 6 to 14 amino acid residues. A separate set of amplification and joining reactions was carried out for each size of CDR. After joining, the H and L chain sets were ligated into the display vector pHEN I and each set was separately electroporated into competent bacterial cells. DNA sequencing showed that randomised CDR3 sequences of defined lengths had been successfully incorporated into the heavy chains (Figure 1B). Based on the number of primary transformants, phages were mixed proportionally to prevent over-representation of any of the synthetic CDR3s in the synthetic sublibrary. The naïve and randomised sublibraries were similarly combined to yield the final repertoire consisting of approximately 2 × 109 primary transformants. This required a total of 190 separate electroporations. Sequencing the DNA of 70 random clones revealed that approximately 65% had the potential to express in-frame scFvs. The completed library thus contained combinatorial pairings of naïve H and L chains as well as sets of synthetically randomised H chains paired with natural chicken L chains.
Figure 1.

Amino acid sequences of VH CDR3s. Amino acid sequences of scFv clones showing the VH CDR3s and their flanking regions in the naïve (A) and the semi-synthetic (B) repertoires which were combined to make up the final Nkuku recombinant antibody library. The clones from the naïve library were randomly picked while representatives from each predetermined size class are shown in the case of the synthetic repertoire. Identical amino acid residues are shown by dashes (-) while dots (.) indicate spaces introduced to align sequences.
Selection of specific antibodies
To test its viability as a source of antibody fragments, the entire library was subjected to four rounds of selection using several model haptens, proteins and viruses. Rescued phage particles obtained after each round of panning were then tested in a polyclonal phage ELISA to determine whether enrichment for binders had taken place with each successive selection step. An increase in specific ELISA signals was evident with the phage pools obtained after panning on the conjugated haptens fluorescein (F-BSA; Figure 2A) and BSA-coupled 4-hydroxy-3-iodo-5-nitrophenylacetic acid (N-BSA; Figure 2B). Figure 2C,2D,2E,2F also shows binding to the proteins keyhole limpet haemocyanin (KLH), turkey egg lysozyme (TEL), cytochrome C (CYT) and porcine thyroglobulin (THY). Antibody pools reactive in ELISA with purified cucumber mosaic virus (CMV), AHSV and BTV (Figure 2G,2H,2I) were also obtained. No enrichment was obtained during selections performed on maltose binding protein (not shown).
Figure 2.
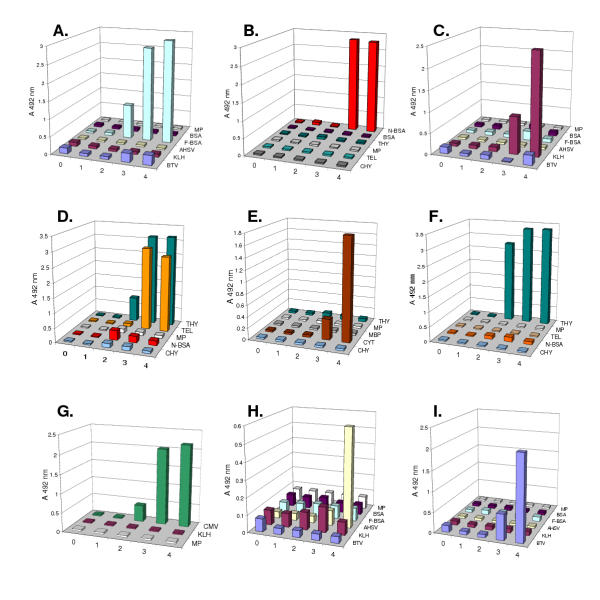
Polyclonal phage ELISA. Polyclonal phage ELISAs showing the increase in specific signal with successive rounds of panning. Phages were selected by panning on immobilised proteins, haptens and viruses. Pools released from the affinity matrix after each round of selection were amplified, purified by precipitation and tested for binding to the homologous target and several heterologous antigens: BSA = bovine serum albumin, MP = 2% milk powder in PBS, MBP = maltose binding protein, F-BSA = fluorescein-conjugated bovine serum albumin, N-BSA = 4-hydroxy-3-iodo-5-nitrophenylacetic acid conjugated to bovine serum albumin, AHSV = purified African horsesickness virus particles, BTV = purified bluetongue virus particles, CMV = purified cucumber mosaic virus particles, CYT = cytochrome C, THY = porcine thyroglobulin, CHY = chymotrypsinogen, KLH = keyhole limpet haemocyanin, TEL = turkey egg lysozyme. The homologous antigens in each case were: A) F-BSA, B) N-BSA, C) KLH, D) TEL, E) CYT, F) THY, G) CMV, H) AHSV and I) BTV. Figures on the X axis indicate the panning round of which the output was tested for polyclonal binding. The figure 0 refers to the unpanned library.
Antigen-specific phage pools selected on some of the viruses and proteins were chosen for further characterisation. Individual phage clones were picked and tested in ELISA for their ability to recognise antigen. Using an ELISA absorbance of greater than 0.2 as the criterion for binding, 99% of the bacterial clones tested secreted fusion phages that specifically recognised CMV. With BTV, 76% were positive and with AHSV the figure was 61%. Using the protein KLH as the target, 44% of the clones were specific binders. In the absence of exhaustive sequence data, however, these figures do not necessarily imply that each binder represented a unique clone. While phage titres were not normalised for each ELISA determination, as a rule, no reactivity with unrelated antigens was discernible indicating that improved binding to the target with each round of panning was due to specific enrichment and not an increase in "stickiness" and/or phage titre. One exception was that the polyclonal phage displayed antibodies selected on TEL showed an unexpected cross-reactivity with porcine thyroglobulin (Figure 2D). Selecting single clones, however, made it possible to isolate a phage displayed antibody that recognised only TEL and another that recognised both TEL and thyroglobulin (not shown).
In an attempt to establish whether any of the binding antibodies were derived from the component of the library that included the synthetically randomised VH CDR3s, DNA coding for several virus-specific binders was sequenced, namely three each that recognised BTV and AHSV and five that bound to CMV. Based on the nucleotide sequences used to introduce the synthetic CDR as well as the presence of only NNK codons in the CDR3 itself, one clone from each of these groups could be shown to have originated from the synthetic component of the final library (Figure 3). No two L or H chains were identical and (surprisingly) one clone (C31) that bound to CMV lacked its entire VH region which indicates that its total binding activity resides in the VL region. A number of phage antibody clones specific for BTV and CMV were tested for their ability to bind as soluble scFvs. Soluble fragment production was induced using IPTG. Of fourteen BTV binders, only two showed ELISA reactivity in both forms. With CMV, the figure was 85 out of 96, but since their DNA sequences were not determined, this figure may reflect multiple copies of fewer actual binders. For BIACORE affinity determinations, the scFvs were directly tested for their ability to bind in soluble form. After affinity purification, yields of soluble fragments ranged from 4 ng to 2,5 μg per millilitre of culture supernatant.
Figure 3.

Amino acid sequences of CDRs of binding antibodies. Amino acid sequences of VH and VL CDRs of antibodies selected from the Nkuku library. Asterisks (*) denote clones which have high probability of having been derived from the portion of the library into which synthetic CDR3s were introduced. The entire H chain of clone C31 was absent, but the fragment bound to CMV particles in ELISA (not shown). The antigens recognised by each of the individual clones are indicated in the figure. Dots (.) indicate spaces introduced to align sequences, x denotes ambiguous bases. (Genbank accession numbers: Lca12, AY366194; Hca12, AY366195; C6, 366196; E8-10, AY366197; E8-3, AY366198; H2, AY366199; C23, AY366200; C24, AY366201; C26, AY366202; C29, AY366203; C31, AY366204)
Sandwich ELISA to detect orbiviruses
African horsesickness is an economically important disease in southern Africa and elsewhere. It is caused by AHSV, an orbivirus (family Reoviridae) containing a characteristic double-stranded RNA segmented genome within a double-shelled capsid. AHSV can be detected by using a sandwich ELISA in which the virus particle is first captured on an affinity matrix of immobilised antibodies and then detected by a second layer of virus-specific antibodies followed by labelled anti-immunoglobulin reagents [33,34]. Phage antibodies are particularly suitable in this format since the detecting signal is amplified by the multiple copies of pVIII, the major capsid protein of the fusion phage. To determine whether the chicken antibody library could provide phage antibodies that could act as the second antibody in the sandwich, round three output phages (Figure 2H) were screened by panning on 2.5 μg/ml of AHSV instead of the usual concentration of 20 μg/ml. This was done in an attempt to exclude low or medium affinity binders by using a limiting amount of target antigen. Only three virus-specific scFvs were found after this high-stringency round of selection. The clone that gave the highest signal (Figure 3; clone Lca12) was chosen for use as a detecting antibody in the sandwich ELISA. For this assay, virus particles were trapped from suspension using virus-specific rabbit IgG and detected using the phage displayed scFv, an anti-pVIII mouse monoclonal antibody (mAb) and a labelled anti-mouse IgG. At a concentration of 40 ng/ml virus, equivalent to 2 ng/well, the assay produced an absorbance at 492 nm of greater than 0.5 (Figure 4A). A phage antibody (Figure 3; clone H2) selected from the fourth round pool of BTV binders was used in a similar assay to detect BTV particles (Figure 4B).
Figure 4.
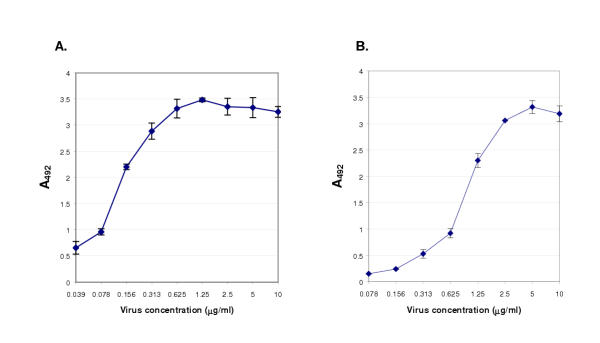
Sandwich ELISA for virus detection. Double antibody sandwich ELISA absorbance values obtained in which dilutions of (A) AHSV or (B) BTV particles were trapped from suspension using virus-specific rabbit IgG and detected using phage-displayed scFvs (clone Lca12 for AHSV, clone H2 for BTV; see Figure 3) as the second antibody in the sandwich. Binding phages were detected with a pVIII-specific mAb and a peroxidase-labelled anti-mouse IgG immunoglobulin. Each point represents the average of three determinations. Error bars show one standard deviation from the mean.
Inhibition ELISA
The specificity of an antigen-antibody interaction can be shown by inhibiting the binding with a different antibody directed against the same antigen. Such inhibition assays are used to detect antibodies against BTV for the purposes of regulating livestock exports. To determine whether BTV-specific scFvs could replace the traditional mAbs usually used in these tests, BTV particles were immobilised by adsorption to a microtitre plate well and immune- and control sera from rabbits or sheep were added before introducing a BTV-specific scFv (Figure 3; clone H2). The binding of the antibody fragment was inhibited by virus-specific antibodies in the sera, the degree of inhibition being dependent on the serum dilution. Rabbit antiserum directed against both the homologous serotype 10 as well as the heterologous serotype 4 showed inhibition. Similarly, sheep antisera raised against three heterologous serotypes (2, 4 and 9) could inhibit the interaction. Non-immune sera from either species had no effect on the ELISA signal and filamentous phage displaying no scFvs did not bind to the immobilised antigen (Figure 5).
Figure 5.
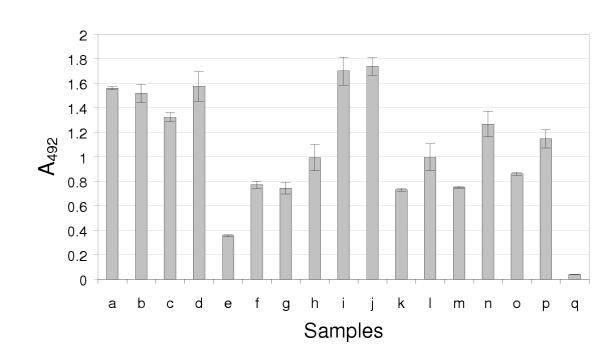
Inhibition ELISA. Inhibition of binding of the phage displayed scFv H2 in a competitive ELISA. Purified BTV serotype 10 particles were immobilised directly on a plastic surface. The samples to be tested for inhibition were then added, followed by the phage displayed scFv H2, an anti-pVIII monoclonal antibody and an enzyme-labelled anti-mouse IgG immunoglobulin. The shaded bars depict the average absorbance value of two determinations while the upper and lower values are shown by the error bars. Test samples used in the inhibition step were: (a) 2% solution of fat-free milk; (b-d) three different non-immune rabbit sera; (e) 1/9 and (f) 1/81 dilutions of anti-BTV serotype 4 rabbit immune serum; (g) 1/9 and (h) 1/81 dilutions of anti-BTV serotype 10 rabbit antiserum; (i) 1/9 and (j) 1/81 dilutions of non-immune sheep serum; (k) 1/9 and (l) 1/81 dilutions of sheep antiserum directed against BTV serotype 2; (m) 1/9 and (n) 1/81 dilutions of sheep antiserum against BTV serotype 4; (o) 1/9 and (p) 1/81 dilutions of BTV serotype 9-specific sheep antisera. The background binding of non-fusion phage to the immobilised BTV serotype 10 is shown by (q).
Affinity of selected scFvs
To determine whether binding affinities of Nkuku-derived scFvs had the potential to be in the same range as those obtained from other large libraries, scFvs directed against three antigens of widely different molecular weights were chosen for BIACORE analysis. These were fluorescein (586 Da), cytochrome C (12.4 kDa) and KLH (400 kDa). All determinations were done in duplicate using separate preparations of scFvs. The experimental results were fitted to the BIAevaluation 1:1 Langmuir binding model. Visual comparison and the calculated χ2 values indicated an acceptable fit to the model. The high affinities of these three binders (nanomolar or less; Table 2) show that they compare well with antibody fragments derived from naïve libraries of human origin [5,7].
Table 2.
Kinetic and binding parameters for three scFvs selected from the Nkuku library1.
| scFv | ka (1/Ms) | kd (1/s) | KD (M) |
| anti-F-BSA | 7 × 106 | 1.68 × 10-2 | 2.4 × 10-9 |
| anti-CYT | 4.29 × 106 | 3.64 × 10-3 | 3.5 × 10-10 |
| anti-KLH | 2.41 × 106 | 3.64 × 10-3 | 2.83 × 10-9 |
1Association and dissociation rate constants were measured using BIACORE. Each value represents the average of two independent determinations on the same chip surface.
Discussion
Most recombinant antibody libraries are based on human immunoglobulin genes. While this is obviously desirable in human therapy, for veterinary diagnostic tests or other in vitro applications, specific binding reagents could be obtained from any animal source. With veterinary and agricultural applications in mind, we were able to construct a diverse phagemid-based chicken scFv library consisting of approximately two billion clones. Several other large antibody libraries have been given a variety of names (e.g. "Nissim", "Vaughan", "HuCal", "n-CoDer" etc). Accordingly, we suggest for our library the name "Nkuku", derived from the Zulu word "nkukhu", which means "chicken".
To maximise the range of potential paratopes [3], the Nkuku library makes use of the natural L and H chains of the chicken immunoglobulin repertoire together with a selection of synthetically randomised H chain CDR3s of defined sizes. The success of this diversification strategy was shown empirically by obtaining scFvs that recognised a number of haptens, proteins and viruses. Isolating a variety of target-specific phage antibodies further demonstrates the suitability of the chicken as a donor of immunoglobulin genes for use in constructing non-immune antibody libraries [15]. Two different approaches to the combinatorial joining of H and L chains were used. The use of amplification primers that included the linker sequences [21] was not feasible for incorporating randomised H chain CDR3s owing to the impractically long oligonucleotides required (not shown). For constructing the synthetically randomised sublibrary, the classical strategy of employing a separate linker sequence [32] was therefore used. For similar reasons, it was also not possible to reliably include synthetic CDR3s of longer than 14 residues. The final antibody pool therefore relies on the chicken's natural H chain repertoire to provide these longer CDRs. Producing the antibody library was nevertheless straightforward, since all amplification steps were facilitated by the relatively few PCR primers required.
The amino acids comprising the CDR3 of the human immunoglobulin H chain show the most variability and usually make the dominant contribution to binding [35-37]. In the widely distributed human antibody library made by Nissim and co-workers [4], it is this region that has been artificially randomised. Sequencing the DNA of several unselected clones from our chicken library revealed VH CDR3 regions ranging in size from 6 to 19 residues. The CDR3 regions of the L chain were from 5 to 13 amino acids long, also showing wide variability. Since chicken IgY has essentially the same arrangement of CDRs as mammalian IgG, we emulated the Nissim library approach by incorporating our synthetically randomised sequences into the VH CDR3 region [4]. To verify that the randomised VH CDR3 sequences were used in our functional antibodies, the DNA coding for 11 antibody clones that had been selected by panning on three different immobilised viruses was sequenced. It was expected that in the clones originating from the artificially introduced CDR3 sequences, all residues flanking the CDR would be identical to those of the primers used to introduce them. In addition, since NNK codons were used for randomisation, every third base in the synthetic CDR should be either G or T. On the basis of these criteria, three out of the 11 clones appeared to originate from the synthetically introduced repertoire. All L chains were found to be different. In the case of both BTV and AHSV, one out of three binding clones probably originated from the synthetic repertoire while with CMV the proportion was one in five. We also found that one of the antibodies that bound to CMV was lacking its entire H chain (Figure 3; clone C31). Its binding in ELISA was nonetheless essentially indistinguishable from that of several other intact phage displayed scvFs (not shown). It is unlikely that the absent H chain represents a sequencing artefact since the entire insert was sequenced in both directions using primers that anneal to the vector. Extensive gene conversion in chicken L chains has been observed before [21] and raises the intriguing possibility that it may be possible to develop a functional single domain antibody library from only the chicken L chain.
Not all chicken library antibodies that bound as phage displayed fragments could be shown to bind in ELISA when expressed as scFvs. This is not unexpected and could be due either to low expression levels or to a low intrinsic affinity when the fragment is used outside the phage display context. The phage particle with its displayed antibody fragment can nevertheless be a useful ELISA reagent when used as the second antibody in a sandwich format. This was shown in the immunoassy (Figure 4A) designed to detect AHSV particles immobilised on a rabbit IgG affinity matrix using clone Lca12 (Figure 3). An analogous assay for the structurally similar BTV, but using fusion phages from a human antibody library [4], has been described previously [25]. The ability of Nkuku library-derived fusion phages to be incorporated into an assay for viral antibodies as opposed to antigens was shown by the BTV-specific fusion phage designated H2 which could be inhibited by immune rabbit and sheep sera (Figure 5). Inhibition was not confined to antibodies directed against the homologous serotype, indicating that H2 probably recognises an antigenic determinant that occurs on more than one viral serotype. Alternatively, it may bind to a unique region that is located in close proximity to one or more cross-reactive epitopes, such that it is functionally inhibited by heterologous antibodies. Our results show that the Nkuku library can be used to obtain potentially useful and specific assay reagents without resorting to hybridomas. In the case of viruses possessing a multiplicity of serotypes such as the orbiviruses AHSV and BTV [38], recombinant technology may be the only practical way of obtaining antibodies that can identify each individual serotype.
A DNA clone that codes for a particular recombinant antibody is a virtually inexhaustible source which can be unequivocally identified by its nucleotide sequence. Critical immunoassay reagents can therefore be fully characterised. In addition, it is often possible to engineer or evolve phage antibodies showing improved stability or affinity [39]. Most of the antibodies described here bound strongly in ELISA without further modification and at least one (F10) could survive at least one freeze-thaw cycle. This may not always be the case and if the aim is to develop immunoassays for use under field conditions e.g. in Africa, it may be necessary to select for temperature-stable variants. The use of chicken frameworks may be advantageous for this purpose since they have evolved to function at a body temperature approximately 5°C higher than that of humans.
Conclusions
On the basis of the evaluation described here, this chicken-derived "Nkuku" phage library is a potentially versatile source of recombinant antibody fragments which should find many applications in areas such as immunodiagnostics [40] and proteomics [41]. Unless steps are taken to humanise them [21], such avian antibodies are unlikely to be useful for human therapeutics. Human V genes may, however, sometimes lack the intrinsic features required to produce antibodies that can recognise all epitopes with high affinity [42]. For in vitro use, large non-immune phage libraries based on avian donors such as the one described here may therefore prove to be a useful additional source of binding reagents.
Methods
Antigens and immunochemical reagents
Attenuated AHSV serotype 3 and BTV serotype 10 (both members of the genus Orbivirus, family Reoviridae) were obtained originally from the vaccine factory (now Onderstepoort Biological Products Limited) at the then Veterinary Research Institute. The viruses were multiplied in BHK (BTV) or CER (AHSV) cells and purified by sucrose density-gradient centrifugation [43]. Rabbit and sheep sera were supplied by the Biochemistry and Virology Divisions of the Onderstepoort Veterinary Institute. The sheep sera are used routinely as reference standards by the OIE World Reference Centre for bluetongue virus. Purified cucumber mosaic virus particles were a generous gift from Dr G. Pietersen, Plant Protection Research Institute, Roodeplaat. KLH, F-BSA, porcine thyroglobulin, TEL and cytochrome C were from Sigma. N-BSA was prepared from the succinimide ester of 4-hydroxy-3-iodo-5-nitrophenylacetic acid -CAP-OH (NIP-CAP-OSU; Genosys) and BSA fraction V according to the method described by the Medical Research Council (Cambridge, UK) in the instructions issued with their "Nissim" antibody library. B62-FE2, a mAb specific for an epitope on the phage coat protein VIII of filamentous male-specific phages, was from Progen Biotechnik (Heidelberg) while a horseradish peroxidase (HRP)-labelled anti-M13 mAb was purchased from Amersham Pharmacia Biotech (UK). HRP labelled rabbit-anti-mouse immunoglobulins were supplied by Dako (Denmark). A murine hybridoma (Mycl-9E10; ECACC 85102202) producing a mAb (9E10) specific for the c-myc epitope tag, was obtained from the European Collection of Cell Cultures (CAMR, UK). The hybridoma was cultured in protein-free hybridoma medium (Invitrogen Corporation).
Construction of the recombinant antibody library
Five different 5-week old white leghorn hens were killed by overdosing with pentobarbitone and their bursae removed (approved by the Animal Ethics Committee, Onderstepoort Veterinary Institute). Total RNA was extracted with TRI-Reagent (Molecular Research Center, Cincinnati, OH, USA). Oligo d(T)16 primer, RNase inhibitor, MuLV reverse transcriptase (all from the Perkin Elmer Gene Amp RNA PCR core kit, Roche), 10x Ex Taq buffer and dNTPs (both from TaKaRa, Japan) were used for cDNA synthesis. The reaction products were used directly as cDNA templates to construct two different scFv gene sublibraries; the first was a naïve library while the second included a synthetically randomised H-chain CDR3 region. In both sublibraries the VH and VL genes were joined by a (Gly4-Ser)3 linker [30], but two different strategies were followed. For the naïve sublibrary two sets of primers were used in the primary amplification. The VH and VL sequences were first amplified using primer sets Sfi1L/NEWLVarH and NEWLVarL/LCNOT1 (Table 1). Amplification reactions consisted of 8 μl 10x TaKaRa buffer, 2.5 pmol of each primer, 2.5 units (U) of TaKaRa Ex Taq enzyme, the 20 μl reverse transcription product (above) and water to a final volume of 100 μl. PCR was for 30 cycles at 94°C for 1 minute (min), 60°C for 1 min, 72°C for 1 min followed by a final extension period of 1 min at 72°C. The primers NEWLVarH and NEWLVarL contained overlapping linker sequences to allow the two genes to be spliced by overlap extension [21,31]. Prior to splicing, the amplified genes were purified using a QIAquick PCR purification kit (QIAGEN, Germany). Further purification entailed electrophoresis on a 2% agarose gel (Seakem ME) prepared in 1 × TAE supplemented with a final concentration of 10 μg/ml crystal violet [44] followed by extraction from the gel using a QIAquick gel extraction kit (QIAGEN). Splicing was in 100 μl reaction volumes comprising 10 μl 10x TaKaRa buffer, 8 μl TaKaRa dNTPs, 100 ng VL chain DNA, 100 ng VH chain DNA, 2.5 U TaKaRa Ex Taq DNA polymerase, 3 U Promega Pfu DNA polymerase [45] and water to a final volume of 100 μl. The reaction components were transferred from ice to a preheated (94°C) thermal cycler. This was followed by 15 cycles at 94°C for 1 min, 60°C for 1 min, 72°C for 2 min and a final extension at 72°C for 5 min. Pullthrough amplification reactions consisted of 10 μl 10x TaKaRa PCR buffer, 8 μl TaKaRa dNTPs, 20 pmol Sfi1 primer, 20 pmol LCNOT1 primer, 2 μl spliced product, 2.5 U Ex Taq DNA polymerase and water to a final volume of 100 μl. Reaction mixtures were transferred from ice to a thermal cycler block at 94°C. A preheating step of 1 min at 94°C was followed by 25 cycles of 94°C for 1 min, 60°C for 1 min, 72°C for 2 min and a final extension at 72°C for 5 min. The product was gel purified as above. The joined VH and VL genes were digested overnight at 50°C with 20 U Sfi1 (Roche) per μg DNA in 100 μl buffer M (Roche) supplemented with 0.1 mg/ml acetylated BSA. For overnight cutting with Not1 at 37°C, digestion conditions were adjusted by the addition of 1.5 μl of 5 M NaCl, 6 μl 1 M Tris (pH 8.0), 0.5 μl 10 mg/ml acetylated BSA, 20 U Not1 (Roche) per μg DNA and water to final volume of 150 μl. The digested DNA was gel purified as above.
Table 1.
Nucleotide sequences of DNA primers used for amplifying and sequencing chicken immunoglobulin genes
| Primer | Sequence |
| GSfor | 5' GGGGCCACGGGACCGAAGTC 3' |
| GSrev2 | 5' CGCTGACACCGAGGAC 3' |
| LCNOT1 | 5' TGATGGTGGCGGCCGCATTGGGCTG 3' |
| Sfi1L | 5' GTCCTCGCAACTGCGGCCCAGCCGGCCCTGATGGCGGCCGTGACG 3' |
| RandVH * | 5' GACTTCGGTCCCGTGGCCCCATGCGTCGAT[MNN]n TTTGGCGCAGTAGTAGGTGCCGGTGTCCTC 3' |
| OP52 | 5' CCCTCATAGTTAGCGTAACG 3' |
| M13rev | 5' CAGGAAACAGCTATGAC 3' |
| NEWLVarL | 5' TCAGGTGGAGGTGGCTCTGGCGGAGGCGGATCGGCGCTGACTCAGCCGTCCTCGG 3' |
| NEWLVarH | 5' CCGCCAGAGCCACCTCCACCTGAACCGCCTCCACCGGAGGAGACGATGACTTCGG 3' |
* M = A/C, N = A/C/G/T, n = 5–13
The phagemid vector pHEN I (obtained from the Medical Research Council, UK) was purified from an overnight TG1 bacterial culture using a QIAGEN Plasmid Midi Kit. Restriction enzyme digestion conditions were as described for the insert, but with six units of enzyme per μg of vector DNA. The digested vector was purified in the same way as the insert, but using a 1% agarose gel. Ligations were incubated overnight at 16°C using 100 ng cut vector, 40 ng insert, 1 μl 10x ligation buffer (Roche), 0.5 U T4 DNA ligase and water to a final volume of 10.5 μl. The ligated product was first purified using a QIAquick PCR purification kit followed by a diffusion desalting step [46].
The desalted DNA was electroporated into Epicurian Coli Electroporation-Competent TG1 cells (Stratagene, USA) using a Biorad Gene Pulser II electroporator set on 1700 V, 200 Ω and 25 μF with 0.1 cm electroporation cuvettes. After electroporation the bacterial cells were immediately transferred to 1 ml SOC medium. After 1 hour (h) at 37°C in a shaking incubator they were plated onto 243 × 243 mm TYE agar plates (15 g agar, 8 g NaCl, 1 g tryptone, 5 g yeast in 1 l double distilled deionised water) supplemented with 100 μg/ml ampicillin and 2% glucose. Additional serial dilutions were plated to determine the library size. After an overnight incubation at 30°C the bacteria were scraped off the plates in 2x TY (16 g tryptone, 10 g yeast extract, 5 g NaCl dissolved in 1 l double distilled deionised water) supplemented with 100 μg/ml ampicillin and 2% glucose (2x TY A/G). The bacterial suspensions were stored as 15% glycerol stocks at -70°C. Ligations and electroporations were repeated until the required number of primary clones was obtained.
For the sublibrary containing synthetically randomised VH CDR3 areas, the VH gene, the linker sequence and the VL genes were amplified separately prior to joining all three components by overlap extension [32]. The VH gene was prepared as described for the naïve library. Nine different subrepertoires were constructed, each with a different synthetically randomised VH CDR3 region ranging in size from six to 14 amino acid residues. The primer on the 5' end of VH gene (Sfi1L) was the same for all nine subrepertoires, whereas nine different primers were used on the 3' end (Rand6VH, Rand7VH etc.; see Table 1). Amplification of the joined genes after SOE [31] was performed by using primer set Sfi1L/LCNOT1. The linker fragment was amplified with primer set GSfor/GSrev2 using a DNA template obtained from a sequenced clone in the naïve library. All the methods and conditions as used for the naïve sublibrary were implemented except for the SOE reactions where 40 ng VL DNA, 40 ng VH DNA and 54 ng of linker per reaction were joined.
Phages were rescued from a pool of TG1 bacteria harbouring representative amounts of all the sublibraries by the addition of helper phage M13KO7. The phages were recovered from the supernatant by precipitation with a 1/5 volume 20% PEG in 2.5 M NaCl. The precipitated phages displaying antibodies were resuspended in PBS and stored at -70°C in 15% glycerol.
Antibody selection by panning
Immunotubes (Nunc Maxisorp) were coated overnight at 4°C with 100 μg/ml of the relevant protein or conjugated hapten dissolved in PBS. For selections on virus particles, Nunc Polysorp tubes were coated with 20 μg/ml of purified virus. All subsequent panning steps were performed at room temperature. The tubes were blocked for 1 h with 2% fat free milk powder (MP). After washing twice with PBS, approximately 5 × 1012 library phage particles that had been preincubated for 30 min in 2% MP and 0.1% Tween-20 were added to each immunotube. Tubes were first rotated for 30 min before a stationary incubation of 90 min and then washed 20 × with PBS containing 0.1% Tween 20 (PBS/T) followed by a further 20 washes with PBS. Phage displayed antibodies were released by incubating for 10 min with 1.0 ml 100 mM triethylamine (pH 12). The eluate was neutralised by the addition of 0.5 ml 1 M TRIS-HCl (pH 7.4). The neutralised eluate was used to reinfect exponentially growing TG1 cells prior to plating on TYE plates containing 2% glucose and 100 μg/ml ampicillin. After an overnight incubation at 30°C the bacteria were collected and the phagemids rescued by the addition of M13KO7 helper phage (helper phage:bacteria = 20:1). Four such rounds of selection were usually performed. To screen monoclonal phage antibodies, individual clones were rescued by transferring inocula from bacterial colonies to the wells of sterile 96 well tissue culture plates containing 100 μl/well 2x TY A/G. Bacteria were grown overnight at 30°C with shaking at 250 RPM. The next day, a 96 well inoculation device (Sigma: Cat. No R-2508) was used to transfer cells from the master plate to a fresh plate that contained 150 μl 2x TY A/G per well. After a 2.5 h incubation at 250 RPM and 37°C, a 50 μl volume of 2x TY A/G that contained 2 × 109 pfu M13KO7 was added to each well. The plate was incubated at 37°C for 30 min without shaking. After centrifugation for 10 min at 600 × g the supernatant fluids were removed and replaced with 150 μl of 2 X TY that contained 100 μg/ml ampicillin and 25 μg/ml kanamycin. The plate was then incubated overnight at 30°C and 250 RPM. After centrifugation for 10 min at 600 × g, the supernatant fluids containing phage displayed antibodies were collected for testing in ELISA. To screen for monoclonal soluble scFvs, colonies were grown overnight in microtitre plates as above. Cells were then inoculated into wells containing 100 μl of 2x TY A/G, with the glucose concentration decreased to 0.1%. The cells were incubated at 37°C, with shaking, for 3 h after which 50 μl of 2x TY that contained 100 μg/ml ampicillin and 3 mM isopropyl-β-D-thiogalactopyranoside was added to each well. After incubating for a further 16 h at 30°C and 250 RPM the plates were centrifuged as above and the supernatant fluids were tested in ELISA.
Polyclonal phage ELISA
Immunoplates (Nunc Polysorp or Maxisorp) were coated overnight at 4°C with 50 μl/well of the relevant antigens suspended in PBS. Subsequent steps were performed at room temperature. Blocking was for 1 h with 300 μl/well of 2% MP in PBS. Phage displayed antibodies were concentrated 25 × by PEG precipitation and then diluted 1/100 prior to mixing with an equal volume of PBS containing 4% MP and 0.2% Tween 20. After three washes with PBS/T, the wells were incubated for 2 h with 50 μl/well of the phage displayed antibodies. The plate was washed three times and incubated for 1 h with 50 μl/well of an HRP/anti-M13 mAb conjugate (Amersham) diluted 1/5000 in PBS/T containing 2% MP. After a final wash, 50 μl/well of chromogen consisting of 1 mg/ml o-phenylene diamine and 0.5 μl/ml of a 30% (v/v) hydrogen peroxide in 0.1 M citrate buffer (pH 4.5) was added. Absorbance was monitored at 450 nm with the final reading being taken at 492 nm after stopping the reaction with an equal volume of 2 N H2SO4.
Monoclonal fusion phage and scFv ELISAs
The protocol described above was used to show binding of individual scFvs fused to phage particle clones, except that the supernatant fluids from centifuged microtitre plates were diluted 1:1 with PBS containing 0.2% Tween 20 and 4% MP. For selecting individual soluble scFvs, the detergent was omitted from the diluent for this and all subsequent steps. Bound phage displayed scFvs were detected by incubating for 1 h with 50 μl/well of 100 ng/ml mAb B62-FE2 in PBS/T that contained 2% MP followed by incubation for 1 h with 50 μl/well of HRP anti-mouse conjugate diluted 1/1000 in the same buffer. The mAb 9E10 which recognises the c-myc epitope tag was used when detecting soluble scFvs.
DNA sequencing
Phagemid DNA was isolated by means of a QIAprep Spin Miniprep Kit (QIAGEN) from 5 ml overnight cultures of single clones grown at 30°C and 250 rpm in 2x TY A/G. Primers OP52 and M13 rev were used (Table 1). Automated sequencing was done by the Molecular Biology Division, Onderstepoort Veterinary Institute. Sequences were analysed using Staden [47] and Wisconsin GCG [48] software packages.
Inhibition ELISA
A Polysorp (Nunc) microtitre plate was coated overnight at 4°C with 50 μl/well of 10 μg/ml BTV-10 in PBS and blocked as above. Blocking and washing steps were as before except that all incubations were at 37°C. After blocking, the plate was incubated for 1 h with 50 μl/well of the different sera diluted 1/9 in PBS containing 2% MP followed by an incubation of 1 h with 50 μl/well of phage antibody H2. PEG precipitated phages were first diluted in PBS to the pre-precipitation volume and then mixed 1:1 with PBS containing 4% MP and 0.2% Tween 20. Phage antibodies were detected using the pVIII-specific mAb B62-FE2 as described above.
Sandwich ELISA
A Polysorp microtitre plate was coated for 1 h at 37°C with 50 μl/well of 10 μg/ml purified rabbit-IgG in PBS against either BTV-10 or AHSV-3. Blocking and washing steps were as described for the inhibition ELISA. The plate was incubated for 1 h at 37°C with 50 μl/well of the various dilutions of virus in PBS containing 2% MP. All subsequent steps from the addition of antibody B62-FE2 onwards were done as described for the inhibition ELISA, but the incubation times were shortened to 45 minutes.
Expression and purification of soluble scFvs
Individual bacterial clones were grown overnight in 2x TY A/G at 37°C. A dilution of 1/100 was made in the same medium and the bacteria were allowed to grow until the OD600 reached a value of 0.9. The cells were collected by centrifugation and resuspended in a one-fifth volume of 2x TY containing 100 μg/ml ampicillin and 1 mM IPTG. After overnight incubation at 30°C, followed by centrifugation, scFvs were affinity-purified from the supernatant fluid using an anti c-myc tag mAb. Six milligrams of mAb 9E10 IgG was immobilised on AminoLink Plus gel (Pierce) according to the manufacturer's instructions. Medium containing the scFvs was passed over the column at least three times. After washing with 14 ml PBS, the bound scFvs were eluted with 5 ml 100 mM triethylamine and neutralised with 2.5 ml 1 M Tris pH 7.4. The eluted scFvs were concentrated by centrifugation in an Ultrafree Biomax unit (10K NMWL, Millipore) to approximately 300 μl. The concentration was determined spectrophotometrically with an A280 of 1.0 corresponding to 0.7 mg/ml scFv and 1 mg/ml = 40 nM scFv.
Surface plasmon resonance
Kinetic binding constants were determined by surface plasmon resonance using a BIACORE X instrument (BIACORE, Uppsala, Sweden). Experiments were performed at 25°C using HBS-EP running buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005% surfactant P20) throughout. Bound scFvs were removed with 10 μl of 0.1 M glycine pH 2.5. Fluorescein-biotin (Sigma) was diluted in PBS to 10 pg/ml and allowed to bind to the surface of a streptavidin coated chip (Biacore). A blank flow cell was used as a reference surface. The immobilised hapten produced a signal of 12 RU. Anti-fluorescein scFvs ranging from 0.2 to 3.3 nM diluted in HBS-EP were injected over the chip at a flow rate of 30 μl/min for 70 seconds and allowed to dissociate for the same time. Horse heart cytochrome C (Sigma) was covalently bound to the dextran surface of a CM5 chip via its primary amine groups (BIAapplications handbook, BIACORE). A volume of 35 μl of cytochrome C (10 μg/ml diluted in 10 mM acetate buffer, pH 5.5) was injected and unreacted ester groups were blocked with 1 M ethanolamine-HCl, pH 8.5. These conditions resulted in 3,200 RU being immobilised. In the control flow cell 2,000 RU of chicken egg lysozyme was immobilised under the same conditions. Two different batches of anti-cytochrome C scFvs were injected at concentrations ranging from 2.9 nM to 17 pM. KLH was similarly immobilised using 35 μl of the protein at a concentration of 15 μg/ml in 10 mM acetate buffer, pH 4.5. After blocking unreacted groups, 10 μl of 25 mM NaOH was injected to remove unbound KLH. This resulted in 10,523 RU being bound. The second flow cell was left empty. Again two different batches of anti-KLH scFvs were injected, at concentrations ranging from 0.5 nM to 10 nM. All kinetic analyses were done with BIAevaluation software according to the 1:1 Langmuir model using values obtained after subtracting the reference signal.
Abbreviations
AHSV African horsesickness virus
BSA bovine serum albumin
BTV bluetongue virus
CDR complementarity determining region
CMV cucumber mosaic virus
CYT cytochrome C
D diversity (region of immunoglobulin gene)
EDC N-ethyl-N'-(dimethylaminopropyl) carbodiimide
F-BSA fluorescein conjugated to bovine serum albumin
H heavy (chain)
h hour
HRP horseradish peroxidase
J joining (region of immunoglobulin gene)
KLH Keyhole limpet haemocyanin
L light (chain)
mAb monoclonal antibody
min minute
MP fat free milk powder
NHS N-hydroxysuccinimide
N-BSA 4-hydroxy-3-iodo-5-nitrophenylacetic acid conjugated to bovine serum albumin
PBS/T PBS containing 0.1% Tween 20
pfu plaque-forming units
RPM revolutions per minute
RU resonance units (in BIACORE analysis)
scFv single chain variable fragment
TEL turkey egg lysozyme
U unit (of enzyme)
V variable (region of immunoglobulin gene)
2x TY A/G 2x TY supplemented with 100 μg/ml ampicillin and 2% glucose
Authors' contributions
WvW helped conceptualise the study and was largely responsible for the physical construction and evaluation of the library. TM and CM assisted in library construction, screening and evaluation by ELISA. FJ and JF were responsible for identifying, cloning and purifying the scFvs used in the BIACORE analysis. JF was responsible for bioinformatics and was assisted by DM in carrying out the kinetic analyses. DHduP conceived the project, participated in the design of the library, supervised all stages of the study and wrote the major portion of the manuscript. The authors have read and approved the final manuscript.
Acknowledgments
Acknowledgements
This work was supported by the Innovation Fund of the South African Department of Arts, Culture, Science and Technology. We thank the Medical Research Council (Cambridge, United Kingdom) for the pHEN display vector.
Contributor Information
Wouter van Wyngaardt, Email: wouter@moon.ovi.ac.za.
Teresiah Malatji, Email: T.Malatji@bowman.co.za.
Cordelia Mashau, Email: cordelia@moon.ovi.ac.za.
Jeanni Fehrsen, Email: jeanni@moon.ovi.ac.za.
Frances Jordaan, Email: frances@moon.ovi.ac.za.
Dubravka Miltiadou, Email: dubravka@moon.ovi.ac.za.
Dion H du Plessis, Email: dion@moon.ovi.ac.za.
References
- Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J Mol Biol. 1991;222:581–597. doi: 10.1016/0022-2836(91)90498-u. [DOI] [PubMed] [Google Scholar]
- Kang AS, Barbas CF, III, Janda KD, Benkovic SJ, Lerner RA. Linkage of recognition and replication functions by assembling combinatorial antibody Fab libraries along phage surfaces. Proc Natl Acad Sci USA. 1991;88:4363–4366. doi: 10.1073/pnas.88.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenboom HR, Winter G. By-passing immunisation. Human antibodies from synthetic repertoires of germline VH gene segments rearranged in vitro. J Mol Biol. 1992;227:381–388. doi: 10.1016/0022-2836(92)90894-p. [DOI] [PubMed] [Google Scholar]
- Nissim A, Hoogenboom HR, Tomlinson IM, Flynn G, Midgley C, Lane D, Winter G. Antibody fragments from a 'single pot' phage display library as immunochemical reagents. EMBO J. 1994;13:692–698. doi: 10.1002/j.1460-2075.1994.tb06308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan TJ, Williams AJ, Pritchard K, Osbourn JK, Pope AR, Earnshaw JC, McCafferty J, Hodits RA, Wilton J, Johnson KS. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nature Biotechnology. 1996;14:309–14. doi: 10.1038/nbt0396-309. [DOI] [PubMed] [Google Scholar]
- Sheets MD, Amersdorfer P, Finnern R, Sargent P, Lindquist E, Schier R, Hemingsen G, Wong C, Gerhart JC, Marks JD, Lindqvist E. Efficient construction of a large nonimmune phage antibody library: the production of high-affinity human single-chain antibodies to protein antigens. Proc Natl Acad Sci USA. 1998;95:6157–6162. doi: 10.1073/pnas.95.11.6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Haard HJ, Van Neer N, Reurs A, Hufton SE, Roovers RC, Hendrikx P, Bruïne AP, Arends J-W, Hoogenboom HR. A large non-immunized human Fab fragment phage library that permits rapid isolation and kinetic analysis of high affinity antibodies. J Biol Chem. 1999;274:18218–30. doi: 10.1074/jbc.274.26.18218. [DOI] [PubMed] [Google Scholar]
- Gerhart JC, Marks JD, Lindqvist E. Efficient construction of a large nonimmune phage antibody library: the production of high-affinity human single-chain antibodies to protein antigens. Proc Natl Acad Sci USA. 1998;95:6157–6162. doi: 10.1073/pnas.95.11.6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knappik A, Ge L, Honegger A, Pack P, Fischer M, Wellnhofer G, Hoess A, Wölle J, Plückthun A, Virnekäs B. Fully synthetic human combinatorial libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides. J Mol Biol. 2000;296:57–86. doi: 10.1006/jmbi.1999.3444. [DOI] [PubMed] [Google Scholar]
- Ellmark P, Ottosson C, Borrebaeck CA, Malmborg Hager AC, Furebring C. Modulation of the CD40-CD40 ligand interaction using human anti-CD40 single-chain antibody fragments obtained from the n-CoDeR phage display library. Immunology. 2002;106:456–463. doi: 10.1046/j.1365-2567.2002.01473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridder R, Schmitz R, Legay F, Gram H. Generation of rabbit monoclonal antibody fragments from a combinatorial phage display library and their production in the yeast Pichia pastoris. Biotechnology N Y. 1995;13:255–60. doi: 10.1038/nbt0395-255. [DOI] [PubMed] [Google Scholar]
- Ghahroudi MA, Desmyter A, Wyns L, Hamers R, Muyldermans S. Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett. 1997;414:521–526. doi: 10.1016/S0014-5793(97)01062-4. [DOI] [PubMed] [Google Scholar]
- O'Brien PM, Aitken R, O'Neil BW, Campo MS. Generation of native bovine mAbs by phage display. Proc Natl Acad Sci USA. 1999;96:640–645. doi: 10.1073/pnas.96.2.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Kilpatrick J, Whitelam GC. Sheep monoclonal antibody fragments generated using a phage display system. J Immunol Methods. 2000;236:133–146. doi: 10.1016/S0022-1759(99)00227-6. [DOI] [PubMed] [Google Scholar]
- Davies EL, Smith JS, Birkett CR, Manser JM, Anderson-Dear DV, Young JR. Selection of specific phage-display antibodies using libraries derived from chicken immunoglobulin genes. J Immunol Methods. 1995;186:125–135. doi: 10.1016/0022-1759(95)00143-X. [DOI] [PubMed] [Google Scholar]
- Reynaud CA, Anquez V, Dahan A, Weill JC. A single rearrangement event generates most of the chicken immunoglobulin light chain diversity. Cell. 1985;40:283–91. doi: 10.1016/0092-8674(85)90142-4. [DOI] [PubMed] [Google Scholar]
- Reynaud CA, Anquez V, Grimal H, Weill JC. A hyperconversion mechanism generates the chicken light chain preimmune repertoire. Cell. 1987;48:379–88. doi: 10.1016/0092-8674(87)90189-9. [DOI] [PubMed] [Google Scholar]
- Reynaud CA, Dahan A, Anquez V, Weill JC. Somatic hyperconversion diversifies the single Vh gene of the chicken with a high incidence in the D region. Cell. 1989;59:171–83. doi: 10.1016/0092-8674(89)90879-9. [DOI] [PubMed] [Google Scholar]
- Thompson CB, Neiman PE. Somatic diversification of the chicken light chain gene is limited to the rearranged light chain segment. Cell. 1987;48:369–78. doi: 10.1016/0092-8674(87)90188-7. [DOI] [PubMed] [Google Scholar]
- Yamanaka HI, Inoue T, Ikeda-Tanaka O. Chicken monoclonal antibody isolated by a phage display system. J Immunol. 1996;157:1156–1162. [PubMed] [Google Scholar]
- Andris-Widhopf J, Rader C, Steinberger P, Fuller R, Barbas CF., III Methods for the generation of chicken monoclonal antibody fragments by phage display. J Immunol Methods. 2000;242:159–181. doi: 10.1016/S0022-1759(00)00221-0. [DOI] [PubMed] [Google Scholar]
- Cary SP, Lee J, Wagenknecht R, Silverman GJ. Characterization of superantigen-induced clonal deletion with a novel clan III-restricted avian monoclonal antibody: exploiting evolutionary distance to create antibodies specific for a conserved VH region surface. J Immunol. 2000;164:4730–4741. doi: 10.4049/jimmunol.164.9.4730. [DOI] [PubMed] [Google Scholar]
- Griffiths AD, Williams SC, Hartley O, Tomlinson IM, Waterhouse P, Crosby WL, Kontermann RE, Jones PT, Low NM, Allison TJ, Prospero TD, Hoogenboom HR, Nissem A, Cox JPL, Harrison JL, Zacolo M, Gherardi E, Winter G. Isolation of high affinity human antibodies directly from large synthetic repertoires. EMBO J. 1994;13:3245–60. doi: 10.1002/j.1460-2075.1994.tb06626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sblattero D, Bradbury A. Exploiting recombination in single bacteria to make large phage antibody libraries. Nat Biotechnol. 2000;18:75–80. doi: 10.1038/71958. [DOI] [PubMed] [Google Scholar]
- Van Wyngaardt W, Du Plessis DH. Selection of an scFv phage antibody that recognizes bluetongue virus from a large synthetic library and its use in ELISAs to detect viral antigen and antibodies. Onderstepoort J Vet Res. 1998;65:125–131. [PubMed] [Google Scholar]
- Griep R, Van Twisk C, Shots A. Selection of beet necrotic yellow vein virus single chain Fv antibodies from a semi-synthetic combinatorial antibody library. European J Plant Pathol. 1999;105:147–156. doi: 10.1023/A:1008727326113. [DOI] [Google Scholar]
- Harper K, Toth RL, Mayo MA, Torrance L. Properties of a panel of single chain variable fragments against potato leafroll virus obtained from two phage display libraries. J Virol Methods. 1999;81:159–168. doi: 10.1016/S0166-0934(99)00071-3. [DOI] [PubMed] [Google Scholar]
- Williamson RA, Burioni R, Sanna PP, Partridge LJ, Barbas CF, III, Burton DR. Human monoclonal antibodies against a plethora of viral pathogens from single combinatorial libraries. Proc Natl Acad Sci USA. 1993;90:4141–4145. doi: 10.1073/pnas.90.9.4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Marks JD. Applying phage antibodies to proteomics: selecting single chain Fv antibodies to antigens blotted on nitrocellulose. Anal Biochem. 2000;286:119–128. doi: 10.1006/abio.2000.4788. [DOI] [PubMed] [Google Scholar]
- Huston JS, Levinson D, Mudgett-Hunter M, Tai M-S, Novotný J, Margolies MN, Ridge RJ, Bruccoleri RE, Haber E, Crea R, Oppermann H. Protein engineering of antibody binding sites: Recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc Natl Acad Sci USA. 1988;85:5879–5883. doi: 10.1073/pnas.85.16.5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- Clackson T, Hoogenboom HR, Griffiths A, Winter G. Making antibody fragments using phage display libraries. Nature. 1991;352:624–628. doi: 10.1038/352624a0. [DOI] [PubMed] [Google Scholar]
- Du Plessis, DH van Wyngaardt W, Bremer CW. An indirect sandwich ELISA utilising F(ab')2 fragments for the detection of African horsesickness virus. J Virol Methods. 1990;29:279–289. doi: 10.1016/0166-0934(90)90055-K. [DOI] [PubMed] [Google Scholar]
- Hamblin C, Mertens PP, Mellor PS, Burroughs JN, Crowther JR. A serogroup specific enzyme-linked immunosorbent assay for the detection and identification of African horse sickness viruses. J Virol Methods. 1991;31:285–292. doi: 10.1016/0166-0934(91)90166-W. [DOI] [PubMed] [Google Scholar]
- Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol. 1987;196:901–917. doi: 10.1016/0022-2836(87)90412-8. [DOI] [PubMed] [Google Scholar]
- Kabat EA, Wu TT. Identical V region amino acid sequences and segments of sequences in antibodies of different specificities. J Immunol. 1991;147:1709–1719. [PubMed] [Google Scholar]
- Kabat EA, Wu TT, Reid-Miller M, Perry HM, Gottesman KS, Foeller C. Variable region heavy chain sequences. In Sequences of proteins of Immunological Interest, NIH Publication No 91-3242, Technical Information Service (NTIS) 1991.
- Roy P. Bluetongue virus proteins. J Gen Virol. 1992;73:3051–3064. doi: 10.1099/0022-1317-73-12-3051. [DOI] [PubMed] [Google Scholar]
- Wörn A, Plückthun A. Stability engineering of antibody single chain Fv fragments. J Mol Biol. 2001;305:989–1010. doi: 10.1006/jmbi.2000.4265. [DOI] [PubMed] [Google Scholar]
- Sapats SI, Heine HG, Trinidad L, Gould GJ, Foord AJ, Doolan SG, Prowse S, Ignjatovic J. Generation of chicken single chain antibody variable fragments (scFv) that differentiate and neutralize infectious bursal disease virus (IBDV) Arch Virol. 2003;148:497–515. doi: 10.1007/s00705-002-0931-2. [DOI] [PubMed] [Google Scholar]
- Liu B, Huang L, Sihlbom C, Burlingame A, Marks JD. Towards proteome-wide production of monoclonal antibody by phage display. J Mol Biol. 2002;315:1063–73. doi: 10.1006/jmbi.2001.5276. [DOI] [PubMed] [Google Scholar]
- Lantto J, Lindroth Y, Ohlin M. Non-germline encoded residues are critical for effective antibody recognition of a poorly immunogenic neutralization epitope on glycoprotein B of cytomegalovirus. Eur J Immunol. 2002;32:1659–1669. doi: 10.1002/1521-4141(200206)32:6<1659::AID-IMMU1659>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Huismans H, Van Der Walt N, Cloete M, Erasmus BJ. Isolation of a capsid protein of bluetongue virus that induces a protective immune response in sheep. Virology. 1987;157:172–179. doi: 10.1016/0042-6822(87)90326-6. [DOI] [PubMed] [Google Scholar]
- Rand KN. Crystal violet can be used to visualize DNA bands during gel electrophoresis and to improve cloning efficiency. Tech Tips Online. 1996;1:4:T40022. [Google Scholar]
- Yamanaka HI, Kirii Y, Ohmoto H. An improved phage display antibody cloning system using newly designed PCR primers optimised for Pfu DNA polymerase. J Biochem. 1995;117:1218–1227. doi: 10.1093/oxfordjournals.jbchem.a124847. [DOI] [PubMed] [Google Scholar]
- Atrazhef AM, Elliot JF. Simplified desalting of ligation reactions immediately prior to electroporation into E. coli. Biotechniques. 1996;21:1024. doi: 10.2144/96216bm12. [DOI] [PubMed] [Google Scholar]
- Staden R. The Staden package. In: Griffin AM, Griffin HG, editor. In Methods in Molecular Biology. Vol. 25. Totawa, NJ: Humana Press Inc; 1994. pp. 9–170. [DOI] [Google Scholar]
- Anon Program manual for the Wisconsin Package, Version 8 September 1994. Genetics Computer Group, 575 Science Drive, Madison, Wisconsin, USA, 53711.