

Abstract
Background
A valuable weapon in the arsenal available to yeast geneticists is the ability to introduce specific mutations into yeast genome. In particular, methods have been developed to introduce deletions into the yeast genome using PCR fragments. These methods are highly efficient because they do not require cloning in plasmids.
Results
We have modified the existing method for introducing deletions in the yeast (S. cerevisiae) genome using PCR fragments in order to target point mutations to this genome. We describe two PCR-based methods for directing point mutations into the yeast genome such that the final product contains no other disruptions. In the first method, site-specific genomic (SSG) mutagenesis, a specific point mutation is targeted into the genome. In the second method, random domain-localized (RDL) mutagenesis, a mutation is introduced at random within a specific domain of a gene. Both methods require two sequential transformations, the first transformation integrates the URA3 marker into the targeted locus, and the second transformation replaces URA3 with a PCR fragment containing one or a few mutations. This PCR fragment is synthesized using a primer containing a mutation (SSG mutagenesis) or is synthesized by error-prone PCR (RDL mutagenesis). In SSG mutagenesis, mutations that are proximal to the URA3 site are incorporated at higher frequencies than distal mutations, however mutations can be introduced efficiently at distances of at least 500 bp from the URA3 insertion. In RDL mutagenesis, to ensure that incorporation of mutations occurs at approximately equal frequencies throughout the targeted region, this region is deleted at the same time URA3 is integrated.
Conclusion
SSG and RDL mutagenesis allow point mutations to be easily and efficiently incorporated into the yeast genome without disrupting the native locus.
Background
Exogenous DNA introduced into yeast cells recombines at high frequency with homologous sequences in the genome. This property has been exploited to provide powerful tools for generating specific changes in the yeast genome. For example, a deletion allele generated on a plasmid can be used to delete the wild-type allele in the genome [1]. In addition, deletion alleles can be synthesized by PCR and directly targeted to the genome, replacing the ORF with a selectable marker [2-5]. In fact, a collection of approximately 5900 deletion mutants have been constructed by this method, with each mutant deleted for a separate non-essential yeast ORF [6].
Alleles containing point mutations are often more useful than complete deletion alleles for defining the functions of particular domains or elements within a gene. Point mutations can be easily introduced into cloned genes carried on plasmids (reviewed in [7]). However, genes carried on plasmids may not be regulated identically to genes at their native locus. For example, distant regulatory sequences not present in the plasmid may affect expression of the native gene [8-10], and the chromatin structure on a plasmid may not be identical to the structure at the native locus [11]. In addition, expression of cloned genes can be affected by the broad range of factors that regulate plasmid copy number [12-14].
Most methods for integrating mutant alleles into the chromosome require either a plasmid-borne copy of the mutant allele [15] or introduction of marker alleles and other non-native sequence in the region of the mutation [16]. Recently an elegant method for introducing point mutations into the genome was devised by Resnick and colleagues that does not require cloning of the mutant allele [17]. This method was termed delitto perfetto (Italian slang for perfect murder) because the mutation is introduced into the genome without leaving any remnants of non-native sequence. The delitto perfetto method involves two sequential transformations. First, yeast is transformed with a PCR fragment that targets two marker genes, URA3 and Kanr to a specific site in the genome. Second, the Ura+ Kanr isolates are re-transformed with an approximately 140 bp double-stranded oligonucleotide that flanks the URA3-Kanr insertion site and contains a mutation near its center. Transformants are selected that replace the marker genes with the mutated oligonucleotide.
Here we report two methods for targeting mutations efficiently into the yeast genome. These methods are related to delitto perfetto, however they substitute a PCR fragment for the long oligonucleotide. In the first method, termed site-specific genomic (SSG) mutagenesis, a point mutation is introduced at a targeted nucleotide in the genome. In the second method, termed random domain-localized (RDL, pronounced "riddle") mutagenesis, mutations are introduced at random within a single domain of a gene. For both methods, a single intermediate strain can be used to create many different mutant alleles.
Results
Site-specific genomic (SSG) mutagenesis in the USC2 region of the IME1 promoter
SSG mutagenesis involves two sequential transformations (Fig 1). For the first transformation, a PCR fragment containing URA3 is integrated into the yeast genome at a site 20–500 bp from the intended site of mutation; for the second transformation, URA3 is replaced with a PCR fragment containing the mutation near one end of the fragment (see Methods).
Figure 1.
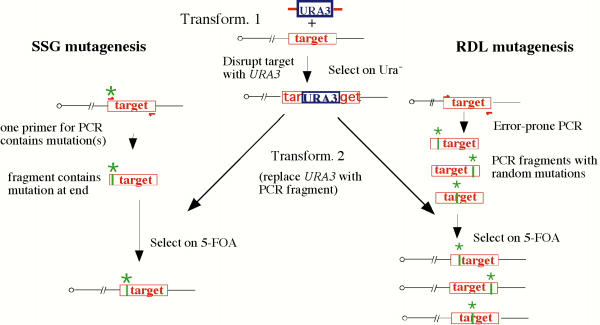
Introduction of mutations into the genome by site-specific genomic (SSG) and random domain-localized (RDL) mutagenesis. Both methods involve two sequential transformations. The first transformation (Transform. 1 in center of diagram) disrupts the targeted sequence with a PCR fragment containing URA3. The left and right ends of the fragment share 40 bp of homology to either side of the insertion site. In SSG mutagenesis (left side of figure), the fragment is inserted into the target region, and in RDL mutagenesis (right side of figure) the fragment replaces the target sequence. Transformants are selected on medium lacking uracil. The second transformation (Transform. 2) replaces URA3 with a PCR fragment containing one or more mutations (represented by an asterisk). In SSG mutagenesis, the mutations are at one end of the fragment and were introduced on the primer. In RDL mutagenesis, the mutations are introduced by error-prone PCR at random in the fragment. Transformants are selected on medium containing 5-fluoroorotic acid (5-FOA) to select for loss of URA3.
As a first demonstration of SSG mutagenesis, we introduced a mutation in the UCS2 region of the IME1 promoter, a region critical for transcriptional control of this gene [18]. For the first transformation, URA3 was inserted 36 bp from the intended site of the mutation. In this trial, >100 Ura+ isolates were identified, of which 26 were analyzed by diagnostic PCR. Of these 26 isolates, seven were integrated at the correct location; the remaining 19 transformants were probably gene conversion events at the ura3-1 locus [19]. For the second transformation, the URA3 strain generated in the first step was retransformed with a 707 bp PCR fragment amplified from wild-type genomic DNA (Fig. 2A). As discussed below, it was necessary to cotransform this fragment with a plasmid containing another marker, in this case a high copy plasmid bearing the LEU2 marker. One end of the PCR fragment corresponded to sequences 46 bp upstream of the URA3 insertion site and contained a single mutation 10 bp from this end. The other end corresponded to sequences 661 bp downstream of the URA3 insertion site. After screening transformants on FOA, six FOAr colonies were identified, of which four had deleted URA3. As revealed by diagnostic PCR and subsequent sequencing of the region, all four of these isolates had incorporated the single base pair mutation. Thus point mutations can be efficiently introduced into the genome by two sequential transformations with PCR fragments.
Figure 2.
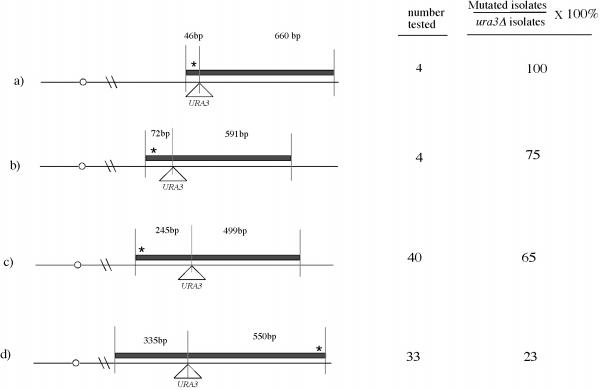
SSG mutagenesis at four sites. The diagrams on the left represent the PCR fragment used in the second transformation (thick horizontal bar) and the targeted site on the chromosome (thin horizontal line). The triangle below each chromosome represents the site that URA3 is inserted in the first transformation. The numbers above each PCR fragment are the base pairs between the URA3 insertion site and the ends of the fragment The first data column shows the number of ura3Δ transformants, the second column shows the percentage of ura3Δ transformants that contained the mutation. In all cases, the mutations were 10–20 bp from the end of the fragment.
In the above experiment, a single bp mutation was inserted. In a second trial, we attempted to introduce two mutations within a 3 bp region (Fig 2B). In this trial, the site of URA3 insertion (in the first transformation) was 463 bp from the site of insertion used in the first trial. In the second transformation, mutations were targeted 49 and 51 bp from the URA3 site. For this second trial, four FOAr colonies were recovered, and diagnostic PCR revealed that all of these isolates were deleted for URA3. These PCR also indicated that three of the four FOAr isolates contained both mutations. The fourth isolate contained the wild-type sequence.
Requirement for co-transformation in SSG mutagenesis
For the second transformation described above, the PCR fragment containing the mutation was co-transformed with a plasmid vector containing a selectable marker (in this case LEU2). Transformants were first selected for the presence of the plasmid and then either screened or selected for loss of URA3. To determine whether this cotransformation was necessary, we also plated transformed cells from the first SSG mutagenesis trial directly on FOA medium. After five days of growth, we analyzed FOAR colonies for growth on Ura- medium and for loss of URA3 (by diagnostic PCR). Of 102 colonies growing on FOA medium, all were phenotypically Ura-, but none contained a deletion of URA3. We observed similar results in other trials. Thus, transformants must be allowed to grow before they can be screened or selected on FOA. Interestingly, when the Leu- colonies were replica-plated to FOA medium, only a fraction of cells in any of the colonies were FOAR. Thus generation of FOAr cells may be a relatively late event during the growth of the colony.
Introduction of mutation at sites distant from marker
In the two SSG mutageneses described above, mutations were incorporated 36–51 bp from the URA3 insertion site. We next asked whether mutations could be incorporated at greater distances from the URA3 site. For this purpose, in the second transformation we used PCR fragments in which the end containing the mutation(s) was either 250 bp upstream (Fig. 2C) or 539 bp downstream from the URA3 insertion site (Fig 2D). In both of the experiments, as in Fig 2B, the mutated primer contained two mutations within a 3 bp region. In these experiments, we isolated 30–40 independent ura3Δ isolates from a single transformation (Fig 2, first data column). Of these ura3Δ isolates, a significant fraction were found to incorporate the mutation (Fig. 2, second column). The yield of ura3Δ transformants in the experiments shown in Fig. 2A and 2B was lower than in the experiments shown in Fig. 2C and 2D. This difference may result from the long region of homology on both sides of the insertion site in the latter experiments.
Comparing all four of the SSG mutageneses shown in Fig. 2, the frequency of incorporating mutations among the ura3Δ isolates was highest for the mutation closest to URA3 and lowest for the mutation farthest from this marker (Fig. 2, second column). These results suggest that only a portion of the PCR fragment is incorporated into the chromosome. However, the four experiments shown in Fig. 2 differed in several ways, including the site of mutation and the size of the PCR fragment. For this reason, the relationship between the frequency of incorporating mutations and the distance from URA3 was investigated further, as described in the next section.
Incorporation of a multiply-mutated PCR fragment into the genome
Substrates containing multiple polymorphisms are useful for mapping regions of heteroduplex [20]. To directly test the hypothesis that the frequency of incorporating an SSG mutation depends on the distance of this mutation from URA3, we constructed a plasmid (pS660) carrying a 750 bp region of the IME1 promoter that contained 9 mutations (Fig. 3A, i). This plasmid was used as a template to amplify the multiply-mutated UCS2 region, and the resulting PCR fragment was transformed into a strain containing URA3 inserted in this region (Fig. 3A, ii). The UCS2 region was amplified and sequenced in ten independent ura3Δ transformants. Consistent with the results shown in Fig. 2, the frequency of incorporating a particular mutation decreased dramatically with the distance of the mutation from the site of URA3 integration (Fig. 3B, circles). For example, mutations close to the URA3 site were incorporated at 100% efficiency, whereas mutations >400 bp from this site were incorporated in less than 20% of the transformants. Although only 10 transformants were sequenced, the difference between the frequency of incorporating the mutation nearest to the marker and the frequency of incorporating the mutation farthest from the marker is significant (P < 0.005). To verify that the gradient of incorporation frequency was determined by the URA3 insertion site, we transformed the same multiply-mutated PCR fragment into a strain containing a URA3 insertion at a different position in UCS2 (Fig 3A, iii). As in the previous experiment, mutations near the URA3 site incorporated at 100% efficiency, and the efficiency of incorporation dropped with increasing distance from the site (Fig. 3B, triangles). Examining the sequence of 20 transformants yielded further confirmation that the frequency of incorporating mutations decreased as the distance from the marker increased. Each sequence revealed an uninterrupted tract of mutations, starting at the site nearest the marker and extending in both directions for variable distances from this site.
Figure 3.
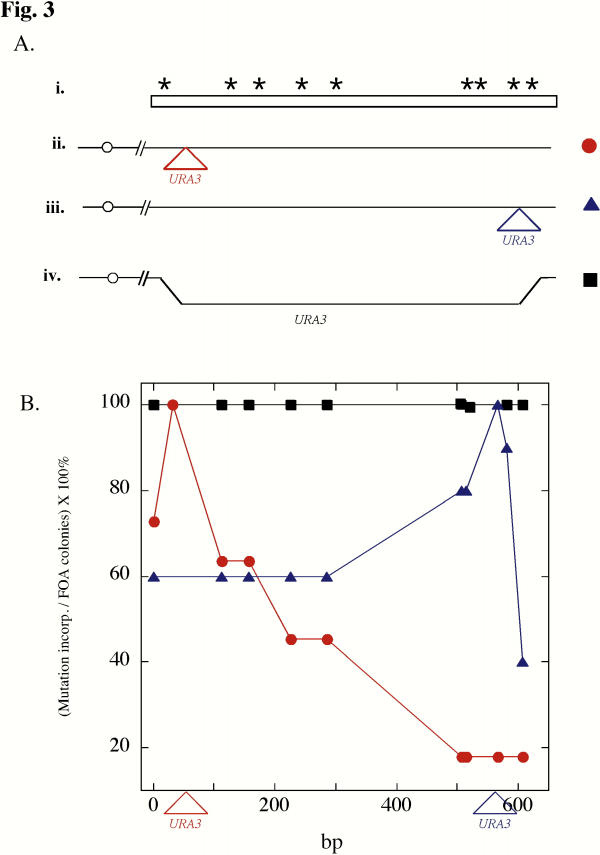
Incorporation of multiply-mutated PCR fragment. A) Diagram of a multiply-mutated PCR fragment (i) and the chromosomal region of different strains (ii, iii, and iv), each of which contain URA3 integrated at a different position. The approximate sites of each of the nine mutations are represented by asterisks. The site of URA3 insertion in the first two strains is indicated by triangles, and the region replaced by URA3 in the third strain is indicated by a displaced line. The URA3 strains diagrammed in (ii) and (iii) contained a mutation 20 bp from the URA3 insertion that was not present in the PCR fragment, thus there were actually 10 polymorphisms between the PCR fragment and the chromosome. B) The x-axis represents a linear map of the targeted region of the genome. Triangles below the x-axis show the site of the URA3 insertions for strain 1 (red triangle) and strain 2 (blue triangle). The y-axis shows the frequency of incorporation for each mutation after transformation of the strains diagrammed in A (ii–iv) with the PCR fragment diagrammed in A (i). Strain 1, red circles; strain 2, blue triangles; strain 3, black squares).
Effect of genome deletions on the frequency of incorporating mutations
As described above, only a portion of the PCR fragment is incorporated into the target region of the genome. One means to ensure that the majority of a PCR fragment is incorporated into the genome is to limit the homology between the fragment and the chromosome to the ends of the fragment. To limit homology to the fragment ends, we modified the method for creating strains in the first transformation such that the targeted region of the chromosome was deleted at the same time that URA3 is inserted in the genome.
To verify that the entire deleted region would be incorporated when homology was limited to the ends, we generated a strain in which the entire UCS2 sequence was replaced with URA3 (Fig 3A, iv). This strain was transformed with the multiply-mutated UCS2 fragment described above. The fragment contained 200–300 bp of homology on either side of the deletion. Six FOAR colonies were chosen, and the UCS2 region from these isolates were amplified and then sequenced. We found that all transformants had deleted URA3 and incorporated all 9 mutations (Fig. 3B, open circles). Thus deletion of genomic sequence in the first transformation allowed mutations to be introduced efficiently throughout the deleted region.
Random domain-localized (RDL) mutagenesis
Based on the results described in the previous section, we developed a variation of SSG mutagenesis, termed random domain-localized (RDL or "riddle") mutagenesis, to produce random mutations constrained within one domain of a gene at its native locus. In brief, RDL mutagenesis, like SSG mutagenesis involves two sequential transformations (Fig 1A, right side). The first transformation deletes the targeted region and replaces it with URA3. The second transformation replaces URA3 with a randomly-mutagenized copy of the targeted region. These randomly-mutagenized fragments were synthesized under error-prone PCR conditions such that each PCR fragment will contain on average one mutation (see Methods).
As an initial test of RDL mutagenesis, we targeted the 750 bp UCS region of the IME1 promoter for RDL mutagenesis. After the second transformation, we screened 60 FOAR isolates by diagnostic PCR. We found that 37 of these isolates had replaced the URA3 gene with the PCR fragment. The UCS2 region in eight of these isolates was sequenced. We found that all of these isolates contained from 1–4 mutations in the UCS2 region, and each isolate contained different mutations. Thus RDL mutagenesis is an efficient method for targeting mutations at random to a single domain of the yeast genome.
Discussion
New methods for directing genomic mutations in yeast
We report two methods for efficiently targeting mutations to the yeast genome using PCR fragments. In the first method, site-specific genomic (SSG) mutagenesis, the PCR fragment is synthesized using a primer containing a mutation. In the second method, random domain-localized (RDL) mutagenesis, the PCR fragment is synthesized under error-prone conditions. Because SSG and RDL mutagenesis do not leave marker genes or other non-native sequences near the mutation, these methods permit study of the effect of mutations separate from the potentially confounding effects of surrounding non-native sequence (see Background).
SSG mutagenesis is similar to the delitto perfetto method for introducing genomic site-specific mutations using long oligonucleotides [17]. The principal advantage of SSG mutagenesis over delitto perfetto, in addition to not requiring synthesis of long oligonucleotides, is that a single strain generated in the first transformation can be used to integrate mutations over a range of at least 500 bp on either side of the insertion site. This increased range allows the same strain to be used to make many different mutations. For example, we introduced one mutation that was 550 bp to the 3' side of URA3 and another mutation that was 250 bp to the 5' side of URA3. Another difference between SSG mutagenesis and delitto perfetto is that the latter method uses a tandem array of URA3 and kanr as the selectable markers, whereas SSG mutagenesis uses only URA3.
Co-transformation is required for SSG and RDL mutagenesis
In both SSG and RDL mutagenesis, replacement of URA3 with a PCR fragment requires cotransformation with a yeast plasmid and then selection for the plasmid prior to selection on FOA medium. It is possible that a phenotypic lag occurs before the concentration of Ura3p enzyme decreases to a level sufficient to allow growth on FOA medium. However, we found that only a fraction of the cells in any colony growing on the Leu- medium are FOAR. This latter result suggests that replacement of URA3 or loss of the extruded URA3 fragment occurs during growth of the colony.
Heteroduplex formation may extend for variable distances from the ends of the fragment
Our results indicate that when a large region of a PCR fragment is homologous to the genome only a portion of this fragment is incorporated [21]. Specifically, we found that sequences close to the site of URA3 insertion are incorporated at very high efficiencies, but sequences increasingly distant from URA3 are incorporated at progressively lower efficiencies. This gradient of genetic exchange was observed in experiments using singly-mutated fragments and also in experiments using multiply-mutated fragments. One explanation for this gradient would be that the site of physical exchange (crossover) often occurs at sequences between the URA3 insertion site and the mutation; hence, the frequency of such crossovers increases proportionately with the distance between the insertion site and the mutation. However, an abundance of evidence indicates that recombination initiates at double-strand breaks in yeast [22]. Thus a more likely explanation for the gradient of mutation incorporation is that recombination initiates at only one end of the fragment and heteroduplex formation (or repair) extends from this end for variable distance [22].
Recombination may initiate at either one end or the other of the fragment
The frequency of incorporating mutations does not decrease to zero at sites increasingly distant from the site of URA3 insertion. Instead, we observed a biphasic dependence of incorporating mutations with the distance of the mutation from the marker. For example, in an experiment where the 600 bp multiply mutated fragment was targeted to the genome, across the range of mutations from 20 bp – 300 bp from the marker, the frequency of incorporating mutations decreased almost two-fold in proportion to the distance of the mutation from the marker. In contrast, across the range from 300 bp to 600 bp from the marker, the frequency of incorporating mutations did not change.
One explanation for the biphasic relationship between the efficiency of incorporating mutations and the distance from the URA3 marker is suggested by the proposal that recombination between a linear fragment and the chromosome involves invasion of only one of the two ends [21]. We propose that either end of the PCR fragment can invade first, and that heteroduplex extends a variable distance from this end, first through the homologous region on the end of the fragment that initiates recombination, then through the non-homologous regions (URA3), and finally through the homologous region on the other end of the fragment. If this is the case, then the frequency of incorporating a given mutation will depend on which end invades first. For example, if the end containing the mutation initiates recombination, then all molecules that delete URA3 must incorporate this mutation into heteroduplex. In contrast, when the non-mutated end initiates recombination, then the frequency of incorporating a given mutation into heteroduplex depends on how far the mutation is from the marker.
As a result of the recombinogenic nature of DNA ends, mutations can be incorporated efficiently in SSG mutagenesis at least 550 bp distal to the URA3 insertion site and possibly at much greater distances. Indeed, for SSG mutagenesis, because the number of ura3Δ isolates increased as the region of homology increased, we recovered many independent mutants containing a mutation incorporated 550 bp from URA3. In RDL mutagenesis, because the entire targeted region is deleted in the first transformation, any recombination event that replaces URA3 must also incorporate the entire targeted region.
Effect of DNA ends on the frequency of incorporating mutations
The frequency of incorporating mutations at a given end may depend on its sequence. For example, when we transformed a multiply-mutated fragment into a strain, the frequency of incorporating mutations at one end was approximately was four-fold higher than the frequency of incorporating mutations at the other end. Furthermore, the frequency of incorporating mutations at these two ends did not depend on whether the PCR fragment was transformed into a strain containing URA3 near the right or left end of the homologous region. The simplest interpretation of this result is that one end of the fragment initiates recombination at a higher frequency than the other end. The frequency of recombination at a specific region may be influenced by the chromatin structure of the region, the binding of proteins to this region, or the distance of the region from replication origins, transcriptional start sites, or other chromosomal elements [23-25]. In addition, fragment ends with high GC content may integrate into the chromosome more efficiently than ends with lower GC content [19]. Indeed, for the fragment described above, the end that incorporated mutations at high frequency had 50% GC nucleotides over its final 20 bp, whereas the other end had only 40% GC content through this region.
Conclusion
In summary, both random domain-localized and site-specific mutations can be introduced directly into the genome by methods that are as rapid and efficient as introducing these same mutations on plasmids. These methods can be used to analyze the effect of genomic mutations on transcriptional regulatory elements, replication origins, centromeres, and telomeres. The potential scope of SSG and RDL mutagenesis may extend beyond the uses described in this study; for example, these methods can also be used to introduce mutations into ORFs. Even essential genes could be mutated by this method, provided that the URA3 insertion is placed downstream of the gene. Furthermore, SSG mutagenesis could potentially be used to introduce other types of genetic changes into the yeast genome, including insertion of epitope tags and construction of small deletions.
Methods
Yeast strains
SSG mutagenesis was performed on the strain SH773 (MATα ade2 can1:ADE2:CAN1 his3-11,15 leu2-3,112 trp1-3'del ura3-1) [26] and RDL mutagenesis on the strain SH2867 (MATα ade2 can1:ADE2:CAN1 his3-11,15 leu2-3,112 trp1-3'del ura3-1 rme1Δ::LEU2 ime1Δ-lacZ:LEU2). Both strains are in the W303 background. SH2867 was constructed in two steps from SH1521, which contains an ime1Δ-lacZ(URA3) allele [27]. In the first step, the ime1Δ-lacZ(URA3) allele was converted to an ime1Δ-lacZ(LEU2) allele by transforming SH1521 with a PCR fragment containing LEU2 and 40 bp tails homologous to the sequences surrounding URA3. In the second step, the resulting strain (SH2675) was crossed to an rme1Δ::LEU2 strain (SH1337) in the same genetic background; and the resulting diploid was sporulated and dissected to generate SH2867.
PCR amplification and mutation analysis
All primers used in this study are shown in Table 1 (see additional data files). PCRs contained 1 μM of primer, 0.1 mM dNTPs, 1.5 mM MgCl2, 10 mM Tris-HCl (pH 8.3), 50 mM KCL, and 3 units/100 μl reaction of TaqI polymerase (Promega). PCRs to amplify DNA from purified DNA (0.1 μg) were heated to 95°C for 5 min., and then 10 cycles of 94°C for 30 sec., 54°C for 1 min., 72°C for 1 min./kb and another 20 cycles of: 94°C for 30 sec., 65°C for 1 min., and 72°C for 1 min./kb. An elongation step at 72°C for 10 min was added at the end of the reaction. URA3 and LEU2 alleles were amplified from pRS306 and pRS305 respectively [28]. PCRs using genomic DNA as template utilized yeast lysed by microwave radiation as described previously [19] and http://sgi.bls.umkc.edu/honigberglab/. PCR fragments were purified by Wizardprep columns (Promega) prior to yeast transformation, and by Qiaquick columns (Qiagen) prior to sequencing. DNA sequencing was performed by the University of Missouri-Columbia DNA Core Sequencing facility.
The PCR was also employed to identify genomic mutations (diagnostic PCR). Diagnostic PCRs to detect large insertions and deletions in the yeast genome utilized primers flanking the targeted region. Diagnostic PCRs to detect point mutations consisted of two parallel amplification reactions. In the first reaction, one primer was homologous to sequence immediately 5' to the mutation and its final nucleotide corresponded to the mutation; in the second reaction this primer was replaced with a primer differing only in the final nucleotide, which corresponded to the original sequence.
Error-prone PCR
An error-prone PCR [29] were used to create mutant alleles of UCS2. A UCS2 allele containing multiple mutations was generated in a PCR containing 0.1 mM MnCl2 and 2.4 mM MgCl2. UCS2 alleles containing an average of one mutation per fragment were generated in a PCR containing 0.01 mM MnCl2 and 4.0 mM MgCl2. The optimal conditions for both of these PCRs was determined by performing error prone PCR over a range of MnCl2 concentrations, digesting the resulting 1.05 kb fragment with XbaI and HindIII, ligating the resulting 752 bp fragment into these same sites in pRS306 [28], and sequencing 5–6 representative clones from each reaction. The clone containing 9 mutations was designated pS660.
Site-specific genomic (SSG) mutagenesis
SSG mutagenesis involves two sequential transformations (Fig. 1, center). For the first transformation, we amplified a fragment containing URA3. The 60 bp primers used to synthesize this fragment had 20 bp matching URA3 at the 3' end and 40 bp matching either side of the targeted insertion point at the 5' end. Thus the ends of the PCR fragment were homologous to either side of the insertion point, and the middle of the fragment contained the entire URA3. SH773 was transformed with this PCR fragment [30], and transformants were selected as Ura+ colonies. Integration of URA3 at the targeted site was confirmed by diagnostic PCR.
For the second transformation (Fig 1, left side), the URA3 strain created in the first transformation was co-transformed with a high-copy LEU2 plasmid (YEp351) and a 600–900 bp PCR fragment spanning the URA3 insertion site. This PCR fragment was amplified from wild-type genomic DNA using two 25–40 bp primers. One of these primers contained 1–2 bp mutations near its center.
Transformants were selected by two methods. In the first method, approximately 20% of the transformation mixture (1 × 108 cells) was placed in 25 ml of Leu- liquid medium and grown for 40 hours, and then 1 × 108 cells were plated on medium containing 5-fluoroorotic acid (FOA) and incubated for three days to select for isolates that deleted the URA3 marker. In the second method, the transformation mixture was plated on Leu- medium at a density of approximately 10,000 colonies/plate on 10 plates. After 3 days of growth at 30°C, the Leu- plates were replica-plated to FOA. Papillations growing after 24 hours were tested by diagnostic PCR for deletion of URA3 and incorporation of the mutations. Isolates that had deleted URA3 were amplified from the genome and sequenced. The first method recovers transformants efficiently and requires fewer plates, but the second method ensures that all transformants derive from independent transformation events. For this reason, all data in Fig. 1 and Fig. 2 were derived by the second method.
Random domain-localized (RDL) mutagenesis
Like SSG mutagenesis, RDL mutagenesis requires two sequential transformations. The first transformation is similar to SSG mutagenesis, except that the region targeted for mutagenesis is deleted and replaced with URA3. In the second transformation, the strain created by the first transformation was co-transformed with a high-copy TRP1 plasmid (pTV3) and PCR fragments that span the URA3 insertion site. These fragments were generated by error-prone PCR such that they contain on average one mutation per fragment. A slightly larger PCR fragment amplified from genomic DNA was used as the template for error-prone PCR. Transformants were initially selected on Trp- medium and then screened or selected on FOA medium as described above for SSG mutagenesis. FOAr isolates that replaced URA3 with a PCR fragment were identified by diagnostic PCR.
Authors' contributions
MG carried out many of the experiments in the study and contributed to the development of the method. SMH conceived of the study, participated in its design and coordination, and prepared the manuscript. MK contributed to the design of the method and the interpretation of results.
Supplementary Material
Table 1; short description of data: primers used in this study
Acknowledgments
Acknowledgements
We thank Sarah Piccirillo and Shawn Atkinson for technical assistance. Research was funded by NIH grant R01-GM58013 to SMH and grants from the Israel Science Foundation and the Kurt Lion Foundation to MK.
Contributor Information
Misa Gray, Email: graymi@umkc.edu.
Martin Kupiec, Email: martin@post.tau.ac.il.
Saul M Honigberg, Email: hongibergs@umkc.edu.
References
- Rothstein R. Targeting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Methods Enzymol. 1991;194:281–301. doi: 10.1016/0076-6879(91)94022-5. [DOI] [PubMed] [Google Scholar]
- Baudin A, Ozier -K, Denouel A, Lacroute F, Cullin C. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res. 1993;21:3329–3330. doi: 10.1093/nar/21.14.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz M, Muir R, Lim E, J. M, Weber S, Heitman J. Gene disruption with PCR products in Saccharomyces cerevisiae. Gene. 1995;158:113–117. doi: 10.1016/0378-1119(95)00144-U. [DOI] [PubMed] [Google Scholar]
- Amberg DC, Botstein D, Beasley EM. Precise gene disruption in Saccharomyces cerevisiae by double fusion polymerase chain reaction. Yeast. 1995;11:1275–1280. doi: 10.1002/yea.320111307. [DOI] [PubMed] [Google Scholar]
- Wach A. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast. 1996;12:259–265. doi: 10.1002/(SICI)1097-0061(19960315)12:3<259::AID-YEA901>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Guldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kotter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang CY, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Botstein D, Shortle D. Strategies and applications of in vitro mutagenesis. Science. 1985;229:1193–1201. doi: 10.1126/science.2994214. [DOI] [PubMed] [Google Scholar]
- Weiler KS, Wakimoto BT. Heterochromatin and gene expression in Drosophila. Annu Rev Genet. 1995;29:577–605. doi: 10.1146/annurev.ge.29.120195.003045. [DOI] [PubMed] [Google Scholar]
- Labrador M, Corces VG. Setting the boundaries of chromatin domains and nuclear organization. Cell. 2002;111:151–154. doi: 10.1016/S0092-8674(02)01004-8. [DOI] [PubMed] [Google Scholar]
- Emerson BM. Specificity of gene regulation. Cell. 2002;109:267–270. doi: 10.1016/S0092-8674(02)00740-7. [DOI] [PubMed] [Google Scholar]
- Thoma F. Protein-DNA interactions and nuclease-sensitive regions determine nucleosome positions on yeast plasmid chromatin. J Mol Biol. 1986;190:177–190. doi: 10.1016/0022-2836(86)90291-3. [DOI] [PubMed] [Google Scholar]
- Scott-Drew S, Wong CM, Murray JA. DNA plasmid transmission in yeast is associated with specific sub- nuclear localisation during cell division. Cell Biol Int. 2002;26:393–405. doi: 10.1006/cbir.2002.0867. [DOI] [PubMed] [Google Scholar]
- Holmes SG, Mitchell Smith M. Replication of minichromosomes in Saccharomyces cerevisiae is sensitive to histone gene copy number and strain ploidy. Yeast. 2001;18:291–300. doi: 10.1002/1097-0061(20010315)18:4<291::AID-YEA668>3.3.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Hegemann JH, Klein S, Heck S, Guldener U, Niedenthal RK, Fleig U. A fast method to diagnose chromosome and plasmid loss in Saccharomyces cerevisiae strains. Yeast. 1999;15:1009–1019. doi: 10.1002/(SICI)1097-0061(199907)15:10B<1009::AID-YEA396>3.3.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Boeke JD. In vitro mutagenesis and plasmid shuffling: from cloned gene to mutant yeast. Methods Enzymol. 1991;194:302–318. doi: 10.1016/0076-6879(91)94023-6. [DOI] [PubMed] [Google Scholar]
- Kitazono AA, Tobe BT, Kalton H, Diamant N, Kron SJ. Marker-fusion PCR for one-step mutagenesis of essential genes in yeast. Yeast. 2002;19:141–149. doi: 10.1002/yea.806. [DOI] [PubMed] [Google Scholar]
- Storici F, Lewis LK, Resnick MA. In vivo site-directed mutagenesis using oligonucleotides. Nat Biotechnol. 2001;19:773–776. doi: 10.1038/90837. [DOI] [PubMed] [Google Scholar]
- Sagee S, Sherman A, Shenhar G, Robzyk K, Ben-Doy N, Simchen G, Kassir Y. Multiple and distinct activation and repression sequences mediate the regulated transcription of IME1, a transcriptional activator of meiosis- specific genes in Saccharomyces cerevisiae. Mol Cell Biol. 1998;18:1985–1995. doi: 10.1128/mcb.18.4.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray M, Honigberg SM. Effect of chromosomal locus, GC content and length of homology on PCR- mediated targeted gene replacement in Saccharomyces. Nucleic Acids Res. 2001;29:5156–5162. doi: 10.1093/nar/29.24.5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS, Petes TD. Expansions and contractions of the genetic map relative to the physical map of yeast chromosome III. Mol Cell Biol. 1988;8:595–604. doi: 10.1128/mcb.8.2.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, McGill C, Shafer B, Strathern JN. Ends-in vs. ends-out recombination in yeast. Genetics. 1993;135:973–980. doi: 10.1093/genetics/135.4.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilera A, Chavez S, Malagon F. Mitotic recombination in yeast: elements controlling its incidence. Yeast. 2000;16:731–754. doi: 10.1002/1097-0061(20000615)16:8<731::AID-YEA586>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Abdullah MF, Borts RH. Meiotic recombination frequencies are affected by nutritional states in Saccharomycescerevisiae. Proc Natl Acad Sci U S A. 2001;98:14524–14529. doi: 10.1073/pnas.201529598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TC, Lichten M. Factors that affect the location and frequency of meiosis-induced double-strand breaks in Saccharomyces cerevisiae. Genetics. 1995;140:55–66. doi: 10.1093/genetics/140.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RH, Honigberg SM. Nutritional regulation of late meiotic events in Saccharomyces cerevisiae through a pathway distinct from initiation. Mol Cell Biol. 1996;16:3222–3232. doi: 10.1128/mcb.16.6.3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purnapatre K, Piccirillo S, Schneider BL, Honigberg SM. The CLN3/SWI6/CLN2 pathway and SNF1 act sequentially to regulate meiotic initiation in Saccharomyces cerevisiae. Genes Cells. 2002;7:675–691. doi: 10.1046/j.1365-2443.2002.00551.x. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell RC, Joyce GF. Mutagenic PCR. PCR Methods Appl. 1994;3:S136–S140. doi: 10.1101/gr.3.6.s136. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96. doi: 10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table 1; short description of data: primers used in this study