

Through a series of follow up experiments, the authors have come to understand that the genotype and therefore the in vivo phenotype of the Δbbk46/vector B. burgdorferi clone (1470), the data for which are shown in Table 4, were incorrect. In the publication, the mutant clone was reported to contain all of the B. burgdorferi plasmids of the wild type parent clone; however, this finding was interpreted in error and the authors now understand that in addition to lacking gene bbk46, the mutant clone also lacks plasmid lp28-1. It has been reported previously that B. burgdorferi clones lacking lp28-1 are unable to evade the host immune response resulting in the inability of the spirochetes to persist in mice [1–5]. Because the Δbbk46/vector B. burgdorferi clone (1470) lacks lp28-1, this suggests that the persistence phenotype (lack of spirochete reisolates in the tissues of infected mice 3 weeks post inoculation) shown in Table 4 is a result of the missing lp28-1 plasmid rather than the bbk46 gene. The authors have just completed careful re-derivation of the Δbbk46/vector B. burgdorferi clone (1607), genotype analysis and analysis of the phenotype of the new clone in mouse infectivity. The authors now find that the new Δbbk46/vector B. burgdorferi clone (1607), which contains all of the B. burgdorferi plasmids of the wild-type parent clone, demonstrates wild type serology and tissue reisolation in the 9 out of 9 mice inoculated with 1×104 spirochetes. These data confirm that the persistence phenotype reported for the Δbbk46/vector B. burgdorferi clone (1470) in Table 4 of the publication is due to the loss of the lp28-1 plasmid and not the deletion of the bbk46 gene. The authors have confirmed that the reported genotypes of all other clones in the publication are correct.
Table 4. The bbk46 gene is dispensable for B. burgdorferi mouse infection by needle inoculation.
| Clone | Serologya | Positive reisolation of spirochetes from mouse tissuesb | ||
| Ear | Bladder | Joint | ||
| Δbbk46/vector | 9/9 | 9/9 | 9/9 | 9/9 |
| Δbbk46/bbk46 + | 5/6 | 5/6 | 5/6 | 5/6 |
Determined 3 weeks post inoculation by serological response to B. burgdorferi total protein lysate.
Number of mice positive for spirochete reisolation/ number of mice analyzed. NA, not applicable.
The conclusion that the bbk46 gene is critical for the ability of the spirochete to evade the humoral immune response and persistently infect mice is incorrect. Therefore, all text in the Abstract, Author Summary, Introduction, Results, and Discussion relating to the infectivity phenotype of the original bbk46 deletion mutant is invalid. Furthermore, as the in vivo-expressed bbk46 gene identified in the IVET screen was incorrectly reported to be a novel virulence factor critical for Borrelia burgdorferi persistence in mice the corrected title for this publication should read “In Vivo Expression Technology Identifies Novel Borrelia burgdorferi Genes Expressed during an Active Murine Infection.”
The conclusions of this publication which remain valid are the development and application of in vivo expression technology (IVET) in B. burgdorferi to identify B. burgdorferi genes that are expressed during an active murine infection. Specifically, 289 non-identical B. burgdorferi in vivo-expressed sequences were identified that mapped to distinct classes of putative regulatory sequences across the genome including 80 sequences that mapped to canonical promoter positions upstream of annotated open reading frames. Characterization of the BbIVET identified candidate in vivo-expressed promoter sequence, Bbive162, which mapped immediately upstream of the uncharacterized BBK46 open reading frame on virulence plasmid lp36, revealed more than 100-fold induction during mammalian infection compared to in vitro growth. Furthermore, bbk46 expression was demonstrated to be RpoS-independent. Finally, the bbk46 open reading frame was found to be unable to produce detectable amounts of protein during in vitro growth and recombinant BBK46 protein was non-immunoreactive with immune serum from mice infected with B. burgdorferi.
In addition, the ORF associated with Bbive clone 290 in Table 3 should read BB0775 rather than BB0755 as originally listed and BB0181 should be noted as a homolog of FlgK.
Table 3. B. burgdorferi in vivo-expressed candidate genes organized by functional category.
| Bbive clonea | Replicon | ORFb | Protein designation, Annotated functionc |
| Cell division | |||
| 289 | chromosome | BB0715 | FtsA cell division protein |
| Cell envelope | |||
| 15 | chromosome | BB0213 | Putative lipoprotein |
| 94 | chromosome | BB0760 | Gp37 protein |
| 175 | lp54 | BBA36 | Lipoprotein |
| 271 | lp54 | BBA57 | Lipoprotein |
| 297 | lp25 | BBE16 | BptA |
| 151 | lp28-2 | BBG01 | Putative lipoprotein |
| 267 | lp38 | BBJ34 | Putative lipoprotein |
| 269 | lp38 | BBJ51 | VlsE paralog, pseudogene |
| 162 | lp36 | BBK46 | Immunogenic protein P37, authentic frameshift |
| 77 | cp32-8, cp32-3, cp32-7, cp32-9, lp56, cp32-4,cp32-6, cp32-1 | BBL28, BBS30, BBO28, BBN28, BBQ35, BBR28, BBM28, BBP28 | Mlp lipoprotein family |
| DNA replication | |||
| 274 | chromosome | BB0111 | DnaB replicative helicase |
| 226 | chromosome | BB0632 | RecD exodeoxyribonuclease V, alpha chain |
| 152 | lp28-3 | BBH13 | RepU replication machinery |
| Energy metabolism | |||
| 62 | chromosome | BB0057 | Gap glyceraldehyde-3-phosphate dehydrogenase, type 1 |
| 34 | chromosome | BB0327 | Glycerol-3-phosphate O acyltransferase |
| 44 | chromosome | BB0368 | NAD(P)H-dependent glycerol-3-phosphate dehydrogenase |
| 47 | chromosome | BB0381 | Trehalase |
| 81 | chromosome | BB0676 | Phosphoglycolate phosphate |
| Fatty acid and phospholipid metabolism | |||
| 85 | chromosome | BB0704 | AcpP acyl carrier protein |
| Motility and chemotaxis | |||
| 14 | chromosome | BB0181 | FlgK flagellar hook-associated protein |
| 29 | chromosome | BB0293 | FlgB flagellar basal body rod |
| 290 | chromosome | BB0775 | Flagellar hook-basal body complex protein |
| 65 | chromosome | BB0551 | CheY-1 chemotaxis response regulator |
| 222 | chromosome | BB0568 | Chemotaxis response regulator protein-glutamate methylesterase |
| Prophage function | |||
| 295 | cp32-8, cp32-7, cp32-1, cp32-3, cp32-6, cp32-4 | BBL23, BBO23, BBP23, BBS23, BBM23, BBR23 | Holin BlyA family |
| Protein fate | |||
| 193 | chromosome | BB0031 | LepB signal peptidase I |
| Protein synthesis | |||
| 202 | chromosome | BB0113 | RpsR ribosomal protein S18 |
| 216, 217 | chromosome | BB0485 | RplP ribosomal protein L16 |
| 58 | chromosome | BB0495 | RpsE 30S ribosomal protein S5 |
| 59 | chromosome | BB0496 | 50S ribosomal protein L30 |
| 219 | chromosome | BB0503 | RplQ ribosomal protein L17 |
| 232 | chromosome | BB0660 | GTP-binding Era protein |
| 288 | chromosome | BB0682 | TrmU tRNA (5-methylaminomethyl-2-thiouridylate)-methyltransferase |
| Regulation | |||
| 208 | chromosome | BB0379 | Protein kinase C1 inhibitor |
| 50 | chromosome | BB0420 | Hk1 histidine kinase |
| Nucleoside salvage | |||
| 148 | lp25 | BBE07 | Pfs protein, pseudogene |
| Transcription | |||
| 1 | chromosome | BB0389 | RpoB DNA-directed RNA polymerase, beta subunit |
| 287 | Chromosome | BB0607 | PcrA ATP-dependent DNA helicase |
| 84 | chromosome | BB0697 | RimM 16S rRNA processing protein |
| Transport | |||
| 204 | chromosome | BB0318 | MglA methylgalactoside ABC transporter ATP-binding protein |
| 46 | chromosome | BB0380 | MgtE Mg2+ transport protein |
| Unknown | |||
| 56 | chromosome | BB0049 | Hypothetical protein |
| 69 | chromosome | BB0063 | Pasta domain protein |
| 2 | chromosome | BB0102 | Conserved hypothetical |
| 8 | chromosome | BB0138 | Conserved hypothetical |
| 13 | chromosome | BB0176 | ATPase family associated with various cellular activities |
| 23 | chromosome | BB0265 | Conserved hypothetical |
| 212 | chromosome | BB0428 | Conserved hypothetical |
| 52, 53 | chromosome | BB0429 | Conserved hypothetical |
| 220 | chromosome | BB0504 | Conserved hypothetical |
| 67 | chromosome | BB0562 | Conserved hypothetical |
| 223 | chromosome | BB0577 | Conserved hypothetical |
| 71 | chromosome | BB0592 | Caax amino protease family |
| 73 | chromosome | BB0619 | DHH family phosphoesterase function |
| 96 | chromosome | BB0799 | Conserved hypothetical |
| 240 | cp26 | BBB27 | Unknown essential protein |
| 145, 146 | lp25 | BBE0036 | Hypothetical protein |
| 147 | lp25 | BBE01 | Conserved hypothetical |
| 265, 266 | lp38 | BBJ30 | Conserved hypothetical |
| 171 | lp38 | BBJ36 | Conserved hypothetical |
| 75,173 | lp38 | BBJ46 | Conserved hypothetical |
| 129, 136, 296 | cp32-8, cp32-1, cp32-7, lp56, cp32-9 | BBL41, BBP40, BBO42, BBQ48, BBN41 | Conserved hypothetical |
| 130, 249 | cp32-8, cp32-1, cp32-6 | BBL42, BBP41, BBM41 | Conserved hypothetical |
| 117, 177 | cp32-6, lp56, cp32-9, cp32-8, cp32-3, cp32-1, cp32-4 | BBM18, BBQ25, BBN18, BBL18, BBS18,BBP18, BBR18 | Conserved hypothetical |
| 182, 183 | lp56 | BBQ41 | PF-49 protein |
| 188 | lp56 | BBQ84.1 | Conserved hypothetical |
| 189 | lp56, lp28-3, lp17 | BBQ89, BBH01, BBD01 | Conserved domain protein |
| 244 | cp32-4, cp32-3, cp32-6, p56, cp32-8, cp32-9 | BBR05, BBN05, BBM05, BBQ12, BBL05, BBO05, BBP05 | Lyme disease protein of unknown function |
In some cases two or more Bbive clones shared overlapping, non-identical sequence, as indicated by multiple Bbive clone numbers.
ORF, open reading frame that maps just downstream and in the same orientation to the Bbive sequence.
Annotation described by Fraser et al. [45].
Please see a corrected version of the article below.
Labandeira-Rey M, Seshu J, Skare JT (2003) The absence of linear plasmid 25 or 28-1 of Borrelia burgdorferi dramatically alters the kinetics of experimental infection via distinct mechanisms. Infect Immun 71: 4608–4613.
Labandeira-Rey M, Skare JT (2001) Decreased infectivity in Borrelia burgdorferi strain B31 is associated with loss of linear plasmid 25 or 28–1. Infect Immun 69: 446–455.
Lawrenz MB, Wooten RM, Norris SJ (2004) Effects of vlsE complementation on the infectivity of Borrelia burgdorferi lacking the linear plasmid lp28-1. Infect Immun 72: 6577–6585.
Purser JE, Norris SJ (2000) Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proc Natl Acad Sci U S A 97: 13865–13870.
Zhang JR, Hardham JM, Barbour AG, Norris SJ (1997) Antigenic variation in Lyme disease borreliae by promiscuous recombination of VMP-like sequence cassettes. Cell 89: 275–285.
In Vivo Expression Technology Identifies Novel Borrelia burgdorferi Genes Expressed during an Active Murine Infection
Tisha Choudhury Ellis1, Sunny Jain1, Angelika K. Linowski1, Kelli Rike1, Aaron Bestor2, Patricia A. Rosa2, Micah Halpern1, Stephanie Kurhanewicz1 and Mollie W. Jewett1*
1Burnett School of Biomedical Sciences, University of Central Florida College of Medicine, Orlando, Florida, United States of America
2Laboratory of Zoonotic Pathogens, Rocky Mountain Laboratories, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Hamilton, Montana, United States of America
*Mollie.Jewett@ucf.edu
Abstract
Analysis of the transcriptome of Borrelia burgdorferi, the causative agent of Lyme disease, during infection has proven difficult due to the low spirochete loads in the mammalian tissues. To overcome this challenge, we have developed an In Vivo Expression Technology (IVET) system for identification of B. burgdorferi genes expressed during an active murine infection. Spirochetes lacking linear plasmid (lp) 25 are non-infectious yet highly transformable. Mouse infection can be restored to these spirochetes by expression of the essential lp25-encoded pncA gene alone. Therefore, this IVET-based approach selects for in vivo-expressed promoters that drive expression of pncA resulting in the recovery of infectious spirochetes lacking lp25 following a three week infection in mice. Screening of approximately 15,000 clones in mice identified 289 unique in vivo-expressed DNA fragments from across all 22 replicons of the B. burgdorferi B31 genome. The in vivo-expressed candidate genes putatively encode proteins in various functional categories including antigenicity, metabolism, motility, nutrient transport and unknown functions. Candidate gene bbk46 on essential virulence plasmid lp36 was found to be highly induced in vivo and to be RpoS-independent. The bbk46 gene was dispensable for B. burgdorferi infection in mice. Our findings highlight the power of the IVET-based approach for identification of B. burgdorferi in vivo-expressed genes, which might not be discovered using other genome-wide gene expression methods. Further investigation of the novel in vivo-expressed candidate genes will contribute to advancing the understanding of molecular mechanisms of B. burgdorferi survival and pathogenicity in the mammalian host.
Author summary
Lyme disease is caused by tick-bite transmission of the pathogenic spirochete Borrelia burgdorferi. An increased understanding of how B. burgdorferi survives throughout its infectious cycle is critical for the development of innovative diagnostic and therapeutic protocols to reduce the incidence of Lyme disease. One of the major difficulties blocking this effort has been genome-wide identification of the B. burgdorferi genes that are expressed in the mammalian host environment. Using in vivo expression technology (IVET) in B. burgdorferi for the first time, we have identified B. burgdorferi genes that are expressed during an active murine infection. We demonstrate that candidate gene bbk46, encoded on essential linear plasmid 36, is highly expressed in vivo and, unlike some other known B. burgdorferi in vivo-induced genes, is not RpoS regulated. The bbk46 gene, however, was not found to be required for B. burgdorferi infection in mice. Further studies focused on analysis of the novel B. burgdorferi in vivo-expressed genes identified using IVET will provide insight into the ability of this pathogen to survive in the mammalian host.
Introduction
Lyme disease is a multi-stage inflammatory disease caused by the pathogenic spirochete Borrelia burgdorferi, which is transmitted by the bite of an infected tick [1]. B. burgdorferi has an enzootic life cycle that requires persistence in two disparate environments, the arthropod vector and the mammalian host. B. burgdorferi is well adapted to modulate its expression profile in response to the different conditions encountered throughout its infectious cycle [2]. Although the specific environmental signals that induce changes in spirochete gene expression are not fully defined, it has been reported that changes in temperature, pH, the presence or absence of mammalian blood, as well as changes in bacterial growth rate, can affect patterns of gene expression [2–8]. DNA microarray analysis and proteomics have been used to examine changes in the global expression profile of B. burgdorferi grown under in vitro conditions that partially mimic the tick and mouse environments [3–5]. A rat dialysis membrane chamber (DMC) implant model, together with microarray technology, has been used to help identify B. burgdorferi genes expressed in response to mammalian host-specific signals [7–10]. Although the data reported in these studies provide insight into the molecular mechanisms of gene regulation, they may not fully reflect the patterns of B. burgdorferi gene expression during an active mammalian infection. Furthermore, transcriptome analysis of B. burgdorferi during murine infection has proven difficult given that spirochete loads in the blood and tissues are too low to recover sufficient spirochete RNA for direct microarray analysis [11].
In vivo expression technology (IVET) is a gene discovery method used to identify transcriptionally active portions of a microbial genome during interaction of the microorganism with a particular environment or host organism [12,13]. In this system, the environment itself directly selects for upregulated bacterial loci [14]. The IVET selection system functions on the premise that deletion of a biosynthetic gene can lead to attenuation of growth and persistence of a pathogen in the host environment. This attenuation can be complemented by expression of the biosynthetic gene driven by promoters that are transcriptionally active in vivo. Thus, in the environment of interest, in vivo transcriptionally active promoters can be selected from a genomic library of DNA fragments cloned upstream of the essential biosynthetic gene [12–15]. IVET is a sensitive and versatile method for identification of in vivo-expressed genes that has been used with pathogenic bacteria and fungi in a wide variety of host environments and has identified a number of previously uncharacterized virulence genes [14–18].
Using IVET we have developed and applied a genome-wide genetic screening approach to identify B. burgdorferi genes that are expressed during an active murine infection. This is the first time that an IVET strategy has been applied to B. burgdorferi Furthermore, this approach resulted in the identification of novel B. burgdorferi in vivo-expressed genes.
Results
Mammalian host-adapted spirochetes demonstrate a 100-fold decrease in ID50 relative to in vitro grown spirochetes. It is clear that B. burgdorferi modulates its gene expression profile at different stages of the infectious cycle [2]. The infectious dose of wild-type B. burgdorferi varies depending on the environment from which the spirochetes are derived. For example, the 50% infectious dose (ID50) of spirochetes derived from partially fed ticks has been found to be two orders of magnitude lower than that of log phase in vitro grown B. burgdorferi [19]. In order to quantitatively assess the impact that adaptation to the mammalian environment has on B. burgdorferi infectivity the 50% infectious dose (ID50) of spirochetes derived directly from the mammalian host was determined and compared to that of log phase in vitro grown spirochetes. B. burgdorferi are only transiently present in the blood of immunocompetent mice [20], whereas spirochetes persist longer in the blood of immunocompromised mice [21]. Therefore, the blood of severe combined immunodeficiency (scid) mice infected with B. burgdorferi was used as a source of spirochetes adapted to the mammalian environment. Strikingly, an inoculum containing approximately eight in vivo-derived spirochetes was able to infect five out of six mice, whereas, 5,000 in vitro grown spirochetes were required to obtain this level of infectivity (Table 1). The ID50 for in vivo-derived spirochetes was found to be less than eight organisms. In contrast, the ID50 for in vitro grown spirochetes was calculated to be 660 organisms. These data indicate that mammalian host-adapted spirochetes are 100-fold more infectious than in vitro grown spirochetes, likely due to appropriate coordinate expression of in vivo-expressed genes important for murine infectivity.
Table 1. In vivo-adapted B. burgdorferi are highly infectious.
| In vivo grown spirochetes | In vitro grown spirochetes | ||
| Spirochete dose | Number of mice infecteda/number of mice analyzed | Spirochete dose | Number of mice infecteda/number of mice analyzed |
| 8×102 | 6/6 | 5×104 | 6/6 |
| 8×101 | 6/6 | 5×103 | 6/6 |
| 8×100 | 5/6 | 5×102 | 2/6 |
| 5×101 | 1/6 | ||
| 5×100 | 0/6 | ||
| ID50 b | <8 spirochetes | ID50 b | 660 spirochetes |
Mouse infection was determined 3 weeks post inoculation by serological response to B. burgdorferi proteins and reisolation of spirochetes from ear, bladder and joint tissues.
The ID50 was calculated according to method of Reed and Muench [83].
The B. burgdorferi IVET system is a robust method for selection of B. burgdorferi sequences that are expressed during murine infection. We have developed a genome-wide genetic screening method to identify B. burgdorferi genes that are expressed during mouse infection using an in vivo expression technology (IVET) approach [12,13]. The in vivo expression technology vector, pBbIVET, carries the B. burgdorferi bmpB Rho-independent transcription terminator sequence [22] repeated in triplicate (3XTT), to prevent any read-through promoter activity from the pBSV2* Borrelia shuttle vector backbone, followed by the promoter-less in vivo-essential pncA gene (Figure 1) [23,24]. Spirochetes lacking linear plasmid (lp) 25 are non-infectious in mice and severely compromised in the tick vector [25–31]. The pncA gene, located on lp25, encodes a nicotinamidase that is sufficient to restore murine infectivity to B. burgdorferi lacking the entire lp25 plasmid [24]. Genetic transformation of low-passage, infectious B. burgdorferi occurs at low frequency and efficiency hampering introduction of a complex DNA library into an infectious background [32,33]. Because B. burgdorferi clones lacking lp25 and lp56 demonstrate increased transformability [34], we isolated a clonal derivative of the low-passage infectious clone A3 that lacks both lp25 and lp56. This clone was designated A3 68-1 [35]. Clones A3 and A3 68-1 were transformed by electroporation with 20 µg of a Borrelia shuttle vector. The transformation frequency and efficiency of A3 68-1 was determined relative to that of the A3 parent. As expected, genetic transformation of A3 68-1 occurred at a high frequency and efficiency. We recovered approximately 2,000 transformants/ml in clone A3 68-1, whereas no transformants were recovered with a parallel transformation of clone A3 (data not shown). In order to test the function of the B. burgdorferi IVET system, the promoter for the in vivo essential ospC gene [36], was cloned in front of the promoter-less pncA gene in pBbIVET (Figure 1), creating plasmid pBbIVET-ospC p. This plasmid, along with pBbIVET alone, was transformed into the non-infectious, low-passage, highly transformable B. burgdorferi clone A3 68-1. All clones were tested for their abilities to infect groups of 6 C3H/HeN mice at an infectious dose 100 (ID100) of 1×104 spirochetes [37], indicating the presence or absence of an active promoter sufficient to drive expression of pncA thereby restoring infectivity. Spirochetes were reisolated from the ear, bladder and joint tissues of 5/6 mice infected with B. burgdorferi harboring pBbIVET-ospC p. No spirochetes were reisolated from mice (0/6) infected with B. burgdorferi carrying the promoter-less pBbIVET alone. Together these data demonstrated that our promoter trap system functioned with a known in vivo active promoter.
Figure 1. Schematic representation of the pBbIVET vector.
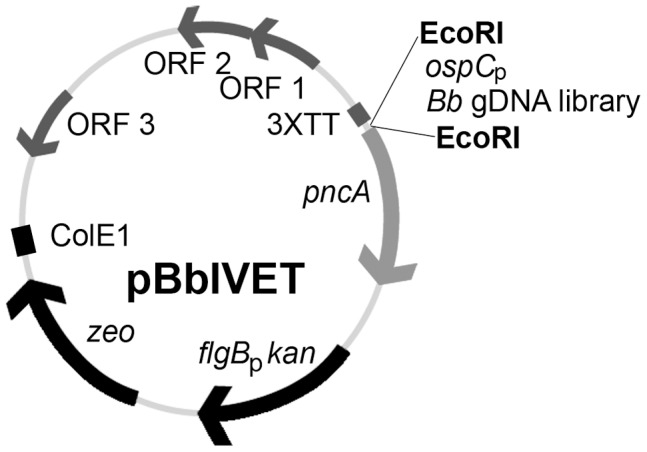
Features of this vector include: 3XTT, the transcriptional terminator sequence for bmpB [22] repeated in triplicate; pncA, promoterless pncA gene; flgB p kan, kanamycin resistance cassette; zeo, zeocin resistance marker; ColE1, E. coli origin of replication; ORFs 1, 2, 3, B. burgdorferi cp9 replication machinery. The EcoRI restriction site was used to clone the B. burgdorferi control in vivo-expressed promoter, ospC p, as well as the B. burgdorferi (Bb) gDNA library, in front of the promoterless pncA gene. The pBbIVET vector was derived from the B. burgdorferi shuttle vector pBSV2* [80].
Screening for B. burgdorferi genes expressed during murine infection. A B. burgdorferi genomic DNA library using an average DNA fragment size of approximately 200 bps was constructed upstream of the promoter-less pncA gene (Figure 1) in the pBbIVET vector in E. coli, yielding approximately 30,000 independent clones. A small subset of individuals from the 30,000 clone library in E. coli was analyzed by PCR and DNA sequencing and determined to carry non-identical B. burgdorferi DNA fragments. The strategy used to construct the pBbIVET library allowed the DNA fragments to be cloned in either the forward or reverse orientation relative to the pncA gene. Therefore, a library of 30,000 clones each harboring a unique 200 bp DNA fragment represented approximately 2X coverage of the 1.5 Mb genome of B. burgdorferi. Although the initial analysis of the transformation efficiency of B. burgdorferi clone A3 68-1 demonstrated that each transformation of 20 µg of a single purified plasmid into this genetic background yielded approximately 10,000 transformants, this transformation efficiency was not achieved when 20 µg of complex library plasmid DNA was transformed into A3 68-1. Forty four transformations of the library plasmid DNA resulted in recovery of approximately 15,000 individual clones in B. burgdorferi A3 68-1, representing an IVET library in B. burgdorferi with approximately 1X coverage of the spirochete genome. As described for the pBbIVET library in E. coli, a subset of individuals from 15,000 clone library in B. burgdorferi were analyzed and found to carry non-identical B. burgdorferi DNA fragments.
Like the BbIVET system described here, many IVET strategies are based upon complementation of auxotrophy. For microorganisms other than B. burgdorferi these strategies have allowed negative selection against “promoter-less” clones in minimal medium in which the auxotroph mutants are unable to grow [38]. B. burgdorferi lacking pncA are not attenuated for growth in the complex, undefined B. burgdorferi medium, BSKII. Moreover, there is currently no minimal medium available that supports the growth of wild-type B. burgdorferi. Therefore, the BbIVET system did not include negative selection against “promoter-less” clones in vitro. 179 mice were infected with pools from the B. burgdorferi IVET library of approximately 100 clones each, with each clone at a dose of 1×104 spirochetes resulting in a total dose of 1×106 spirochetes per mouse. Three weeks post inoculation, mice were sacrificed and ear, heart, bladder and joint tissues were harvested for reisolation of infectious spirochetes. 175 out of 179 mice became infected with B. burgdorferi as determined by reisolation of spirochetes from at least two or more of the tissue sites analyzed (Table 2). However, due to the potentially stochastic nature of the kinetics of infection [39] and/or tissue-specific promoter activity of distinct B. burgdorferi genomic fragments not all four tissue sites from all 175 infected mice were found to be positive for spirochete reisolation. Nonetheless, the recovery of live spirochetes from infected mouse tissues suggested that these spirochetes harbored in vivo active promoter(s) in the pBbIVET plasmid sufficient to drive expression of the in vivo-essential pncA gene to restore spirochete mouse infectivity. Total genomic DNA was isolated from each pool of reisolated spirochetes from each of the four mouse tissues and the pBbIVET plasmid DNA rescued in E. coli. Colony PCR using primers targeting the genomic DNA insert region of the pBbIVET vector was performed on 24 of the resulting E. coli colonies from each plasmid rescue transformation. No reisolated spirochetes were found to harbor a pBbIVET plasmid lacking a genomic DNA fragment insert. The amplified inserts were analyzed by restriction digest using a cocktail of the A/T-rich restriction enzymes to identify those DNA fragments with distinct restriction patterns, suggesting that these fragments represent different in vivo active promoters. Up to eleven non-identical restriction digest patterns were detected for every subset of 24 E. coli transformants carrying pBbIVET DNA that were analyzed (Figure S1). The DNA fragments corresponding to each distinct restriction digest pattern were further analyzed by DNA sequencing and the identities of the sequences determined by microbial genome BLAST analysis. Screening of approximately 15,000 BbIVET clones through mice resulted in the identification of 289 non-identical B. burgdorferi in vivo-expressed (Bbive) DNA fragments from across the chromosome and all 21 plasmid replicons of the B. burgdorferi B31 segmented genome (Table 2). Although the 1∶1 molar ratio of insert to vector used to generate the pBbIVET library did not preclude insertion of more than one fragment into each clone, only 20 out of the 289 clones were found to harbor two distinct DNA fragments. Of these clones the 3′ DNA fragment, proximal to the pncA ORF, was assumed to be the active promoter and was included in the subsequent analyses.
Table 2. The B. burgdorferi IVET system selects for in vivo-active promoters.
| Number of BbIVET clones screened | Positive reisolation of infectious spirochetes from mouse tissuesa | Number of unique genomic fragments recovered | |||
| Ear | Heart | Bladder | Joint | ||
| ∼15,000 | 175/179 | 173/179 | 172/179 | 174/179 | 289 |
Number of mice positive for spirochete reisolation/ number of mice analyzed. Four mice were reisolate-negative for all tissues analyzed. Three mice were reisolate-negative for the bladder tissue. One mouse was reisolate-negative for the heart and joint tissues. One mouse was reisolate-negative for the heart tissue.
B. burgdorferi in vivo expressed promoters map to distinct classes of putative regulatory sequences across the genome. Genomic mapping of the 289 unique Bbive promoters identified in this genetic screen demonstrated that 67% of the sequences mapped to sense DNA in the same direction as annotated open reading frames, 27% mapped to antisense DNA in the opposite direction to annotated open reading frames and 6% mapped to intergenic regions lacking annotated open reading frames. Of the large percentage of sense sequences, 41%, which represented 28% of the total Bbive sequences, mapped to regions just upstream of and in the same orientation to annotated open reading frames, suggesting that these sequences are promoters for the associated open reading frames and that these open reading frames are candidate in vivo-expressed genes. The remaining 59% of the sense sequences, which represented 39% of the total Bbive sequences, mapped within annotated open reading frames, suggesting the possibility for promoter elements within B. burgdorferi genes. Similar findings of putative transcriptional start sites within genes and operons have been reported for other bacterial pathogens [40,41]. The sequences that mapped to putative promoter locations in the genome and the genes associated with these promoters were prioritized for further analysis. Among the list of 80 sequences, 9 promoter regions were represented by two overlapping genomic DNA fragments. Five of these overlapping sequence pairs shared the same 3′ end, suggesting that the sequences belonging to each pair contained the same promoter. Whereas, the other four overlapping sequence pairs harbored distinct 3′ ends, suggesting that each sequence contained a unique promoter. The 71 in vivo-expressed candidate genes have been annotated to encode proteins in various functional categories including: cell division, cell envelope, replication, metabolism, motility, protein synthesis, transport and unknown functions (Table 3).
IVET identified candidate gene bbk46 on virulence plasmid lp36. Linear plasmid 36 is required for B. burgdorferi mouse infection; however, the genetic elements on lp36 that contribute to this phenotype have not been fully defined [37]. IVET identified a candidate in vivo-expressed promoter sequence, Bbive162, which mapped to lp36. This sequence was found to be 60 bp long, with 48 bp immediately upstream of and in the same direction as the BBK46 open reading frame (Figure 2), suggesting that the bbk46 gene may be expressed during mammalian infection and may contribute to the essential role of lp36 in B. burgdorferi infectivity. Therefore, the bbk46 gene was selected for further analysis.
Figure 2. The nucleotide and putative amino acid sequence of the BBK46 open reading frame.

The reverse complement of nucleotides 28391 to 29474 on lp36, encompassing bbk46 (Genbank GeneID: 1194234) and its putative promoter sequence. The nucleotide sequence of Bbive162 is underlined. The putative ribosome binding site is shown in bold italics. The putative BBK46 amino acid sequence is shown in bold. The stop codons at nucleotides 625 and 820 are highlighted in gray. The position of the inserted FLAG-epitope tag sequence is indicted with a star (*). The position of the inserted cMyc-epitope tag sequence is indicated with a number sign (#).
Expression of the bbk46 gene is induced during murine infection. Our BbIVET screen identified gene bbk46 as a putative in vivo-expressed gene. The BbIVET screen was designed to identify B. burgdorferi DNA fragments that are expressed in vivo and did not discriminate between those promoters that are specifically induced in vivo and those promoters that are expressed both in vitro and in vivo. Therefore, quantitative reverse transcription PCR (qRT-PCR) was used to validate the expression of bbk46 in vivo and to determine whether bbk46 expression was upregulated in vivo compared to in vitro. Total RNA was isolated from bladder tissue collected from mice infected with 1×105 wild-type B. burgdorferi three weeks post-inoculation as well as log phase in vitro grown spirochetes. RNA was converted to cDNA using random hexamer primers and the mRNA level of each target gene was measured relative to the constitutive recA gene using quantitative PCR. The gene expression levels of flaB and ospC were also measured as control constitutively-expressed and in vivo-induced genes, respectively. These data demonstrated that although bbk46 was expressed during in vitro growth, expression of this gene was increased more than 100-fold during mammalian infection (Figure 3A). Consistent with their known patterns of gene regulation, flaB expression was relatively unchanged in vivo compared to in vitro; whereas, ospC demonstrated a nearly 1000-fold increase in expression in vivo compared to in vitro (Figure 3A). Moreover, the relative amount of bbk46 expression during in vitro growth was found to be approximately 10-fold more than that of ospC. Whereas, the in vivo expression levels of genes bbk46, ospC and flaB were similar.
Figure 3. Expression of the bbk46 gene is upregulated during murine infection and is RpoS-independent.

Total RNA was isolated from bladder tissue collected from (A) mice infected with 1×105 wild-type B. burgdorferi three weeks post-inoculation (in vivo, gray bars) and from log phase in vitro grown spirochetes (in vitro, white bars) or (B) stationary phase temperature-shifted stationary phase in vitro grown wild-type (white bars) or ΔrpoS (gray bars) B. burgdorferi. RNA was reverse transcribed to cDNA using random hexamer primers. The expression of bbk46, flaB and ospC were quantified using quantitative reverse transcription polymerase chain reaction (qRT-PCR) and a standard curve analysis method. The mRNA levels of the bbk46, flaB and ospC gene transcripts were normalized to that of the constitutive recA gene. The data are expressed as the gene transcript/recA transcript. The data represent the average of triplicate qRT-PCR analyses of 3 biological replicates. Error bars represent the standard deviation from the mean.
RpoS is a global regulator that controls expression of genes expressed during mammalian infection, including ospC [2]. Because bbk46 expression was induced in vivo in a manner similar to that of ospC, we sought to determine whether, like ospC, bbk46 is an RpoS-regulated gene. RNA was isolated from stationary phase temperature-shifted wild-type and ΔrpoS mutant spirochetes, a growth condition previously shown to induce expression of rpoS and rpoS-regulated genes [42]. Quantitative RT-PCR was then performed for genes bbk46, flaB, ospC and recA, as described above. As expected, ospC expression was increased approximately 20 times in the presence compared to the absence of rpoS (Figure 3B). In contrast, bbk46 expression was RpoS-independent under these growth conditions (Figure 3B). Likewise, no RpoS-dependent change in gene expression was detected for flaB. Interestingly, the amount of flaB expression detected in the stationary phase temperature-shifted spirochetes (Figure 3B) was dramatically decreased compared to the amount of flaB expression detected in log phase and in vivo grown spirochetes (Figure 3A), suggesting that flaB is not expressed at the same level under all growth conditions. Together these data demonstrated that bbk46 was highly induced during murine infection and bbk46 expression was not controlled by RpoS during in vitro growth.
The bbk46 open reading frame fails to produce detectable amounts of protein during in vitro growth. The bbk46 gene is a member of paralogous gene family 75, which also includes lp36-encoded genes bbk45, bbk48 and bbk50, all of which are annotated to encode putative P37 immunogenic lipoproteins [43–45] (Figure 4). These genes are located on the right arm of lp36 in B. burgdorferi clone B31, which is a highly variable region among distinct B. burgdorferi isolates [44]. The members of paralogous gene family 75 are conserved within B. burgdorferi isolates but are not present in the relapsing fever spirochetes. The B. burgdorferi clone B31 BBK45, BBK48 and BBK50 proteins are predicted to be 301, 288 and 332 amino acids, respectively. However, B. burgdorferi clone B31 bbk46 is annotated as a pseudogene as a result of an authentic frame shift resulting in a TAA stop codon at nucleotide 625 [43–45], thereby producing a putative 209 amino acid protein. In contrast, the BBK46 homolog in clone N40, BD04, harbors a CAA codon at nucleotide 625, resulting in a glutamic acid residue at amino acid 209 and producing a putative 273 amino acid protein [46]. Sequence analysis of the cloned bbk46 open reading frame confirmed the presence of the TAA stop codon at nucleotide 625 (Figure 2). To experimentally determine the size of the BBK46 protein produced in B. burgdorferi B31 the bbk46 ORF along with a FLAG epitope tag sequence prior to the stop codon at nucleotide 625 and a cMyc epitope tag sequence prior to the stop codon at nucleotide 820 (Figure 2) was cloned into the B. burgdorferi shuttle vector pBSV2G under the control of either the constitutive flaB promoter or the putative endogenous bbk46 promoter. A mutant clone lacking the entire BBK46 open reading frame was constructed by allelic exchange and verified by PCR analysis (Figure 5A and 5B). The bbk46 mutant clone was transformed with the shuttle vectors carrying the epitope tagged bbk46 constructs. BBK46 protein production was assessed in both E. coli and B. burgdorferi. Immunoblot analyses using αFLAG and αcMyc monoclonal antibodies resulted in detection of a FLAG-epitope tagged protein of an approximate molecular mass of 23 kDa, which is the predicted size of the 209 amino acid BBK46 protein, in the E. coli clones carrying both the flaB p-driven and the bbk46 p-driven constructs (Figure 6). Surprisingly, no FLAG epitope tagged protein was detected in either B. burgdorferi clone (Figure 6), although bbk46 gene expression was observed in these clones (data not shown), indicating that the lack of detectable BBK46 protein was not likely the result of a transcription defect. Furthermore, no cMyc epitope tagged protein was detected in either E. coli or B. burgdorferi. Together these data suggested that although the bbk46 ORF is competent to produce a 23 kDa protein in E. coli and the transcript is expressed in B. burgdorferi during in vitro growth, the protein is either not produced or is rapidly turned over in log phase in vitro grown B. burgdorferi.
Figure 4. Amino acid alignment of the putative members of the immunogenic protein P37 family encoded on lp36.

Shown is an amino acid alignment of the P37 protein family members BBK45 (GenBank accession no. NP_045617.2), BBK46 (translated bbk46, Genbank GeneID: 1194234), BBK48 (GenBank accession no. NP_045619.1) and BBK50 (GenBank accession no. NP_045621.1). Amino acids identical to the consensus sequence are shaded. The predicted SpLip lipobox sequence [81] is indicted with five stars. Dashes represent spaces introduced for optimal sequence alignment. The positions of the two stop codons in the bbk46 translation are indicated with arrows. Amino acid sequences were aligned using the CLUSTAL W algorithm in the MEGALIGN program from the DNASTAR Lasergene suite.
Figure 5. Generation of the Δbbk46 mutant and genetic complemented clones in B. burgdorferi.

(A) Schematic representation of the wild-type (WT) and Δbbk46 loci on lp36. The sequence of the entire bbk46 open reading frame was replaced with a flaB p-aadA antibiotic resistance cassette [37,82]. Locations of primers for analysis of the mutant clones are indicated with small arrows and labels P7-P12, P19 and P20. Primer sequences are listed in Table 5. (B) PCR analysis of the Δbbk46 mutant clone. Genomic DNA isolated from WT and Δbbk46/ vector spirochetes served as the template DNA for PCR analyses. DNA templates are indicated across the bottom of the gel image. The primer pairs used to amplify specific DNA sequences are indicated at the top of the gel image and correspond to target sequences as shown in A. Migration of the DNA ladder in base pairs is shown to the left of each image. (C) In vitro growth analysis of mutant clones. A3-68ΔBBE02 (WT), bbk46::flaB p-aadA/ pBSV2G (Δbbk46/ vector) and bbk46::flaB p-aadA/ pBSV2G-bbk46 (Δbbk46/ bbk46+) spirochetes were inoculated in triplicate at a density of 1×105 spirochetes/ml in 5 ml of BSKII medium. Spirochete densities were determined every 24 hours under dark field microscopy using a Petroff-Hausser chamber over the course of 96 hours. The data are represented as the number of spirochetes per ml over time (hours) and is expressed as the average of 3 biological replicates. Error bars indicate the standard deviation from the mean.
Figure 6. BBK46 protein production is detectable in E. coli but not in B. burgdorferi.

Immunoblot analysis of total protein lysate prepared from 1.5×108 B. burgdorferi Δbbk46 (Bb) or E. coli harboring either pBSV2G flaB p-bbk46-FLAG-cMyc (flaB p) or pBSV2G bbk46 p-bbk46-FLAG-cMyc (bbk46 p). Protein lysates were separated by SDS-PAGE and immunoblots performed using anti-FLAG monoclonal antibodies (α FLAG) and anti-cMyc monoclonal antibodies (α cMyc). 300 ng of purified PncA-FLAG [23] and GST-BmpA-cMyc [78] proteins served as positive controls (+) for each antibody. The positions of markers to the left of the panel depict protein standard molecular masses in kilodaltons.
As a putative member of the P37 immunogenic lipoprotein family, BBK46 is predicted to localize to the spirochete outer surface and to be immunogenic during mammalian infection. Therefore, recombinant BBK46, lacking the first 32 amino acids that are predicted to comprise the signal sequence for the lipoprotein, was produced in E. coli as an N-terminal fusion to glutathione S-transferase (GST). To assess the immunogenicity of the BBK46 protein, immunoblot analysis was performed using purified rGST-BBK46 probed with mouse immune serum collected 21 days post inoculation with 1×104 wild-type B. burgdorferi. The rGST-BBK46 protein was found to be non-immunoreactive with mouse immune serum, in contrast to the control antigen BmpA (Figure S2). These data suggest that, if produced in B. burgdorferi, BBK46 is not an immunoreactive antigen. However, these data do not rule out the possibility that the immunogenic epitope is not present or available in the recombinant protein produced in E. coli.
The bbk46 gene is dispensable for B. burgdorferi infection in mice. In vitro growth analysis demonstrated that the bbk46 mutant and complemented clones had no detectable in vitro phenotypes (Figure 5C). Initial characterization of the role of bbk46 in mouse infectivity suggested that this gene may contribute to spirochete persistence in mouse tissues. However, this phenotype was subsequently attributed to the loss of lp28-1 from the bbk46/vector mutant clone [27–29,47,48], rather than the bbk46 gene itself. The Δbbk46/vector B. burgdorferi clone was re-derived and verified to contain all of the plasmids of the parent clone. Therefore, to examine the role of bbk46 in mouse infectivity, groups of 9 or 6 C3H/HeN female mice were needle inoculated with 1×104 Δbbk46/vector or Δbbk46/bbk46 + spirochetes, respectively. Three weeks post inoculation the mice were assessed for B. burgdorferi infection by serology and reisolation of spirochetes from the ear, bladder and joint tissues. All of the mice inoculated with the Δbbk46/vector clone were seropositive for anti-B. burgdorferi antibodies and positive for spirochete reisolation from all tissues examined (Table 4). As expected, 5 out 6 mice inoculated with Δbbk46/bbk46 + complemented spirochetes demonstrated no defect in mouse infectivity (Table 4). Together these findings suggest that the bbk46 gene is not required for B. burgdorferi needle inoculation of mice at a dose of 1×104 spirochetes.
Discussion
In this study we have successfully adapted and applied for the first time an IVET-based genetic screen for use in B. burgdorferi for the purpose of identifying spirochete genes that are expressed during mammalian infection. Historically, genetic manipulation of low passage, infectious B. burgdorferi has been challenged by the low transformation frequencies of these spirochetes, preventing application of classic in vivo genetic screening techniques such as in vivo expression technology (IVET) and signature-tagged mutagenesis (STM) [49] to identify B. burgdorferi genetic elements important for pathogenicity. However, advances in the understanding of the B. burgdorferi restriction modification systems that inhibit transformation [34,50–53] have recently allowed construction and characterization of a comprehensive STM mutant library in infectious B. burgdorferi [54]. The foundation for our strategy for development of IVET in B. burgdorferi was based upon the spirochete's requirement of lp25 for both restriction modification and virulence functions. Spirochetes lacking lp25 are highly transformable but non-infectious in mice [27–29,34]. Restoration of the lp25-encoded pncA gene to lp25− spirochetes restores wild-type infectivity [24] but maintains high transformation frequency. At the time of the development of the pBbIVET system the true start codon of the pncA gene was not defined; therefore, the promoter-less pncA gene construct in the pBbIVET plasmid used an engineered AUG start codon and was missing the first 24 nucleotides of the now defined pncA ORF [23]. Furthermore, this construct was purposefully designed without a ribosome binding site (RBS) and was dependent upon the cloned B. burgdorferi DNA fragments to contain both a promoter and a functional RBS. Although we acknowledge that this requirement may have limited the number of clones identified in our screen, during development of the BbIVET system we found that inclusion of an RBS sequence in the promoterless pncA construct resulted in vector-driven PncA production in the absence of a promoter. Thus, in order to reduce the possibility of recovering false positive clones, the pBbIVET system was designed without an RBS. The enzyme Tsp509I was selected to generate the DNA fragments for the pBbIVET library because the AATT restriction site of this enzyme is present approximately every 58 bp in the B. burgdorferi B31 genome. However, it is possible that DNA fragments generated with this enzyme will not result in sequences that contain a 3′ RBS appropriately distanced from the start codon of the pncA ORF, thereby limiting the number of clones identified in the screen.
Screening of a 15,000 clone B. burgdorferi genomic library in mice identified 289 DNA sequences from across all 22 B. burgdorferi replicons capable of promoting pncA expression resulting in an infectious phenotype. It is likely that the BbIVET screen did not achieve saturation because the number of clones analyzed was only estimated to cover the B. burgdorferi genome one time, under the assumption that each cloned DNA fragment in the library was unique. Analysis of the pBbIVET library in B. burgdorferi suggested that the library was composed of 15,000 unique clones. However, because only a small fraction of the library was examined for the sequences of the DNA fragment inserts, our findings do not rule out the potential that the library was composed of less than 15,000 non-identical clones and therefore, may represent less than 1X coverage of the genome. Of the 175 mice infected with the pBbIVET library, 10% resulted in reisolation of a single clone, 62% resulted in reisolation of two to five unique clones, and 28% resulted in reisolation of six to eleven unique clones. Furthermore, 57% of the 289 Bbive sequences were only recovered once; whereas, 39% of the sequences were recovered two to five times and 4% of the sequences were recovered six to twelve times. These data are indicative of the amount of redundancy in the screen and suggest that although the screen may not have been representative of the entire B. burgdorferi genome, a large percentage of mice became infected with multiple clones and many of the Bbive sequences were recovered more than once.
We found that 71 of the Bbive sequences mapped to canonical promoter positions upstream of annotated open reading frames in the B. burgdorferi genome. Unexpectedly, the well characterized in vivo-expressed ospC promoter was not among these sequences. However, the ospC p was successfully recovered in our functional validation of the BbIVET system, suggesting that the BbIVET screen had not reached complete saturation of the genome and with further screening of the BbIVET library the ospC p sequence may be recovered. Alternatively, given that ospC expression is known to be down-regulated after the initial stages of infection [11,55–58] it is possible that in the context of a mixed infection individual pBbIVET clones carrying the ospC p lack a fitness advantage due to decreased expression three weeks post inoculation and may not be recovered in our screen. This explanation may appear to conflict with the findings reported herein that ospC expression is high at three weeks post inoculation and the ospC p served as a robust positive control promoter for the BbIVET system. However, down-regulation of ospC expression at this time point in infection is a stochastic process that occurs at the level of the individual spirochete and does not occur simultaneously across the entire population [57]. Although at the population level the ospC p is expressed at this time point in our studies, in the context of the BbIVET screen individual clones carrying the ospC p may express reduced amount of pncA and may be out competed by other BbIVET clones carrying stronger promoters.
A subset of the genes identified in the BbIVET screen included known in vivo-expressed genes, which provided validation that our genetic system was working as expected and was sufficiently powerful. The screen recovered the promoter for genes bba36 (Bbive175), bba57 (Bbive271), bbb27 (Bbive240), bbj34 (Bbive267), bbj36 (Bbive171), bbj51 (Bbive269), bb0213 (Bbive15) and bb0760 (Bbive94), all of which have been shown previously to be expressed during mammalian infection [11]. Furthermore, bba57 was recently reported to be up-regulated in vivo and to contribute to pathogenesis in the mouse [59]. The bptA gene encodes a function that has been shown to be required for B. burgdorferi survival in the tick and to contribute to mouse infectivity [30,31]. In addition, Bbive14, 58, 232, 84, 269, 295 and 77 are associated with genes that have been shown to be up-regulated in in vivo-like conditions and/or gene products that are immunogenic in humans and mice [5,8,60,61]. Notably, few in vivo-expressed candidate genes identified using BbIVET were previously observed to be up-regulated in mammalian host-adapted spirochetes derived from growth within rat dialysis membrane chambers (DMCs). Genes identified in our analyses that have also been detected by microarray analysis of DMC grown spirochetes include bba36 [8,10], bbj51 [7,8], bb0551, bbm28 [8], bb0495, and bb0660 [7]. The results of the DMC microarray studies are reported as genes that are significantly up-regulated in DMC-derived spirochetes relative to spirochetes grown in vitro; whereas, the BbIVET screen does not distinguish between genes that are specifically induced in vivo and genes that are expressed both in vitro and in vivo. Furthermore, the environmental cues within the DMCs may not fully reflect those experienced by B. burgdorferi during an active infection. Finally, the BbIVET system specifically selects for promoters that are capable of driving expression of pncA allowing the spirochetes to survive throughout a three week mouse infection. Together, these technical and biological differences between the DMC microarray and BbIVET screen likely contributed to the distinct results obtained from the two methods of gene expression analysis. In addition, few genes that have been previously established to be RpoS-regulated in vitro and/or within DMCs [10,62] were identified by the BbIVET screen. RpoS-regulated genes bba36, bba57, bb0265 and bbh01 [10,62] were among the in vivo-expressed Bbive candidate genes. Similarly, only one putative BosR-regulated gene, bb0592 [63], was identified in the BbIVET screen. Although it is unclear why only a small number of known RpoS-regulated promoters were recovered, the recently identified AT-rich BosR binding site [63] contains the restriction site for the Tsp509I restriction enzyme used to generate the BbIVET library. Therefore, it is possible that the BosR binding sites were subject to cleavage by Tsp509I, perhaps resulting in a limited number of DNA fragments that contained BosR-dependent promoters.
The BbIVET screen was carried out in such a way that both DNA fragments that are expressed in vitro and in vivo, as well as those fragments that are specifically induced in vivo, could be recovered. Therefore, it was not surprising that genes encoding cell division, DNA replication, energy metabolism, protein synthesis and transcription functions were identified, all of which are likely functions essential for spirochete growth under all condition. These findings were consistent with those categories of genes not recovered by genome-wide transposon mutagenesis, suggesting that these genes encode essential functions [54]. The BbIVET screen identified genes that encode proteins in functional categories that may contribute to B. burgdorferi infectivity and pathogenesis including, putative lipoproteins, motility and chemotaxis proteins, transport proteins and proteins of unknown function. Similarly, transposon mutagenesis analysis indicated that motility and chemotaxis genes as well as transport genes are important for B. burgdorferi survival in the mouse [54].
Linear plasmid 36 is known to be critical for B. burgdorferi survival in the mouse; however, the genes on lp36 that contribute to this requirement have not been fully characterized [37]. The recently published comprehensive STM study suggests that many of the genes encoded on lp36 participate in B. burgdorferi infectivity [37,54]. BbIVET identified gene bbk46 on lp36. We found that bbk46 was expressed both in vitro and in vivo. However, bbk46 expression was dramatically induced in spirochetes isolated from infected mouse tissues as compared to spirochetes grown in vitro, suggesting a possible role for this gene in B. burgdorferi infectivity. Conversely, no role for bbk46 in B. burgdorferi mouse infection was detected. Consistent with lack of identification of bbk46 as an RpoS-regulated genes in previous studies of the RpoS regulon [10,64], control of bbk46 expression was found to be RpoS-independent under in vitro growth conditions that typically induce expression of rpoS regulated genes [4,8,42,62]. These findings highlight the power and uniqueness of the IVET-based approach for identification of B. burgdorferi in vivo-expressed genes, which might not be discovered using other genome-wide gene expression methods. Surprisingly, BBK46 protein was not detected in spirochetes expressing FLAG epitope tagged bbk46 under the control of the putative native promoter or the constitutive flaB promoter. Moreover, sera from B. burgdorferi infected mice were non-immunoreactive against recombinant BBK46 protein. In support of these data, no peptide corresponding to BBK46 has been detected in genome-wide proteome analysis of B. burgdorferi under different environmental conditions [65]. Our findings suggest that despite high gene expression, the encoded BBK46 protein is produced at low levels in the spirochete and/or BBK46 is rapidly turned over in the cell. Alternatively, bbk46 may function as an RNA. The molecular nature of the functional product of bbk46 is currently under investigation.
In conclusion, we have developed and applied the IVET technology to B. burgdorferi to identify spirochete genes expressed during mammalian infection. This represents the first use of this system in B. burgdorferi. The power of BbIVET was validated by identification of a subset of genes that have been demonstrated previously to be upregulated in vivo as well as unique B. burgdorferi in vivo-expressed genes that had not been recognized previously. Further analysis of the B. burgdorferi in vivo-expressed genes identified herein will contribute to advancing understanding of the molecular mechanisms of B. burgdorferi survival and pathogenicity in the mammalian host.
Materials and Methods
Ethics statement. The University of Central Florida is accredited by the International Association for Assessment and Accreditation of Laboratory Animal Care. Protocols for all animal experiments were prepared according to the guidelines of the National Institutes of Health and were reviewed and approved by the University of Central Florida Institutional Animal Care and Use Committee (Protocol numbers 09-38 and 12–42).
Bacteria clones and growth conditions. All B. burgdorferi clones used were derived from clone B31 A3. Clone A3 68-1, which lacks lp25 and lp56 [35] was used for the pBbIVET library. The B31 A3 wild-type and rpoS::kan B. burgdorferi clones [71] were used for gene expression experiments. All low-passage B. burgdorferi mutant and complemented clones generated herein were derived from infectious clone A3-68ΔBBE02, which lacks cp9, lp56 and gene bbe02 on lp25 [53]. B. burgdorferi was grown in liquid Barbour-Stoenner-Kelly (BSK) II medium supplemented with gelatin and 6% rabbit serum [72] and plated in solid BSK medium as previously described [73,74]. All spirochete cultures were grown at 35°C supplemented with 2.5% CO2. Kanamycin was used at 200 µg/ml, streptomycin was used at 50 µg/ml and gentamicin was used at 40 µg/ml, when appropriate. All cloning steps were carried out using DH5α E. coli, which were grown in LB broth or on LB agar plates containing 50 µg/ml kanamycin, 300 µg/ml spectinomycin or 10 µg/ml gentamicin.
Generation of the pBbIVET plasmid. The promoterless pncA gene was amplified from B. burgdorferi B31 genomic DNA using primers 1 and 2 (Table 5) and Taq DNA polymerase (New England Biolabs). The EcoRI/XbaI-digested pncA fragment was cloned into EcoRI/XbaI-linearized plasmid pBSV2*TT [23], creating plasmid pBbIVET. The in vivo-expressed ospC promoter with EcoRI ends was amplified from B. burgdorferi B31 genomic DNA using primers 3 and 4 (Table 5) and cloned into the EcoRI-cut, Antarctic phosphatase-treated (New England Biolabs) pBbIVET plasmid in front of the promoterless pncA gene, resulting in plasmid pBbIVET ospC p. All plasmids were analyzed and verified by restriction digest and sequence analysis. The pBbIVET and pBbIVET ospC p plasmids were each transformed by electroporation into A3 68-1 [35] as described [37] and transformants selected in solid BSK medium containing kanamycin and confirmed by PCR using primers 1 and 2 (Table 5). Total genomic DNA was prepared from PCR-positive clones and screened for the presence of the B. burgdorferi plasmid content [71]. The clones that retained the plasmid content of the parent clone were used in further experiments.
Table 5. List of primers used in this study.
| Primer number | Designation | Sequence (5′ – 3′)a |
| 1 | pncA 5′ EcoRI A | cggaattcatgGCACTTATTTTAATAGATATAC |
| 2 | pncA 3′ XbaI | gctctagaTTATATATTAAGCTTACTTTGGCTG |
| 3 | ospC prom 5′ EcoRI | cggaattcTTCTTTTTCATTAATTTGTGCCTCC |
| 4 | ospC prom 3′ EcoRI | cggaattcTTAATTTTAGCATATTTGGCTTTGCTTATGTCG |
| 5 | pUC18R BSV2 | AGCGGATAACAATTTCACACAG |
| 6 | pncA prom 3′ seq | ACTGTTAGATACTGGCAAAGTGCC |
| 7 | bbk46Fup500 | GTTCTTTTATGGAGCAAGCAACTAA |
| 8 | bbk46Rup500 | CGGAAGCCACAAGAGGCGACAGACACTATCTTAGTACCTCTTCTTAGAATCTG |
| 9 | bbk46Fdown500 | GGCGAGATCACCAAGGTAGTCGGCAAATAAATAATACTAATCTTAGATAGCTCAGCTTT |
| 10 | bbk46Rdown500 | CTAGCTTCACTAGTTTCCCTAGA |
| 11 | flaBpaadA F | TGTCTGTCGCCTCTTGTG |
| 12 | flaBpaadA R | TTATTTGCCGACTACCTTGGTG |
| 13 | K465′kpn1fwd | cggggtaccCTTCCAGTGTAGGCTTTAGTTT |
| 14 | K463′FLAGrev | TTAtttatcatcatcatctttataatcTGCCTCAACTGCCTTTCTC |
| 15 | K465′FLAGfwd | gattataaagatgatgatgataaaTAAAATGCTTCAAAGGAAAATTATGAATGG |
| 16 | K463′C-mycSalIrev | acgcgtcgacTTAcagatcttcttcagaaataagtttttgttcATAAGCAGCTTCATATGCTTTATTT |
| 17 | K465′PCR3fwd | CGGGGTACCCTTCCAGTGTAG |
| 18 | K463′PCR3rev | ACGCGTCGACTTACAGATCTTCTTCAGAAATA |
| 19 | Lp3629018F | AGCATTATTTGTACTTCTAGGC |
| 20 | Lp3629013R | ACATACTAGACAACAACAAGTC |
| 21 | flaBF3 | GCATTAACGCTGCTAATCTTAG |
| 22 | flaBR3 | GCATTAATCTTACCAGAAACTCC |
| 23 | recA F | AATAAGGATGAGGATTGGTG |
| 24 | recA R | GAACCTCAAGTCTAAGAGATG |
| 25 | ospC1 F | ACGGATTCTAATGCGGTTTTACCT |
| 26 | ospC1 R | CAATAGCTTTAGCAGCAATTTCATCT |
| 27 | flaBp 5′ KpnI | gggggtaccTGTCTGTCGCCTCTTGTGGCT |
| 28 | flaBp 3′ BamHI | gggggatccGATTGATAATCATATATCATTCCT |
| 29 | bbk46+S 5′ BamHIF | cgggatccATGAATTTAATTGCTAAATTATTTATTTTATCCAC |
| 30 | bbk46-S 5′ BamHIF | cgggatcc ATGTGTAACCTATATGATAATCTTGCAGAC |
| 31 | bbk46 3′ XhoIR | ccgctcgag TTAATAAGCAGCTTCATATGCTTTATTTAG |
Lowercase indicates all non-B. burgdorferi sequence.
Generation of the BbIVET library. Total genomic DNA was isolated from a 250 ml culture of B. burgdorferi B31 clone A3 grown to a density 1×108 spirochetes/ml using the Qiagen genomic DNA buffer set and Genomic-tip 500/G, according to the manufacturer's protocol (Qiagen). A3 genomic DNA was partially digested with Tsp509I (New England Biolabs). The partial digests were electrophoretically separated on a 0.8% agarose gel and the 300 to 500 bp range of DNA fragments extracted and ligated in a 1∶1 molar ratio with EcoRI-digested and Antarctic phosphatase-treated pBbIVET. Library ligations were electroporated into E. coli Top10 cells (Life Technologies) and transformants selected on LB agar containing 50 µg/ml kanamycin, resulting in approximately 30,000 independent clones. Plasmid DNA was isolated from these cells and 20 µg aliquots of the plasmid library were transformed by electroporation into B. burgdorferi A3 68-1, as previously described [74]. One fifth of each transformation was plated on solid BSK medium containing kanamycin. B. burgdorferi pBbIVET colonies were verified to contain B. burgdorferi DNA fragments by PCR using primers 5 and 6 (Table 5) and the number of transformants recovered quantitated. The approximately 15,000 B. burgdorferi clones recovered over 40 transformations were stored in aliquots of pools of approximately 100 BbIVET clones each in 25% glycerol at −80°C.
Selection of B. burgdorferi clones having in vivo-expressed DNA fragments. Each BbIVET pool (∼100 clones) was grown in 10 ml of fresh BSKII medium to a density of 1×108 spirochetes/ml. In groups of approximately 20 animals, 144 6–8 week old C3H/HeN female mice were each inoculated (80% intraperitoneal and 20% subcutaneous) with a dose 1×106 spirochetes of a unique pool of ∼100 BbIVET clones, under the assumption that each clone was present at dose 1×104 spirochetes. A fraction of each inoculum was plated on solid BSK medium and colonies screened for the presence of virulence plasmid lp28-1. Three weeks post inoculation, spirochetes were reisolated from ear, heart, bladder and joint tissues in 10 ml BSKII medium containing 20 µg/ml phosphomycin (Sigma), 50 µg/ml rifampicin (Sigma) and 2.5 mg/ml amphotericin B (Sigma) in 0.2% dimethyl sulfoxide (Sigma). Total genomic DNA was isolated from each spirochete cultures using the Wizard genomic DNA purification kit (Promega) and transformed into chemically competent E. coli DH5α cells and colonies selected on LB agar containing kanamycin to recover the pBbIVET plasmids. Twenty four transformants were chosen at random from each plasmid rescue and colony PCR performed using primers 5 and 6 (Table 5) to amplify the in vivo-expressed DNA fragment. PCR products were subsequently digested with a cocktail of restriction enzymes (DraI, SspI and AseI) and visualized on a 1% agarose gel. Approximately 14,000 E. coli clones were analyzed in this manner. All unique BbIVET fragments, as determined by the restriction digest pattern (Figure S1), were analyzed by direct sequencing of the PCR product using primer 5. Each individual sequence was identified by blastn analysis and mapped to its location in the B. burgdorferi B31 genome.
Deletion of bbk46. We used a PCR-based overlap extension strategy to delete the bbk46 gene. A spectinomycin/ streptomycin resistance cassette, flaBp-aadA [75] with blunt ends, was amplified from genomic DNA isolated from clone ΔguaAB [35] using Phusion High-fidelity DNA polymerase (Thermo Scientific) and primers 11 and 12 (Table 5). The 500 bp flanking region upstream of the bbk46 ORF was amplified from the B. burgdorferi B31 clone A3 genomic DNA using the Phusion High-fidelity DNA polymerase and primers 7 and 8 (Table 5). This introduced a 25 bp sequence at the 3′ end of this fragment that was complementary to the 5′ end of the flaBp-aadA cassette. Similarly, the 500 bp flanking region downstream of the bbk46 ORF was amplified using the primers 9 and 10 (Table 5), which introduced a 5′ sequence of 30 bp that was complementary to the 3′ end of the resistance cassette. The PCR products from the above 3 reactions were mixed in equal volumes and used as a template for a fourth amplification reaction using Phusion High-fidelity DNA polymerase and primers 7 and 10 (Table 5) in order to generate a product containing the resistance cassette flanked by the 500 bp sequences upstream and downstream of the bbk46 ORF. This product was ligated with linear pCR-Blunt using a Zero Blunt PCR cloning Kit (Life technologies), yielding the allelic exchange plasmid pCR-Blunt-Δbbk46-flaBp-aadA. B. burgdorferi A3-68ΔBBE02 was transformed with 20 µg of pCR-Blunt-Δbbk46-flaBp-aadA purified from E. coli as previously described [37]. Streptomycin-resistant colonies were confirmed to be true transformants by PCR using primer pairs 7 and 10 and 11 and 12 (Table 5). Positive Δbbk46-flaBp-aadA clones were screened with a panel of primers [71] for the presence of all of the B. burgdorferi plasmids of the parent A3-68ΔBBE02 clone [53], and a single clone was selected for further experiments.
Complementation of the Δbbk46 mutant. A PCR-based overlap extension strategy was used to create a DNA fragment encompassing the bbk46 gene and putative upstream promoter sequence with the introduction of a FLAG epitope tag immediately upstream of the putative premature stop codon and a cMyc epitope tag immediately upstream of the downstream stop codon. This was done by using Phusion High-fidelity DNA polymerase (New England Biolabs) and the primers pairs 13 and 14, 15 and 16, and 17 and 18 (Table 5). A KpnI restriction site was introduced at the 5′ end of this fragment and a SalI site at the 3′ end. The KpnI+SalI-digested PCR product was ligated into KpnI+ SalI-digested B. burgdorferi shuttle vector pBSV2G [76] and cloned in E. coli. The pBSV2G bbk46 p-bbk46-FLAG-cMyc plasmid structure and sequence were confirmed by restriction digest and DNA sequence analysis. In addition, a 400 bp DNA fragment encompassing the flaB promoter with KpnI and BamHI ends was amplified from B31 A3 genomic DNA using primers 27 and 28 (Table 5). The KpnI+BamHI-digested PCR product was ligated into KpnI+ BamHI-digested B. burgdorferi shuttle vector pBSV2G [76]. The bbk46-FLAG-cMyc gene without the putative bk46 promoter sequence and with BamHI and SalI ends was amplified from pBSV2G bbk46 p-bbk46-FLAG-cMyc plasmid DNA using Phusion High-fidelity DNA polymerase (New England Biolabs) and primers 29 and 18 (Table 5). The BamHI+SalI-digested PCR product was ligated into BamHI+SalI-digested pBSV2GflaB p and cloned in E. coli. The pBSV2G flaB p-bbk46-FLAG-cMyc plasmid structure and sequence were confirmed by restriction digest and DNA sequence analysis. The Δbbk46 mutant was transformed with 20 µg of pBSV2G bbk46 p-bbk46-FLAG-cMyc, pBSV2G flaB p-bbk46-FLAG-cMyc or pBSV2G alone isolated from E. coli and positive transformants selected as previously described [37,77]. The clones that retained the B. burgdorferi plasmid content of the parent clone were selected for use in further experiments.
Immunoblot analysis of BBK46-FLAG-cMyc. Production of the BBK46-FLAG-cMyc protein was examined in both E. coli and B. burgdorferi carrying pBSV2G bbk46 p-bbk46-FLAG-cMyc or pBSV2G flaB p-bbk46-FLAG-cMyc. Total E. coli protein lysates were prepared from 2×109 cells harvested following overnight growth in LB medium at 37°C with aeration. E. coli cells were resuspended and lysed in 200 µl B-PER protein extraction reagent (Pierce), followed by the addition of 200 µl 2× Laemmli sample buffer plus 2-mercaptoethanol (Bio-rad). Total B. burgdorferi protein lysates were prepared from 2×109 spirochetes harvested at mid-log phase. The spirochetes were washed twice in 1 ml cold HN buffer (50 mM Hepes, 50 mM NaCl, pH 7.4) and lysed in 200 µl B-PER protein extraction reagent (Thermo Scientific), followed by the addition of 200 µl 2× Laemmli sample buffer plus 2-mercaptoethanol (Bio-rad). 30 ml of each protein lysate (∼1.5×108 cells) were separated by SDS-PAGE and transferred to a nitrocellulose membrane. 300 ng of PncA-FLAG [23] and GST-BmpA-cMyc [78] proteins served as positive controls. Immunoblot analysis was performed using anti-FLAG monoclonal primary antibody (Genscript) diluted 1∶500 in Tris-buffered saline, pH 7.4 and 0.5% Tween20 (TBST) and goat anti-mouse IgG+IgM-HRP secondary antibody (EMD Millipore) diluted 1∶10,000 in TBST and the signal detected using SuperSignal West Pico chemluminescent substrate kit (Thermo Scientific). The membrane was then stripped using 0.2M NaOH, reblocked using 5% skim milk in TBST and probed with anti-cMyc primary antibody (Genscript) diluted 1∶500 in TBST and goat anti-mouse IgG+IgM-HRP (EMD Millipore) and visualized as described above.
Cloning, purification and seroreactivity analysis of rGST-BBK46. An in-frame glutathinone S-transferase (GST)-BBK46 fusion protein lacking the putative BBK46 signal sequence was generated using primers 30 and 31 (Table 5) and purified, as previously described [78]. Approximately 1 µg of GST-BBK46 was separated by SDS-PAGE, transferred to a nitrocellulose membrane and analyzed by immunoblot for seroreactivity using immune serum collected 3 weeks post inoculation from mice infected with wild-type B. burgdorferi as previously described [78]. Controls included 1 µg of GST alone and total protein lysates generated from BL21 E. coli, B. burgdorferi B31 A3 and E. coli expressing B. burgdorferi bmpA [37] prepared as described above. The membrane was stripped as described above and reprobed with anti-GST primary monoclonal antibody (EMD Millipore) diluted 1∶1000 in TBST and goat anti-mouse IgG+IgM-HRP (EMD Millipore) and visualized as described above.
In vitro growth analysis. Wild-type (A3-68ΔBBE02), Δbbk46/vector and Δbbk46/bbk46 + spirochetes were inoculated in triplicate at a density of 1×105 spirochetes/ml in 5 ml of BSK II medium. Spirochete densities were determined every 24 hours under dark field microscopy using a Petroff-Hausser chamber over the course of 96 hours.
RNA isolation from in vitro grown spirochetes. To obtain in vitro grown log phase spirochetes, wild-type (B31 A3) spirochetes were grown in triplicate in 5 ml of BSKII medium pH 7.5 at 35°C to a density of 3×107 spirochetes/ml. To obtain stationary phase, temperature-shifted spirochetes, wild-type (B31 A3) spirochetes were grown in triplicate in 5 ml of BSKII medium pH 7.5 at 35°C to a density of 3×107 spirochetes/ml, transferred to 25°C for 48 hours and then returned to 35°C for an additional 24–36 hours to a density of 2×108 spirochetes/ml. A total of 1×107 spirochetes were harvested from each culture and total RNA was isolated using TRIzol reagent (Life Technologies) according to the manufacturer's instructions. RNA was resuspended in 100 µl DEPC-treated dH2O. RNA was treated with TURBO DNA-free (Life Technologies) to remove any contaminating genomic DNA. 1 µl of Riboguard (40U/µl) RNAse inhibitor (Epicentre) was added to all samples and RNA stored at −80°C.
RNA isolation from infected mouse tissue. B. burgdorferi-infected mouse bladders (see mouse infection experiments below) were manually macerated on ice using sterile scalpels and transferred to a 2 ml sterile tube containing lysing Matrix D (MP Biomedicals). 1ml of RNA pro solution (FastRNA Pro Green kit, MP Biomedicals) was added to each sample on ice. Tissues were homogenized using a PowerGen High-Throughput Homogenizer (Fisher Scientific) following six cycles of beating for 45 sec and 2 minute incubations on ice. Samples were centrifuged at 13,000 rpm for 5 minutes at 4°C. The upper aqueous phase was transferred to new tubes and incubated for 5 minutes at room temperature. 500 µl of 1-bromo-3-chloropropane (Sigma Aldrich) and 45 µl of 5M sodium acetate were added to each sample and samples were incubated for an additional 5 minutes at room temperature. Samples were centrifuged at 13,000 rpm for 5 minutes at 4°C. The upper aqueous phase was transferred to new tubes and RNA precipitated with the addition of 500 µl of absolute ethanol and 1 µl GlycoBlue (Life technologies). RNA was pelleted by centrifugation at 13,000 rpm for 10 minutes at 4°C. RNA was washed with 70% ethanol in DEPC-treated dH20 and resuspended in 100 µl DEPC-treated dH20. RNA was treated with TURBO DNA-free (Life Technologies) to remove any contaminating genomic DNA. 1 µl Riboguard (40U/µl) RNAse inhibitor (Epicentre) was added to all samples and RNA stored at −80°C.
Gene expression analysis. cDNA was synthesized from 1.0 µg of each RNA sample using the iScript cDNA synthesis kit (Bio-Rad) with random primers according to the manufacturer's instructions. Parallel cDNA reactions were carried out in the absence of reverse transcriptase. Real-time quantitative PCR (qPCR) reactions were prepared using 1 µg of each cDNA and iQ SYBR Green Supermix (Bio-Rad). Using an Applied Biosystems 7500 instrument, samples were assayed for the flaB, recA, ospC and bbk46 transcripts using primers pairs 21 and 22, 23 and 24, 25 and 26, and 19 and 20, respectively (Table 5). Standard curves were generated for each gene target using 100 ng, 10 ng, 1.0 ng, 0.1 ng, and 0.01 ng of B31 A3 B. burgdorferi genomic DNA and the amount of each gene transcript calculated. The recA transcript was used as the endogenous reference to which the transcripts of the other genes were normalized. The bbk46 primers were confirmed to be specific for their gene target. Three biological replicate samples were analyzed in triplicate and normalized to recA mRNA. The data were reported as the average gene transcript/recA transcript for each sample. The amplification of samples lacking reverse transcriptase was similar to that of the no-template control.
Mouse infection experiments. Unless otherwise noted, groups of 6–8 week old C3H/HeN female mice (Harlan) were used for all experiments.
ID50 analysis of mammalian-adapted spirochetes. A single C3H/HeN SCID mouse (Harlan) was inoculated with 2×106 B. burgdorferi B31 A3. Two weeks post infection the infected blood was harvested and used to inoculate groups of six wild-type C3H/HeN (Harlan) mice with 100 µl of undiluted infected blood or 100 µl of infected blood diluted 1∶10 or 1∶100 in BSK-H medium. The number of live spirochetes in the infected blood and therefore the actual spirochete dose in the inoculum was determined by plating the blood in solid BSK medium and quantitating the number of colony forming units (Table 1). In addition, groups of six wild-type C3H/HeN mice (Harlan) were inoculated with 5×104, 5×103, 5×102, 5×101 or 5×100, in vitro grown spirochetes at mid-log phase. The in vitro grown spirochetes were confirmed to harbor all plasmids required for infectivity [71].
Functional validation of the BbIVET system. Groups of 6 mice were needle inoculated as described [77] with 1×104 spirochetes of clone A3 68-1 carrying pBbIVET or pBbIVET-ospC p. Mouse infection was assessed 3 weeks post inoculation by reisolation of spirochetes from ear, bladder and joint tissues as previously described [71,79].
Gene expression studies. Three mice were needle-inoculated intradermally under the skin of the upper back with B. burgdorferi clone B31 A3 at a dose of 1×105 spirochetes. Three weeks post inoculation mouse infection was determined by serology [71,79] and bladders harvested for RNA isolation.
bbk46 mutant infectivity studies. Groups of mice were needle inoculated as described [77] with 1×104 spirochetes with B. burgdorferi clones Δbbk46/vector (nine mice) or Δbbk46/bbk46 + (six mice) at a dose of 1×104 spirochetes. The number of spirochetes inoculated into mice was determined using a Petroff-Hausser counting chamber and verified by colony-forming unit (cfu) counts in solid BSK medium. Twelve colonies per inoculum were verified by PCR for the presence of the virulence plasmids lp25, lp28-1 and lp36 in at least 90% of the individuals in the population. Further, total plasmid content of each inoculum was confirmed to be as expected [37,71,77]. Mouse infection was assessed three weeks post inoculation by serology using total B. burgdorferi lysate, as previously described [37]. Spirochetes were reisolated from the ear, bladder and joint tissues, as previously described [37].
Supporting Information
Representative restriction digest analysis of individual pBbIVET plasmids rescued in E. coli. Colony PCR to amplify the in vivo-expressed DNA fragment was performed on a random subset of twenty four E. coli transformants carrying the rescued pBbIVET plasmids from infected mouse tissues. The PCR products were digested with a cocktail of the restriction enzymes DraI, SspI and AseI and separated on a 1% agarose gel. Numbers across the top of each image identify each non-identical restriction digestion pattern detected for the amplified pBbIVET DNA fragments. Representative data from two mouse tissues (A) and (B) are shown. Migration of the DNA ladders is shown in base pairs on both sides of each image. NTC, PCR no template control.
(TIF)
The BBK46 protein is non-immunogenic in mice. Recombinant GST-BBK46 and GST alone produced in and purified from E. coli, along with total protein lysate from E. coli and B. burgdorferi (Bb lysate) and E. coli producing the B. burgdorferi antigen BmpA (+) were separated by SDS-PAGE and transferred to a nitrocellulose membrane. Immunoblot analysis was performed using immune serum collected from mice infected with wild-type B. burgdorferi and anti-GST monoclonal antibodies (α GST). The positions of markers to the left of the panel depict protein standard molecular masses in kilodaltons.
(TIF)
Acknowledgments
Many thanks to Dr. Andrew Camilli for sharing his expertise and to members of the P. Rosa lab for their helpful suggestions during the initial stages of developing IVET for B. burgdorferi. Thank you to Dr. Daniel Dulebohn for sharing his in vitro temperature-shift induction method. Thank you to Selina Sutchu for technical assistance. Thank you to Dr. Travis Jewett for insightful comments and continued support. Thank you to the UCF NAF animal care staff.
Radolf JD, Caimano MJ, Stevenson B, Hu LT (2012) Of ticks, mice and men: understanding the dual-host lifestyle of Lyme disease spirochaetes. Nat Rev Microbiol 10: 87–99.
Samuels DS (2011) Gene regulation in Borrelia burgdorferi. Annu Rev Microbiol 65: 479–499.
Carroll JA, Garon CF, Schwan TG (1999) Effects of environmental pH on membrane proteins in Borrelia burgdorferi. Infect Immun 67: 3181–3187.
Ojaimi C, Brooks C, Casjens S, Rosa P, Elias A, et al. (2003) Profiling of temperature-induced changes in Borrelia burgdorferi gene expression by using whole genome arrays. Infect Immun 71: 1689–1705.
Tokarz R, Anderton JM, Katona LI, Benach JL (2004) Combined effects of blood and temperature shift on Borrelia burgdorferi gene expression as determined by whole genome DNA array. Infect Immun 72: 5419–5432.
Jutras BL, Chenail AM, Stevenson B (2012) Changes in bacterial growth rate govern expression of the Borrelia burgdorferi OspC and Erp infection-associated surface proteins. J Bacteriol.
Brooks CS, Hefty PS, Jolliff SE, Akins DR (2003) Global analysis of Borrelia burgdorferi genes regulated by mammalian host-specific signals. Infect Immun 71: 3371–3383.
Revel AT, Talaat AM, Norgard MV (2002) DNA microarray analysis of differential gene expression in Borrelia burgdorferi, the Lyme disease spirochete. Proc Natl Acad Sci U S A 99: 1562–1567.
Akins DR, Bourell KW, Caimano MJ, Norgard MV, Radolf JD (1998) A new animal model for studying Lyme disease spirochetes in a mammalian host-adapted state. J Clin Invest 101: 2240–2250.
Theel ES, Jespersen DJ, Harring J, Mandrekar J, Binnicker MJ (2013) Evaluation of an Enzyme Immunoassay for Detection of Histoplasma capsulatum Antigen from Urine Specimens. J Clin Microbiol.
Liang FT, Nelson FK, Fikrig E (2002) Molecular adaptation of Borrelia burgdorferi in the murine host. J Exp Med 196: 275–280.
Mahan MJ, Slauch JM, Hanna PC, Camilli A, Tobias JW, et al. (1993) Selection for bacterial genes that are specifically induced in host tissues: the hunt for virulence factors. Infect Agents Dis 2: 263–268.
Mahan MJ, Slauch JM, Mekalanos JJ (1993) Selection of bacterial virulence genes that are specifically induced in host tissues. Science 259: 686–688.
Slauch JM, Camilli A (2000) IVET and RIVET: use of gene fusions to identify bacterial virulence factors specifically induced in host tissues. Methods Enzymol 326: 73–96.
Jackson RW, Giddens SR (2006) Development and application of in vivo expression technology (IVET) for analysing microbial gene expression in complex environments. Infect Disord Drug Targets 6: 207–240.
Hanin A, Sava I, Bao Y, Huebner J, Hartke A, et al. (2010) Screening of in vivo activated genes in Enterococcus faecalis during insect and mouse infections and growth in urine. PLoS One 5: e11879.
Lee SW, Cooksey DA (2000) Genes expressed in Pseudomonas putida during colonization of a plant-pathogenic fungus. Appl Environ Microbiol 66: 2764–2772.
Mendez J, Reimundo P, Perez-Pascual D, Navais R, Gomez E, et al. (2011) A novel cdsAB operon is involved in the uptake of L-cysteine and participates in the pathogenesis of Yersinia ruckeri. J Bacteriol 193: 944–951.
Binnicker MJ, Jespersen DJ, Rollins LO (2011) Evaluation of the Bio-Rad BioPlex Measles, Mumps, Rubella, and Varicella-Zoster Virus IgG multiplex bead immunoassay. Clin Vaccine Immunol 18: 1524–1526.
Binnicker MJ, Jespersen DJ, Harring JA, Rollins LO, Bryant SC, et al. (2008) Evaluation of two commercial systems for automated processing, reading, and interpretation of Lyme borreliosis Western blots. J Clin Microbiol 46: 2216–2221.
Aucott J, Morrison C, Munoz B, Rowe PC, Schwarzwalder A, et al. (2009) Diagnostic challenges of early Lyme disease: lessons from a community case series. BMC Infect Dis 9: 79.
Ramamoorthy R, McClain NA, Gautam A, Scholl-Meeker D (2005) Expression of the bmpB gene of Borrelia burgdorferi is modulated by two distinct transcription termination events. J Bacteriol 187: 2592–2600.
Jewett MW, Jain S, Linowski AK, Sarkar A, Rosa PA (2011) Molecular characterization of the Borrelia burgdorferi in vivo-essential protein PncA. Microbiology 157: 2831–2840.
Purser JE, Lawrenz MB, Caimano MJ, Howell JK, Radolf JD, et al. (2003) A plasmid-encoded nicotinamidase (PncA) is essential for infectivity of Borrelia burgdorferi in a mammalian host. Mol Microbiol 48: 753–764.
Grimm D, Eggers CH, Caimano MJ, Tilly K, Stewart PE, et al. (2004) Experimental assessment of the roles of linear plasmids lp25 and lp28-1 of Borrelia burgdorferi throughout the infectious cycle. Infect Immun 72: 5938–5946.
Grimm D, Tilly K, Bueschel DM, Fisher MA, Policastro PF, et al. (2005) Defining plasmids required by Borrelia burgdorferi for colonization of tick vector Ixodes scapularis (Acari: Ixodidae). J Med Entomol 42: 676–684.
Labandeira-Rey M, Seshu J, Skare JT (2003) The absence of linear plasmid 25 or 28-1 of Borrelia burgdorferi dramatically alters the kinetics of experimental infection via distinct mechanisms. Infect Immun 71: 4608–4613.
Labandeira-Rey M, Skare JT (2001) Decreased infectivity in Borrelia burgdorferi strain B31 is associated with loss of linear plasmid 25 or 28-1. Infect Immun 69: 446–455.
Purser JE, Norris SJ (2000) Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proc Natl Acad Sci U S A 97: 13865–13870.
Revel AT, Blevins JS, Almazan C, Neil L, Kocan KM, et al. (2005) bptA (bbe16) is essential for the persistence of the Lyme disease spirochete, Borrelia burgdorferi, in its natural tick vector. Proc Natl Acad Sci U S A 102: 6972–6977.
Strother KO, de Silva A (2005) Role of Borrelia burgdorferi linear plasmid 25 in infection of Ixodes scapularis ticks. J Bacteriol 187: 5776–5781.
Samuels DS, Mach KE, Garon CF (1994) Genetic transformation of the Lyme disease agent Borrelia burgdorferi with coumarin-resistant gyrB. J Bacteriol 176: 6045–6049.
Tilly K, Elias AF, Bono JL, Stewart P, Rosa P (2000) DNA exchange and insertional inactivation in spirochetes. J Mol Microbiol Biotechnol 2: 433–442.
Lawrenz MB, Kawabata H, Purser JE, Norris SJ (2002) Decreased electroporation efficiency in Borrelia burgdorferi containing linear plasmids lp25 and lp56: impact on transformation of infectious B. burgdorferi. Infect Immun 70: 4798–4804.
Lawrence KA, Jewett MW, Rosa PA, Gherardini FC (2009) Borrelia burgdorferi bb0426 encodes a 2'-deoxyribosyltransferase that plays a central role in purine salvage. Mol Microbiol 72: 1517–1529.
Tilly K, Krum JG, Bestor A, Jewett MW, Grimm D, et al. (2006) Borrelia burgdorferi OspC protein required exclusively in a crucial early stage of mammalian infection. Infect Immun 74: 3554–3564.
Jewett MW, Lawrence K, Bestor AC, Tilly K, Grimm D, et al. (2007) The critical role of the linear plasmid lp36 in the infectious cycle of Borrelia burgdorferi. Mol Microbiol 64: 1358–1374.
Fischer M, Hlinak A (2000) The lack of binding ability of staphylococcal protein A and streptococcal protein G to egg yolk immunoglobulins of different fowl species (short communication). Berl Munch Tierarztl Wochenschr 113: 94–96.
Fischer H, Dohlsten M, Andersson U, Hedlund G, Ericsson P, et al. (1990) Production of TNF-alpha and TNF-beta by staphylococcal enterotoxin A activated human T cells. J Immunol 144: 4663–4669.
Gruia AD, Fischer S, Smith JC (2003) Molecular dynamics simulation reveals a surface salt bridge forming a kinetic trap in unfolding of truncated Staphylococcal nuclease. Proteins 50: 507–515.
Fischer A, von Eiff C, Kuczius T, Omoe K, Peters G, et al. (2007) A quantitative real-time immuno-PCR approach for detection of staphylococcal enterotoxins. J Mol Med (Berl) 85: 461–469.
Eisermann R, Fischer R, Kessler U, Neubauer A, Hengstenberg W (1991) Staphylococcal phosphoenolpyruvate-dependent phosphotransferase system. Purification and protein sequencing of the Staphylococcus carnosus histidine-containing protein, and cloning and DNA sequencing of the ptsH gene. Eur J Biochem 197: 9–14.
Casjens S, Palmer N, van Vugt R, Huang WM, Stevenson B, et al. (2000) A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs in an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi. Mol Microbiol 35: 490–516.
Casjens SR, Mongodin EF, Qiu WG, Luft BJ, Schutzer SE, et al. (2012) Genome stability of Lyme disease spirochetes: comparative genomics of Borrelia burgdorferi plasmids. PLoS One 7: e33280.
Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, et al. (1997) Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature 390: 580–586.
Schutzer SE, Fraser-Liggett CM, Casjens SR, Qiu WG, Dunn JJ, et al. (2011) Whole-genome sequences of thirteen isolates of Borrelia burgdorferi. J Bacteriol 193: 1018–1020.
Lawrenz MB, Wooten RM, Norris SJ (2004) Effects of vlsE complementation on the infectivity of Borrelia burgdorferi lacking the linear plasmid lp28-1. Infect Immun 72: 6577–6585.
Zhang JR, Hardham JM, Barbour AG, Norris SJ (1997) Antigenic variation in Lyme disease borreliae by promiscuous recombination of VMP-like sequence cassettes. Cell 89: 275–285.
Chiang SL, Mekalanos JJ, Holden DW (1999) In vivo genetic analysis of bacterial virulence. Annu Rev Microbiol 53: 129–154.
Chen Q, Fischer JR, Benoit VM, Dufour NP, Youderian P, et al. (2008) In vitro CpG methylation increases the transformation efficiency of Borrelia burgdorferi strains harboring the endogenous linear plasmid lp56. J Bacteriol 190: 7885–7891.
Jacobs MB, Norris SJ, Phillippi-Falkenstein KM, Philipp MT (2006) Infectivity of the highly transformable BBE02- lp56- mutant of Borrelia burgdorferi, the Lyme disease spirochete, via ticks. Infect Immun 74: 3678–3681.
Kawabata H, Norris SJ, Watanabe H (2004) BBE02 disruption mutants of Borrelia burgdorferi B31 have a highly transformable, infectious phenotype. Infect Immun 72: 7147–7154.
Rego RO, Bestor A, Rosa PA (2011) Defining the plasmid-borne restriction-modification systems of the Lyme disease spirochete Borrelia burgdorferi. J Bacteriol 193: 1161–1171.
Lin T, Gao L, Zhang C, Odeh E, Jacobs MB, et al. (2012) Analysis of an Ordered, Comprehensive STM Mutant Library in Infectious Borrelia burgdorferi: Insights into the Genes Required for Mouse Infectivity. PLoS One 7: e47532.
Liang FT, Jacobs MB, Bowers LC, Philipp MT (2002) An immune evasion mechanism for spirochetal persistence in Lyme borreliosis. J Exp Med 195: 415–422.
Liang FT, Yan J, Mbow ML, Sviat SL, Gilmore RD, et al. (2004) Borrelia burgdorferi changes its surface antigenic expression in response to host immune responses. Infect Immun 72: 5759–5767.
Campbell GL, Fritz CL, Fish D, Nowakowski J, Nadelman RB, et al. (1998) Estimation of the incidence of Lyme disease. Am J Epidemiol 148: 1018–1026.
Young JD (1998) Underreporting of Lyme disease. N Engl J Med 338: 1629.
Kapoor A, Simmonds P, Cullen JM, Scheel TK, Medina JL, et al. (2013) Identification of a pegivirus (GB virus-like virus) that infects horses. J Virol 87: 7185–7190.
Yang X, Popova TG, Hagman KE, Wikel SK, Schoeler GB, et al. (1999) Identification, characterization, and expression of three new members of the Borrelia burgdorferi Mlp (2.9) lipoprotein gene family. Infect Immun 67: 6008–6018.
Yang XF, Hubner A, Popova TG, Hagman KE, Norgard MV (2003) Regulation of expression of the paralogous Mlp family in Borrelia burgdorferi. Infect Immun 71: 5012–5020.
Ouyang Z, Blevins JS, Norgard MV (2008) Transcriptional interplay among the regulators Rrp2, RpoN and RpoS in Borrelia burgdorferi. Microbiology 154: 2641–2658.
Ouyang Z, Deka RK, Norgard MV (2011) BosR (BB0647) controls the RpoN-RpoS regulatory pathway and virulence expression in Borrelia burgdorferi by a novel DNA-binding mechanism. PLoS Pathog 7: e1001272.
Ojaimi C, Mulay V, Liveris D, Iyer R, Schwartz I (2005) Comparative transcriptional profiling of Borrelia burgdorferi clinical isolates differing in capacities for hematogenous dissemination. Infect Immun 73: 6791–6802.
Gjorloff A, Fischer H, Hedlund G, Hansson J, Kenney JS, et al. (1991) Induction of interleukin-1 in human monocytes by the superantigen staphylococcal enterotoxin A requires the participation of T cells. Cell Immunol 137: 61–71.
Bankhead T, Chaconas G (2007) The role of VlsE antigenic variation in the Lyme disease spirochete: persistence through a mechanism that differs from other pathogens. Mol Microbiol 65: 1547–1558.
Dresser AR, Hardy PO, Chaconas G (2009) Investigation of the genes involved in antigenic switching at the vlsE locus in Borrelia burgdorferi: an essential role for the RuvAB branch migrase. PLoS Pathog 5: e1000680.
Lin T, Gao L, Edmondson DG, Jacobs MB, Philipp MT, et al. (2009) Central role of the Holliday junction helicase RuvAB in vlsE recombination and infectivity of Borrelia burgdorferi. PLoS Pathog 5: e1000679.
Yang X, Coleman AS, Anguita J, Pal U (2009) A chromosomally encoded virulence factor protects the Lyme disease pathogen against host-adaptive immunity. PLoS Pathog 5: e1000326.
Lawrenz MB, Hardham JM, Owens RT, Nowakowski J, Steere AC, et al. (1999) Human antibody responses to VlsE antigenic variation protein of Borrelia burgdorferi. J Clin Microbiol 37: 3997–4004.
Elias AF, Stewart PE, Grimm D, Caimano MJ, Eggers CH, et al. (2002) Clonal polymorphism of Borrelia burgdorferi strain B31 MI: implications for mutagenesis in an infectious strain background. Infect Immun 70: 2139–2150.
Barbour AG (1984) Isolation and cultivation of Lyme disease spirochetes. Yale J Biol Med 57: 521–525.
Rosa PA, Hogan D (1992) Colony formation by Borrelia burgdorferi in solid medium: clonal analysis of osp locus variants. In: Munderloh UG, Kurtti TJ, editors. Proceeding of the First International Conference on Tick Borne Pathogens at the Host-Vector Interface. St. Paul, Minnesota: University of Minnesota.
Samuels DS (1995) Electrotransformation of the spirochete Borrelia burgdorferi. In: Nickoloff JA, editor. Methods in Molecular Biology. Totowa, N.J.: Humana Press, Inc. pp. 253–259.
Jewett MW, Byram R, Bestor A, Tilly K, Lawrence K, et al. (2007) Genetic basis for retention of a critical virulence plasmid of Borrelia burgdorferi. Mol Microbiol 66: 975–990.
Elias AF, Bono JL, Kupko JJ, 3rd, Stewart PE, Krum JG, et al. (2003) New antibiotic resistance cassettes suitable for genetic studies in Borrelia burgdorferi. J Mol Microbiol Biotechnol 6: 29–40.
Jewett MW, Lawrence KA, Bestor A, Byram R, Gherardini F, et al. (2009) GuaA and GuaB are essential for Borrelia burgdorferi survival in the tick-mouse infection cycle. J Bacteriol 191: 6231–6241.
Halpern MD, Jain S, Jewett MW (2013) Enhanced detection of host response antibodies to Borrelia burgdorferi using immuno-PCR. Clin Vaccine Immunol 20: 350–357.
Grimm D, Tilly K, Byram R, Stewart PE, Krum JG, et al. (2004) Outer-surface protein C of the Lyme disease spirochete: a protein induced in ticks for infection of mammals. Proc Natl Acad Sci U S A 101: 3142–3147.
Bestor A, Stewart PE, Jewett MW, Sarkar A, Tilly K, et al. (2010) Use of the Cre-lox recombination system to investigate the lp54 gene requirement in the infectious cycle of Borrelia burgdorferi. Infect Immun 78: 2397–2407.
Setubal JC, Reis M, Matsunaga J, Haake DA (2006) Lipoprotein computational prediction in spirochaetal genomes. Microbiology 152: 113–121.
Weisman LE, Thackray HM, Garcia-Prats JA, Nesin M, Schneider JH, et al. (2009) Phase 1/2 double-blind, placebo-controlled, dose escalation, safety, and pharmacokinetic study of pagibaximab (BSYX-A110), an antistaphylococcal monoclonal antibody for the prevention of staphylococcal bloodstream infections, in very-low-birth-weight neonates. Antimicrob Agents Chemother 53: 2879–2886.
Binnicker MJ, Jespersen DJ, Rollins LO (2011) Treponema-specific tests for serodiagnosis of syphilis: comparative evaluation of seven assays. J Clin Microbiol 49: 1313–1317.
Reference
- 1. Ellis TC, Jain S, Linowski AK, Rike K, Bestor A (2013) In Vivo Expression Technology Identifies a Novel Virulence Factor Critical for Borrelia burgdorferi Persistence in Mice. PLoS Pathog 9(8): e1003567. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative restriction digest analysis of individual pBbIVET plasmids rescued in E. coli. Colony PCR to amplify the in vivo-expressed DNA fragment was performed on a random subset of twenty four E. coli transformants carrying the rescued pBbIVET plasmids from infected mouse tissues. The PCR products were digested with a cocktail of the restriction enzymes DraI, SspI and AseI and separated on a 1% agarose gel. Numbers across the top of each image identify each non-identical restriction digestion pattern detected for the amplified pBbIVET DNA fragments. Representative data from two mouse tissues (A) and (B) are shown. Migration of the DNA ladders is shown in base pairs on both sides of each image. NTC, PCR no template control.
(TIF)
The BBK46 protein is non-immunogenic in mice. Recombinant GST-BBK46 and GST alone produced in and purified from E. coli, along with total protein lysate from E. coli and B. burgdorferi (Bb lysate) and E. coli producing the B. burgdorferi antigen BmpA (+) were separated by SDS-PAGE and transferred to a nitrocellulose membrane. Immunoblot analysis was performed using immune serum collected from mice infected with wild-type B. burgdorferi and anti-GST monoclonal antibodies (α GST). The positions of markers to the left of the panel depict protein standard molecular masses in kilodaltons.
(TIF)