

Abstract
Dilated cardiomyopathy (DCM) due to mutations in RBM20, a gene encoding an RNA-binding protein, is associated with high familial penetrance, risk of progressive heart failure and sudden death. Although genetic investigations and physiological models have established the linkage of RBM20 with early-onset DCM, the underlying basis of cellular and molecular dysfunction is undetermined. Modeling human genetics using a high-throughput pluripotent stem cell platform was herein designed to pinpoint the initial transcriptome dysfunction and mechanistic corruption in disease pathogenesis. Tnnt2-pGreenZeo pluripotent stem cells were engineered to knockdown Rbm20 (shRbm20) to determine the cardiac-pathogenic phenotype during cardiac differentiation. Intracellular Ca2+ transients revealed Rbm20-dependent alteration in Ca2+ handling, coinciding with known pathological splice variants of Titin and Camk2d genes by Day 24 of cardiogenesis. Ultrastructural analysis demonstrated elongated and thinner sarcomeres in the absence of Rbm20 that is consistent with human cardiac biopsy samples. Furthermore, Rbm20-depleted transcriptional profiling at Day 12 identified Rbm20-dependent dysregulation with 76% of differentially expressed genes linked to known cardiac pathology ranging from primordial Nkx2.5 to mature cardiac Tnnt2 as the initial molecular aberrations. Notably, downstream consequences of Rbm20-depletion at Day 24 of differentiation demonstrated significant dysregulation of extracellular matrix components such as the anomalous overexpression of the Vtn gene. By using the pluripotent stem cell platform to model human cardiac disease according to a stage-specific cardiogenic roadmap, we established a new paradigm of familial DCM pathogenesis as a developmental disorder that is patterned during early cardiogenesis and propagated with cellular mechanisms of pathological cardiac remodeling.
INTRODUCTION
Dilated cardiomyopathy (DCM) is a progressive and debilitating disease that inevitably leads to heart failure and compromises delivery of life-sustaining nutrients and oxygen to the body (1,2). The irreversible deterioration of cardiac function in DCM frequently leads to significant morbidity and mortality that can be refractory to medical therapy and require cardiac transplantation (3,4). Recognition of DCM as a familial disorder in up to 50% of cases has been the impetus for human genetics investigations to uncover the molecular basis of DCM (5,6). Despite heritability of DCM, development of heart failure is unpredictable into adulthood, suggesting an insidious degenerative process. However, the cellular pathogenesis is not sufficiently understood to explain variable, age-dependent penetrance and provide novel diagnostic, prognostic or therapeutic tools.
A majority of disease-causing or modifying DCM genes initially identified encode proteins involved in cardiomyocyte contraction and cytoskeletal structure. An expanded understanding of the pathobiology of DCM has emerged from genomic strategies, which have linked impaired ion homeostasis and gene regulation (6). Mutations in RBM20, a gene encoding a RNA-binding protein (RBP), were discovered in individuals with early-onset familial DCM, implicating perturbation of posttranscriptional pre-mRNA processing as a distinct molecular basis for human DCM (7). Subsequent studies confirmed that patients with RBM20 mutations manifest disease symptoms at early age and are characterized by a high degree of morbidity and mortality as cardiac systolic function deteriorates towards end-stage heart failure even in young children (8,9). At the molecular level, Rbm20 has been linked to posttranscriptional regulation and the alternative splicing of Titin and Camk2d in cardiac tissue (10,11), yet the temporal sequence of dysregulation during cardiogenesis has not been examined to determine the initial points of divergence and better understand the cellular etiology of this genetic disease.
Conserved throughout evolution, compelling evidence has established RBPs with cardiac function and developmental processes. For example, Rbm24 was recently demonstrated to be enriched in embryonic stem cells (ESC) derived cardiomyocytes and required for sarcomere assembly and heart contractility (12). In addition, Hermes was found to regulate heart development in Xenopus (13). Furthermore, Lin28, DAZL and GRSF1 contribute to maintenance of pluripotency and differentiation of ESCs (14,15) while coordinated teamwork of CELF and MBNL1 proteins are critical during normal cardiac and skeletal muscle development (16). Based on these findings, RNA processing is established as a major regulator of early gene expression machinery during developmental processes and may underlie the progenitor cell contribution to ongoing cardiac homeostasis and tissue renewal (17).
Herein, we hypothesized that Rbm20 is an essential component of the RNA processing machinery during cardiogenesis and is required to regulate cardiac gene expression to pattern normal structural and physiological integrity of newly formed cardiomyocytes. Utilizing the cardiogenic pipeline based on pluripotent stem cells, we aimed to establish an in vitro model of Rbm20-linked DCM to probe DCM development and unmask the initial molecular and cellular dysfunctions independent of confounding physiological contexts. This approach establishes Rbm20 disruption as an early-onset mechanism of human cardiomyopathy with altered early stage cardiogenic gene expression, calcium overload, sarcomeric abnormalities and gene expression patterns of compensated heart failure. Furthermore, the cellular phenotype of Rbm20-deficiency enables a novel approach to apply advanced technology of developmental cell biology towards DCM modeling (18) and employ pharmacological screening to modulate regulatory mechanisms of genetic cardiovascular diseases that are able to halt or reverse dysfunctional cardiogenesis.
RESULTS
Induction of Rbm20 occurs during early embryonic cardiogenesis
Gene expression from natural stages of mouse heart development demonstrates that the molecular network of pluripotent (Pou5f1, Sox2), mesodermal (T-Brachyury, Mesp1) pre-cardiac (Gata4, Tbx5, Nkx2–5) and mature cardiac (Acta1, Tnnt2, Tnt) genes are sequentially expressed during cardiogenesis. In a stage-specific manner, the onset of pre-cardiac gene expression is detected at E8.5 of mouse heart development. Notably, Rbm20 induction was between E7.5 and E8.5 during in vivo cardiogenesis, concomitant with established pre-cardiac and cardiac gene activation (Fig. 1A). Moreover, the expression profile of Rbm20 resembles that of Nkx2-5, a marker for cardiac progenitors. Using mouse embryonic stem cells (mESCs), the expression profile of Rbm20 throughout in vitro cardiogenesis was examined at different time points of embryoid body (EB) differentiation (Days 0–24). Expression of Rbm20 was induced between Days 5 and 9 of ESC differentiation (Fig. 1B). To further validate the in vitro system of ESC-derived cardiomyocytes, a developmental expression profile of pluripotent, mesodermal, pre-cardiac and cardiac markers (Pou5f1, Mesp1, Actc1, Tnni3) during EB differentiation was used as a benchmark. Like in the in vivo roadmap, Pou5f1 expression decreased with the onset of EB differentiation, followed by the transient induction of Mesp1, robust increase in Acta1 and progressive increase in Tnni3 expression with advanced stages of cardiac differentiation (Fig. 1C). To trace cardiac differentiation, a Tnnt2-pGreenZeo reporter system (System Bioscience) was utilized and cardiogenesis was visualized upon induction of the reporter system. Within the EB clusters, temporal and spatial GFP expression was associated with beating areas (starting at ∼Days 10–12). For enrichment of cardiomyocytes, EBs were treated with Zeocin selection medium from Days 12–24. Fluorescence-activated cell sorting (FACS) analysis showed enrichment of the cardiomyocyte population at Day 24 of differentiation upon Zeocin treatment (Fig. 1D).
Figure 1.

Rbm20 is differentially expressed during cardiogenesis (A) Microarray roadmap of in vivo cardiogenesis. Illustration of mouse cardiac development stages from mESC through adult heart and dynamically and spatially differential expression patterns in the left and right ventricles (L: left: R: right). (B) In vitro differentiation of mESCs to EBs demonstrates that Rbm20 is expressed between d5 and d9 using quantitative polymerase chain reaction (qPCR). (C) qPCR analysis of established cardiac genes expressed during ESC differentiation to EBs at different time points. (D) Tnnt2-GFP reporter system in differentiating EBs shows the in vitro activation of troponin verifying in vitro cardiogenesis from d12 to d25. FACS analysis shows the fold change of cardiomyocyte population at d24 upon Zeo selection. *Relative to Eb d5 as a negative control P≤ 0.05.
EBs with knockdown of Rbm20 express pathological forms of unspliced Titin and Camk2d
Tnnt2-pGreenZeo mESCs were infected with four different viral particles containing shRNA sequences targeting the Rbm20 transcript (sh9, sh11, sh12, sh14), differentiated, and characterized at Days 12 and 24 (Fig. 2A). Quantitative real-time PCR (qRT-PCR) results demonstrated that one out of four shRNA target sequences (sh12) significantly down-regulated Rbm20 at Day 10 of ESC differentiation compared with the control infected with non-targeting shRNA (Fig. 2B). To validate the loss-of-function associated with suppression of Rbm20, we assessed the splicing activity of the established targets Titin and Camk2d (10) at Days 12 and 24 (Fig. 2C and D) with the Gapdh gene serving as a loading control. At Day 24, agarose gel electrophoresis revealed accumulation of the unspliced, long isoform (720 bp; ∼80%) of Titin over the smaller isoform (240 bp; ∼20%) in Tnnt2- derived shRbm20 progenitors, whereas the isoform ratio was ∼1:1 in control cardiomyocytes (Fig. 2C). Furthermore, retention of unspliced exons 15 and 16 in the Camk2d transcript resulted in abnormal persistence of isoform A (247 bp; ∼43%) in response to shRbm20 knockdown at Day 24 (Fig. 2D). There were no differences detected in amount of the stage-specific isoforms of Titin and Camk2d at earlier time-points (Fig. 2C and D), confirming the pathological splicing profile of Titin and Camk2d in the presence of decreased Rbm20 during later stages of cardiogenesis.
Figure 2.

Rbm20 knockdown in vitro phenocopies DCM disease expression profiles in vivo. (A) Experimental design to establish Rbm20 knockdown (shRbm20) and characterization of shRbm20-derived EBs at d12 and d24. (B) Structure of the murine Rbm20 gene and position of the shRbm20 viral particles targeting the mRNA of Rbm20 in different exons (9,11,12,14). qPCR of d10 EBs show down regulation of Rbm20 specifically with viral particles targeting exon 12 (SH12) (*P= 0.03) d5 and d10 EBs were used as a negative and positive control of Rbm20 expression (C and D) RT-PCR on EBs derived from shRbm20 samples (SH-12) in biological triplicate (SH1, SH2 and SH3) and control (CTRL1, CTRL2 and CTRL3) at Days 12 and 24 and relative densitometry to detect the splice variants of Titin long isoform (L) (785 bp) and small isoform (S) (385 bp), and Camk2d with the isoforms A (247 bp) and C (118 bp).
shRbm20-deficient cardiogenesis results in defective Ca2+ machinery
We assayed the Ca2+ handling properties of EBs at Day 12 and Tnnt2-selected cells at Day 24 by characterizing spontaneously occurring intracellular Ca2+ transients. In three independent experiments, there were no differences in Ca2+ spike amplitude, area under the curve, time between peaks, or characteristic time of Ca2+ extrusion identified between shRbm20-deficient and control cells at Day 12 (Fig. 3A). However, defective calcium machinery was revealed at d24 of differentiation in the Rbm20-knockdown subpopulation. Notably, the d24 shRbm20-deficient subpopulation exhibited elevated Ca2+ spike amplitude (53.62±7.83 AU versus 32.21±4.34 AU in shRbm20 compared with control , respectively; P<0.05) and area under the curve (253.08±45.62 AU versus, 98.49±13.32 AU, in shRbm20 compared with control, respectively; P<0.05) of individual Ca2+ transients. Furthermore, the time between peaks was longer (1.86±0.15 s versus 1.20±0.21 s, in shRbm20 compared with control, respectively; P<0.05) resulting in slower beating activity. In addition, the characteristic time of Ca2+ extrusion was faster in shRbm20 cardiomyocytes (2.0±0.21 s versus 2.75± 0.15 s, in shRbm20 compared with control, respectively; P<0.05) (Fig. 3B). Molecular characterization of Ca2+ machinery proteins revealed enhanced Serca2a and elevated Camk2d protein levels in shRbm20-deficient subpopulations at Day 24 compared with control cardiac cells (Fig. 3C). Microarray analysis of in vivo cardiogenesis provided a benchmark for the expression profile of Serca2a and Camk2d which demonstrated the activation of both genes at E7.5 and increased expression to a plateau at E8.5 for Serca2a and E12.5 for Camk2d, highlighting temporal modulation of Ca2+ handling protein expression during natural heart development. To define the role of abnormal Ca2+ handling as a mechanism of action for Rbm20 dysregulation, and to test whether a Ca2+ overloaded state is upstream of defective Titin and Camk2d-based DCM, Ca2+ channel blockers were applied. Ca2+ measurements in shRbm20 and control EBs with Ca2+-channel inhibitor verapamil from Days 12–24 demonstrated the ability to completely inhibit Ca2+ transients, yet the pathological expression and splicing profiles of Titin and Camk2d remained unaffected by the modulation of Ca2+ levels (Supplementary Material, Fig. S1).
Figure 3.

Rbm20-dependent calcium handling at d24 of cardiogenesis. (A) Analysis of spontaneous calcium transients (top right) recorded from fluorescent images of Rhod-2 am-labeled derived cardiomyocytes (top left) revealed no differences between shRbm20 (SH) and control (CTRL). (B) shRbm20-derived cardiomyocytes at d24 reveal abnormal Ca2+ handling as demonstrated by enhanced peak amplitude and area under the curve of calcium transients, as well as prolonged time between calcium spikes *denotes P< 0.05. (C) The data represent the mean values from three independent experiments. Western blotting analysis of Serca2a, sarcoplasmatic reticulum calcium 2ATPase, and Camk2d Calcium/calmodulin-dependent protein kinase type II delta chain in shRbm20 and control at d24 of differentiation. Microarray analysis shows the expression profile of Serca2a and Camk2d during in vivo cardiogenesis.
shRbm20-deficient cardiomyocytes exhibit an atypical sarcomeric geometry in both patient-derived and bioengineered samples
We next analyzed the sarcomeric structure of shRbm20-deficient cardiomyocytes by immunostaining and transmission electron microscopy (TEM). Both control and Rbm20-deficient cardiomyocytes showed expression of α-actinin at Day 24 of differentiation. However, compared with controls (n=58), a significantly higher percentage of Rbm20-deficent cardiomyocytes (n=88) had disorganized actinin fibers with punctate and stress fiber architecture (40% control versus 62% shRbm20; P=0.05) (Fig. 4A). TEM in shRbm20 EBs at Day 24 showed elongated and thinner sarcomeres compared with control length [1.93±0.4 µm (n=167) versus 1.67±0.25 µm (n=123)] and width [0.66±0.32 µm (n=135) versus 0.81±0.35 µm (n=110)] (Fig. 4B and C). In order to compare the in vitro phenotype of Rbm20 deficiency to patient-derived pathological samples and sarcomeric structures, cardiac biopsy materials from two related DCM patients carrying a P638L mutation in RBM20 (Fig. 4E) were analyzed and compared with cardiac biopsies from two individuals without DCM (Fig. 4D). The measurements of sarcomere length and width derived from DCM patients compared with controls revealed atypical geometry (length: 1.85±0.23 µm [n=92] versus 1.76±0.12 µm [n=72]; width: 0.7±0.33 µm versus 1.48±0.3 µm; Fig. 5D–F). Thus, the abnormal sarcomere structure within DCM patients was comparable to the in vitro-derived, Rbm20-deficient cardiomyocytes.
Figure 4.

Cardiomyocytes at d24 manifest abnormal sarcomere geometry in Rbm20-deficiency as demonstrated in patient-derived cardiac tissues (A) α-actinin immunostaining images from shRbm20 and wild-type cardiomyocytes at d24. More punctuate staining and enrichment of stress fiber structures in shRbm20 cardiomyocytes compared with wild-type controls. (B) TEM images of shRbm20- and control-derived cardiomyocyte sarcomeres. The red lines indicate the distance between the Z lines used for the measurement of the sarcomere length and the yellow lines indicate the sarcomere width. (C) Violin plot shows the statistically significant difference in sarcomere geometry of shRbm20 versus control samples. Orange dot, median of sarcomeric length and width in shRbm20 and control cardiomyocytes. (D) TEM images of DCM and healthy human sarcomeres. (E) Violin plot shows the statistically significant difference in sarcomere geometry in two human DCM compared with two healthy samples.
Figure 5.

Knockdown of Rbm20 is associated with dysregulation of gene splicing and a limited subset of cardiac genes at d12. (A) Summary of total transcripts analyzed by RNAseq in shRbm20 and control EBs at d12. (B) Barplot for the GO term enrichment analysis for differential genes at Day 12. (C) RNAseq data from shRbm20 and control-derived EBs at d12 (left) and expression profile through in vivo cardiogenesis of the 21 genes differentially expressed in shRbm20 (right). Natural cardiogenesis is represented by nine developmental stages E7.5, to adult stages, and by left (L) and right (R) ventricles when they became distinguishable in the heart, ESC was used as pluripotent stage. Link between differentially expressed genes in shRbm20 cardiomyocytes and cardiac diseases associated. EL, embryonic lethal; IHD, ischemic heart disease; HCM, hypertrophic cardiomyopathy; CHD, congenital heart disease; DCM, dilated cardiomyopathy.
Knockdown of Rbm20 is associated with dysregulation of gene splicing and gene expression within cardiogenic pathways
RNA-seq was performed on EBs at Day 12 of differentiation with or without shRbm20 to uncover the initial corruption due to knockdown of Rbm20 by profiling whole-genome transcriptome. Starting from 50 224 transcripts detected at a developmental stage preceding pathological cellular phenotype, a distinct group of 49 genes were identified as differentially spliced, 12 genes had different isoforms, 11 genes were transcribed with different transcription start-site positions and 21 genes were differentially expressed in control and shRbm20-derived EBs (Fig. 5A and Supplementary Material, Tables S2 and S3). Since Rbm20 is known to regulate gene processing, the differentially-spliced genes were analyzed and noted to have regulatory function at the transcriptional and post-transcriptional level as noted with transcription factors Mef2a and Relb (Supplementary Material, Fig. S3, red label); SF1, involved in spliceosome complex; Tial1, a RNA-binding protein; and Zscan10 and 8, zinc finger proteins (19–23). Focusing the analysis on differentially expressed genes in shRbm20 EBs and comparing them to the natural cardiogenic roadmap identified 21 Rbm20-dependent genes involved in heart development and muscle differentiation (Fig. 5B and C). Furthermore, 16 of the 21 dysregulated genes (76%) have established associations with cardiac pathobiology. Mutations and aberrant gene expression related to Ors1, Shh, Nkx2-5, Pgam2, Myl4, Myl3, Myl7, Tnnt2, Csrp3, Myl2, Pobec2, Rad, Tdgf1, Actc1, Mybpc3 and Cox6a2 are associated with specific cardiac defects including embryonic lethal defects, congenital heart diseases, cardiomyopathies and inflammation of the myocardium (Fig. 5C, Supplementary Material, Fig. S4) (24–28). Notably, 3 of the 16 cardiac genes (Act1, Pgam2 and Myl2) differentially expressed in shRbm20-derived EBs are transcriptionally regulated by Mef2a (Supplementary Material, Fig. S4 red + label) (19,29). By searching TRASFACT motif, 10 differentially expressed genes were identified as predicted targets of Mef2a or Relb transcription factors (Supplementary Material, Fig. S4, blue + label). The expression profiles obtained by RNAseq were validated by qRT-PCR (Supplementary Material, Table S1).
Transcriptome analysis of shRbm20-derived cardiac subpopulations at Day 24 revealed dysregulation of extracellular matrix gene networks
The comparison of transcriptome profiles of control and Rbm20-knockdown groups at Day 24 using RNAseq revealed 240 known genes differentially expressed upon in vitro differentiation. 170 of these genes displayed a dynamic expression profile within the in vivo cardiogenesis roadmap according to the time-course microarray data set using mouse developmental model system (nine time points from mESC to adult heart) (Fig. 6). The gene interaction network (node-edge information), ECM genes from gene ontology (GO) function enrichment analysis, and expression profiles of these 170 genes from both natural cardiogenesis and RNA-seq data were visualized in an integrated circular plot to highlight gene–gene interactions. In the circular plot, the gene–gene interactions were defined by the STRING database based on experiments, co-expression, gene fusion or co-occurrence, etc. The interaction information was retrieved by querying STRING database using the entire gene list as input with the default settings on the database website (http://string-db.org/).
Figure 6.

Rbm20-deficient EBs at d24 revealed dysregulation of gene expression patterns in extracellular matrix networks. Circular plot showed gene interactions, expression patterns in natural cardiogenesis (time-course microarray) and in control and shRbm20-derived cardiomyocytes at Day 24 (RNAseq). Differentially expressed genes are represented by red (up-regulated) and green (down-regulated) dots with curves in the center linking them and representing the interactions between genes (green: interaction connecting to down-regulated genes; red: interaction between up-regulated genes only). The sizes of the gray bubbles surrounding the red and green dots correspond to the numbers of interactions for each gene (i.e. larger bubbles represent more interactions). The inner heatmap represents the expression profiles during natural cardiogenesis. Natural cardiogenesis is characterized by time-course microarray experiment at nine different developmental stages using a mouse model including mESCs, E7.5, E8.5, E9.5, E12.5, E14.5, E18.5, 3 days after birth (D3) and adult (A) stages, starting from E9.5, the heart was dissected by left (L) and right (R) ventricles. At each time point, the heatmap showed the average expression of biological triplicate. The middle heatmap is for the two control samples in RNAseq and outer heatmap for three shRbm20-derived samples. Genes belonging to the ECM are indicated by blue bars on the gray circular band. The names of hub genes in the interaction network with more than 10 interactions are labeled in red.
The hub genes (labeled with red) with more than 10 interacting partners genes included Prodh2, Otc, Ugt2b34, Ambp, Fabp1, Aldob, Apoa1, Spp2, Apob, Hnf4a, Cps1, Alpi and Vtn. GO enrichment analysis on these 170 genes revealed that the most significantly enriched function centered on extracellular matrix (ECM) pathways which encompassed 33 genes differentially expressed across the developmental roadmap (Fig. 6 and Supplementary Material, Fig. S5). Furthermore, most of the ECM genes were upregulated in the Rbm20-knockdown group indicating an acquisition of pathological features in a cell-autonomous model system. Therefore, Rbm20-dependent mechanisms are contributing to dysfunctional cardiac gene expression starting at early stages of cardiogenesis and may be directly altering signaling mechanisms linked to pathological cardiac remodeling.
DISCUSSION
DCM is a debilitating disease that results in progressive deterioration of cardiac function and refractory heart failure with significant morbidity and mortality (2,4). Despite DCM being historically described as idiopathic, genetic mechanisms are now recognized as a major contributor to DCM pathogenesis (30,31). Mutations within RBM20, a gene encoding an RNA-binding motif protein, have been discovered in individuals with severe cases of familial DCM (7–9). In order to facilitate the mechanistic understanding of early-onset DCM pathogenesis, we tested the hypothesis that Rbm20 acts as a crucial component of RNA processing machinery to establish primordial cardiac gene networks underpinning proper cardiac developmental processes required for normal structure and function of nascent cardiomyocytes. A time course microarray analysis of natural heart development identified the induction of Rbm20 gene expression at E8.5 of cardiogenesis, which remained stable throughout maturation of cardiac tissue. Furthermore, the activation of Rbm20 in our in vitro model of cardiogenesis was confirmed between Day 5 and 9 of mESC differentiation. Compared with the dynamic expression of pluripotent, pre-cardiac and cardiac genes analyzed through in vitro cardiogenesis, the Rbm20 expression profile mimics that of gold-standard early cardiac markers, such as Nkx2–5 (32). This expression profile suggests that Rbm20 is contributing to early cardiac gene networks that are not present in pluripotent ESC, yet are maintained throughout adult myocardium. Given the predominant phenotype in patients with RBM20 mutations is associated with cardiomyopathic changes, the molecular mechanisms leading to DCM could be contributing to cellular dysfunction throughout the stages of cardiogenesis corresponding to the induction and expression of the Rbm20 gene.
A previous study showed that cardiomyopathic changes due to a mutation in Rbm20 impaired splicing of the sarcomeric structural protein Titin and the Ca2+ regulator Camk2d (10). Moreover, it was reported that the persistence of the unspliced form of Camk2d is independently linked to the DCM phenotype (33). Since mutations in RBM20 are associated with early-onset DCM, it was hypothesized that this gene is required for proper embryonic heart development. This hypothesis required a stem cell-derived model system to determine molecular dysfunction at specific developmental time points. By knocking down Rbm20 during in vitro cardiogenesis, we confirmed that cardiomyocytes with Rbm20 deficiency detectable at Day 10 exhibit pathological Titin and Camk2d profiles at Day 24 of differentiation. The aberrant persistence of the long Titin isoform (L) and the absence of the short isoform (S) confirmed a pathological link between Rbm20 and Titin in our model system. Furthermore, analysis of Ca2+ transients by fluorescence microscopy in Rbm20-knockdown EBs revealed abnormalities at Day 24 that indicated anomalous Ca2+ handling within the cytoplasm likely due to an aggregation of dysfunctional regulatory pathways including Camk2d. Proportionally, the increased level of the sarcoplasmic reticulum Ca2+ pump, Serca2a, confirmed the increased requirement of Ca2+ turnover in shRbm20 cells. However, chronic treatment of EBs with verapamil, a Ca2+ channel blocker, does not rescue the splicing of Titin and Camk2d, indicating that the Ca2+ phenotype is downstream of Rbm20-dependent splice variants. As the link between Ca2+ machinery alterations and cardiomyopathy has been established (18), herein we demonstrated for the first time an Rbm20-dependent dysregulation of Ca2+ homeostasis. Together, the dysregulation of structural genes and Ca2+ handling machinery are established as pathological features of DCM and can be recapitulated within an in vitro model system.
In addition to the pathological regulation of intracellular Ca2+, ultrastructural analysis of sarcomeres from Tnnt2-selected shRbm20 cardiomyocytes at Day 24 unmasked an elongated, thinner structural geometry. Cardiac tissues derived directly from patients with RBM20 mutations have comparable sarcomeric morphological structure, suggesting a specific phenotype linked to RBM20 both in vivo and in vitro model. Although several forms of DCM have demonstrated sarcomeric disarray (18,34), herein we show for the first time that the altered structure of the sarcomere unit in patient-derived cardiac tissues with Rbm20-dependent DCM can be recapitulated within a cell-autonomous stem cell model system. The structural aberration of the sarcomere units could in part explain the anatomy of the dilated heart in patients with DCM and the retention of the more compliant Titin isoform, N2BA, associated with low passive tension (35,36). Collectively, these structural changes may directly contribute to a reduction of the contractile force in the hearts of DCM patients.
RNA-seq analysis of shRbm20 EBs versus control revealed aberrant splicing of Mef2a and Relb and dysregulation of 16 known genes whose failure has been linked with heart disease (37–45), suggesting that Rbm20 can directly regulate cardiac-dependent RNA processing of transcription factors (21,46). The splice variants of transcription factors such as Mef2a and dysregulation of cardiac gene networks are the first detectable events at Day 12. At this initial time point of molecular divergence, the profiles of genes differentially expressed in shRbm20 EBs resemble an abnormal profile compared with the anticipated cardiogenesis profile derived from the natural roadmap (47). Moreover, the shift from low to high expression of two stage-specific genes, Cdhr2 and Cubn, in shRbm20 EBs at Day 12 highlights the possibility that desynchronized gene patterns may negatively regulate sequential acquisition of innate cardiogenic processes. Furthermore, mapping initial gene expression changes in early cardiogenesis has revealed pathological changes in stem cell-derived cardiac tissue that was previously observed in physiological systems (10). With progression of the disease in a physiological system, the compensatory mechanisms are thought to affect Extracellular matrix (ECM) genes and cardiac remodeling processes, which have also been linked to DCM (10). Herein, gene expression analysis of enriched cardiac tissue at Day 24 demonstrated characteristic changes in ECM genes previously detectable in physiological systems. Although the presence of ECM genes in our RNA-seq analysis was unexpected, these data indicates that cell-autonomous mechanisms linked to Rbm20 may drive cellular phenotypes previously considered by-products of physiological adaptation to disease states. Thus, we now show that dysregulation of a wide range of cardiac-related genes and pathways (e.g. ECM) can be detected and modeled in the absence of physiological compensation using a stem cell-based disease model system.
Collectively, this study demonstrates that Rbm20 is expressed in early cardiogenesis and functions in the patterning of cardiac-gene expression in our stem cell-derived model system through splicing regulation of transcription factors, such as Mef2a, that could directly affect downstream gene expression patterns linked to cardiomyopathic profiles. The “butterfly effect” or small perturbations early in a process that leads to widespread disruption have been observed herein with Rbm20 dysregulation in early stages of cardiogenesis that culminates in derangement of cellular phenotypes involving structural and Ca2+ handling machinery and is sufficient to produce expression profiles that were previously identified only in decompensated physiological systems (10). Therefore, stem cell-based developmental model systems offer a distinctive advantage for genotype/phenotype analysis to decipher between initial disease-causing mechanism and delayed compensatory effects. Ongoing studies within a wide array of DCM genotypes using high-throughput stem cell-based technology will likely be sufficient to reveal alternative strategies to map disease-causing mechanisms linked to progressive heart failure. The dissection of novel developmental targets will accelerate our ability to modulate progenitor cell contribution and optimize innate cardiac regeneration for patients with a genetic underpinning for cardiomyopathic disease processes.
MATERIALS AND METHODS
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless stated otherwise.
In vivo cardiogenesis microarray
The time-course microarray experiments were performed on mouse R1 embryonic stem cells (mESC), mouse whole embryo (E7.5), heart tubes (E8.5), and left and right ventricles (E9.5, E12.5, E14.5, E18.5, newborn (3 days after birth) and adult heart. Gene expression data were computed using RMA algorithm as previously described (47).
Tnnt2-pGreenZeo mESC culture and differentiation
mESCs were cultured in stem cell medium and subsequently differentiated into three-layer EBs) using the Agreewell method (Stemcell Technologies). The Tnnt2-pGreenZeo mESCs were established by infecting mESCs with Tnnt2-pGreenZeo packaged virus particles (System Biosciences, Mountain View, CA). After infection, cellular colonies derived from single cells were PCR screened to assay for the transgene. EB cultures were enriched for Tnnt2-positive cardiomyocytes by treatment with 700 mg/ml Zeocin (Invitrogen) from Days 12 to 24.
All our analyses were performed from plates of cells that achieved 20–30% cardiac induction at d12 based on activation of Tnnt2/GFP.
shRbm20 knockdown
Tnnt2-pGreenZeo mESCs were infected with Mission TRC1 viral particles (Sigma, St. Louis, MO) containing pre-designed shRNA lenti plasmids directed against different exons (exons # 9, 11, 12, and 14) of mouse Rbm20. In addition, a mixture of all four viral particles (shmix) was used. Non-targeting viral particles were used as a control (CTRL). Stable lines for each shRbm20, puromycin-resistant viral particle were established by culturing the cells for 30 days in selection media containing 2 µg/ml puromycin (Invitrogen). The Tnnt2-pGreenZeo mESCs were differentiated into EBs and screened at Days 9 and 10 for the highest level of Rbm20 knockdown by qRT-PCR.
RT-PCR for alternative splicing
RT-PCR was carried out with RNA extracted from shRbm20 and control EBs at Days 12 and 24 using a RNeasy Plus Mini kit (Qiagen, MD). The reverse transcriptase (RT) reaction was performed using an iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA).
Ca2+ imaging
Intracellular Ca2+ dynamics in control and shRbm20-derived EBs at Days 12 and 24 were assessed as previously described.21 Briefly, cells were plated on laminin coated coverslips for >24 h, loaded with the Ca2+-fluorescent probe Rhod-2 AM (Invitrogen) in Tyrode's solution containing 140 mm NaCl, 5.4 mm KCl, 1 mm MgCl2, 10 mm glucose, 1.5 mm CaCl2, and 10 mm HEPES (pH 7.4) and imaged with a Zeiss LSM Live 5 laser confocal microscope (Zeiss). Spontaneous Ca2+ transients were recorded at 37°C. A custom Matlab program (Mathworks) was implemented to calculate the mean amplitude of Ca2+ spikes corresponding to the mean area under the curve of spontaneous Ca2+ transients and the mean time between spikes from 8–12 consecutive Ca2+ cycles. To determine the characteristic time of Ca2+ extrusion (τ), the relaxation phase of each transient (from peak amplitude to baseline) was fitted to the exponential function:
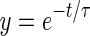 |
where y is fluorescence in AU, t is time, and τ is the time to reach 63% of exponential relaxation. The Ca2+ analysis was performed three times at d12 and d24 and each time the shRbm20 samples were head to head compared with parental cell line expressing innate levels of Rbm20.
Western blotting
At Day 24, Tnnt2-selected EBs were lysed on ice in RIPA protein lysis buffer and samples were processed as previously described (48).
Cell staining
Immunostaining for α-actinin inTnnt2-selected shRbm20 and control-derived EBs was performed at Day 24 of differentiation and images were acquired by Zeiss LSM 510 Axiovert laser confocal microscopy (Zeiss).
Transmission Electron Microscopy
Tnnt2-selected shRbm20, control-derived EBs and human endomyocardial biopsy specimens were collected processed for TEM and examined on a FEI Tecnai G2 12 transmission electron microscope (TEM) operated at 80 kV. Micrographs were taken on a JEOL 1200 EXII electron microscope.49 ImageJ software (NIH) was utilized to measure the length and the width of the sarcomere.
Flow cytometry
GFP expression levels were quantified in Tnnt2-pGreenZeo EBs at d5, d12, and d24 cultured in differentiation media upon Zeocin treatment from d12 to d24. Specifically, the EBs infected with the Tnnt2-pGreenZeo reporter system was subjected to spontaneous differentiation. During this process, beating cardiomyocytes that were GFP-positive begin to appear at d12. At this stage, treatment with Zeocin was initiated until d24 to enrich the culture with Tnnt2-positive cardiomyocytes. Flow cytometry was performed using a Gallios flow cytometer (Beckman Coulter) and data analyzed with the Kaluza 1.2 software (Beckman Coulter). Quantification of green GFP cardiomyocytes was performed at d5, d12 and d24 upon differentiation. GFP values for d12 and d24 were calculated by normalizing their corresponding geometric mean values by the geometric mean value associated with d5. Thus, GFP fluorescence intensity was 2.95 and 6.94-fold higher than d5 values in d12 and d24 differentiated cells, respectively.
RNA sequencing (RNA-seq)
RNA was extracted from shRbm20 and control (EB) at d12 and d24 via a Qiagen RNeasy kit. Sequencing library was prepared using Trueseq and sequenced on Illumina Hiseq 2000. For the details of RNA-seq analysis, pair-end short reads in the FASTQ format from sequencer were first filtered and only high quality reads with Phred scores >33 were kept for the following analysis. After filtering the short reads were aligned or mapped back to human genome (Homo_sapiens_UCSC-hg19) with Bowtie2. The unmapped kept reads were split to segments and mapped to genome with Tophat2 and Bowtie2 for junction searching with coverage-search algorithm on. Discovered splices were indexed and joined. The all the results were reported in the BAM files for the following analysis. The BAM files for each sample were used to assemble transcripts with Cufflinks with Cuffcompare then able to compare transcript assemblies to annotation. The assembled transcripts were merged together using Cuffmerge (49,50). Cuffdiff were used to finds differentially expressed gene and transcripts and detects differential splicing and/or promoter use (51). The results were then imported into R for extracting differential genes and data visualization using R CummerRbund package. The P-value cutoff for differential analysis was 0.05 after FDR control.
Statistical analysis
Heatmap and hierarchical clustering used to order the genes in heatmap and circular plot were performed using R functions. Non-linear model used to fit the curve for gene expression across time points were performed with Spline model. Wilcoxon test were applied to the sarcomere analysis and modified distribution plot were generated using R functions and customized R scripts.
qRT-PCR
Real-time PCR to confirm the RNA-seq results used RNA extracted from Days 12 and 24 control and shRbm20 EBs via a Qiagen RNeasy kit. cDNA synthesis was completed using an iScript cDNA Synthesis kit.
SUPPLEMENTARY MATERIAL
FUNDING
This study was supported by National Institutes of Health New Innovator Award # OD 07015.
Supplementary Material
ACKNOWLEDGEMENTS
A special thanks to the Todd and Karen Wanek Family Program for Hypoplastic Left Heart Syndrome (HLHS). AHA Postdoctoral Fellowship (A.M.F.), the Leducq Foundation, and the Center for Regenerative Medicine at Mayo Clinic. Authors thank Katherine Hartjes and Miranda Lange for critical review of the manuscript.
Conflict of Interest statement. None declared.
References
- 1.Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of Cardiomyopathies. Circulation. 1996;93:841–842. doi: 10.1161/01.cir.93.5.841. [DOI] [PubMed] [Google Scholar]
- 2.Maron B.J., Towbin J.A., Thiene G., Antzelevitch C., Corrado D., Arnett D., Moss A.J., Seidman C.E., Young J.B. Contemporary Definitions and Classification of the Cardiomyopathies: An American Heart Association Scientific Statement From the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 3.Towbin J.A., Lowe A.M., Colan S.D., Sleeper L.A., Orav E.J., Clunie S., Messere J., Cox G.F., Lurie P.R., Hsu D., et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296:1867–1876. doi: 10.1001/jama.296.15.1867. [DOI] [PubMed] [Google Scholar]
- 4.Tsirka A.E., Trinkaus K., Chen S.-C., Lipshultz S.E., Towbin J.A., Colan S.D., Exil V., Strauss A.W., Canter C.E. Improved outcomes of pediatric dilated cardiomyopathy with utilization of heart transplantation. J. Am. Coll. Cardiol. 2004;44:391–397. doi: 10.1016/j.jacc.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 5.Michels V.V., Moll P.P., Miller F.A., Tajik A.J., Chu J.S., Driscoll D.J., Burnett J.C., Rodeheffer R.J., Chesebro J.H., Tazelaar H.D. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N. Engl. J. Med. 1992;326:77–82. doi: 10.1056/NEJM199201093260201. [DOI] [PubMed] [Google Scholar]
- 6.Olson T.M., Chan D.P. In: Moss and Adams’ Heart Disease in Infants, Children, and Adolescents: Including the Fetus and Young Adult. Allen H.D., et al., editors. Vol. 1. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2013. pp. 1235–1246. [Google Scholar]
- 7.Brauch K.M., Karst M.L., Herron K.J., de Andrade M., Pellikka P.A., Rodeheffer R.J., Michels V.V., Olson T.M. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 2009;54:930–941. doi: 10.1016/j.jacc.2009.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li D., Morales A., Gonzalez-Quintana J., Norton N., Siegfried J.D., Hofmeyer M., Hershberger R.E. Identification of novel mutations in RBM20 in patients with dilated cardiomyopathy. Clin. Transl. Sci. 2010;3:90–97. doi: 10.1111/j.1752-8062.2010.00198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wells Q.S., Becker J.R., Su Y.R., Mosley J.D., Weeke P., D'Aoust L., Ausborn N.L., Ramirez A.H., Pfotenhauer J.P., Naftilan A.J., et al. Whole exome sequencing identifies a causal rbm20 mutation in a large pedigree with familial dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2013;6:317–326. doi: 10.1161/CIRCGENETICS.113.000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo W., Schafer S., Greaser M.L., Radke M.H., Liss M., Govindarajan T., Maatz H., Schulz H., Li S., Parrish A.M., et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012;18:766–773. doi: 10.1038/nm.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li S., Guo W., Dewey C.N., Greaser M.L. Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Res. 2013;41:2659–2672. doi: 10.1093/nar/gks1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poon K.L., Tan K.T., Wei Y.Y., Ng C.P., Colman A., Korzh V., Xu X.Q. RNA-binding protein RBM24 is required for sarcomere assembly and heart contractility. Cardiovasc. Res. 2012;94:418–427. doi: 10.1093/cvr/cvs095. [DOI] [PubMed] [Google Scholar]
- 13.Gerber W.V., Vokes S.A., Zearfoss N.R., Krieg P.A. A role for the RNA-binding protein, hermes, in the regulation of heart development. Dev. Biol. 2002;247:116–126. doi: 10.1006/dbio.2002.0678. [DOI] [PubMed] [Google Scholar]
- 14.Yang D.-H., Moss E.G. Temporally regulated expression of Lin-28 in diverse tissues of the developing mouse. Gene Expr. Patterns. 2003;3:719–726. doi: 10.1016/s1567-133x(03)00140-6. [DOI] [PubMed] [Google Scholar]
- 15.Richards M., Tan S.-P., Tan J.-H., Chan W.-K., Bongso A. The transcriptome profile of human embryonic stem cells as defined by SAGE. Stem Cells. 2004;22:51–64. doi: 10.1634/stemcells.22-1-51. [DOI] [PubMed] [Google Scholar]
- 16.Dasgupta T., Ladd A.N. The importance of CELF control: molecular and biological roles of the CUG-BP, Elav-like family of RNA-binding proteins. Wiley Interdiscip. Rev. RNA. 3. 2012:104–121. doi: 10.1002/wrna.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verheggen C., Almouzni G., Hernandez-Verdun D. The ribosomal RNA processing machinery is recruited to the nucleolar domain before RNA Polymerase I during Xenopus laevis development. J. Biol. Chem. 2000;149:293–306. doi: 10.1083/jcb.149.2.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun N., Yazawa M., Liu J., Han L., Sanchez-Freire V., Abilez O.J., Navarrete E.G., Hu S., Wang L., Lee A., et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med. 2012;4:130ra147. doi: 10.1126/scitranslmed.3003552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naya F.J., Black B.L., Wu H., Bassel-Duby R., Richardson J.A., Hill J.A., Olson E.N. Mitochondrial deficiency and cardiac sudden death in mice lacking the MEF2A transcription factor. Nat. Med. 2002;8:1303–1309. doi: 10.1038/nm789. [DOI] [PubMed] [Google Scholar]
- 20.Le Bon A., Montoya M., Edwards M.J., Thompson C., Burke S.A., Ashton M., Lo D., Tough D.F., Borrow P. A role for the transcription factor RelB in IFN-α production and in IFN-α-stimulated cross-priming. Eur. J. Immunol. 2006;36:2085–2093. doi: 10.1002/eji.200535228. [DOI] [PubMed] [Google Scholar]
- 21.Edmondson D.G., Lyons G.E., Martin J.F., Olson E.N. Mef2 gene expression marks the cardiac and skeletal muscle lineages during mouse embryogenesis. Development. 1994;120:1251–1263. doi: 10.1242/dev.120.5.1251. [DOI] [PubMed] [Google Scholar]
- 22.Dember L.M., Kim N.D., Liu K.-Q., Anderson P. Individual RNA recognition motifs of TIA-1 and TIAR have different RNA binding specificities. J. Biol. Chem. 1996;271:2783–2788. doi: 10.1074/jbc.271.5.2783. [DOI] [PubMed] [Google Scholar]
- 23.Rino J., Desterro J.M.P., Pacheco T.R., Gadella T.W.J., Carmo-Fonseca M. Splicing Factors SF1 and U2AF associate in extraspliceosomal complexes. Mol. Cell. Biol. 2008;28:3045–3057. doi: 10.1128/MCB.02015-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie L., Hoffmann A.D., Burnicka-Turek O., Friedland-Little J.M., Zhang K., Moskowitz I.P. Tbx5-Hedgehog molecular networks are essential in the second heart field for atrial septation. Dev. Cell. 2012;23:280–291. doi: 10.1016/j.devcel.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hildreth V., Webb S., Chaudhry B., Peat J.D., Phillips H.M., Brown N., Anderson R.H., Henderson D.J. Left cardiac isomerism in the Sonic hedgehog null mouse. J. Anat. 2009;214:894–904. doi: 10.1111/j.1469-7580.2009.01087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Costa M.W., Guo G., Wolstein O., Vale M., Castro M.L., Wang L., Otway R., Riek P., Cochrane N., Furtado M., et al. Functional characterization of a novel mutation in NKX2–5 associated with congenital heart disease and adult-onset cardiomyopathy. Circ. Cardiovasc. Genet. 2013;6:238–247. doi: 10.1161/CIRCGENETICS.113.000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H., Li J., Wang Y., Yang T. Proteomic analysis of effluents from perfused human heart for transplantation: identification of potential biomarkers for ischemic heart damage. Proteome Sci. 2012;10:21. doi: 10.1186/1477-5956-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schaub M.C., Hefti M.A., Zuellig R.A., Morano I. Modulation of contractility in human cardiac hypertrophy by myosin essential light chain isoforms. Cardiovasc. Res. 1998;37:381–404. doi: 10.1016/s0008-6363(97)00258-7. [DOI] [PubMed] [Google Scholar]
- 29.Schlesinger J., Schueler M., Grunert M., Fischer J.J., Zhang Q., Krueger T., Lange M., Tonjes M., Dunkel I., Sperling S.R. The cardiac transcription network modulated by Gata4, Mef2a, Nkx2.5, Srf, histone modifications, and microRNAs. PLoS Genet. 2011;7:e1001313. doi: 10.1371/journal.pgen.1001313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fatkin D., Otway R., Richmond Z. Genetics of dilated cardiomyopathy. Heart Fail. Clin. 2010;6:129–140. doi: 10.1016/j.hfc.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 31.Hershberger R.E., Lindenfeld J., Mestroni L., Seidman C.E., Taylor M.R., Towbin J.A. Genetic evaluation of cardiomyopathy—a Heart Failure Society of America practice guideline. J. Card. Fail. 2009;15:83–97. doi: 10.1016/j.cardfail.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 32.Lints T., Parsons L., Hartley L., Lyons I., Harvey R. Nkx-2.5: a novel murine homeobox gene expressed in early heart progenitor cells and their myogenic descendants. Development. 1993;119:969. doi: 10.1242/dev.119.3.969. [DOI] [PubMed] [Google Scholar]
- 33.Xu X., Yang D., Ding J.-H., Wang W., Chu P.-H., Dalton N.D., Wang H.-Y., Bermingham J.R., Jr, Ye Z., Liu F., et al. ASF/SF2-Regulated CaMKIIδ alternative splicing temporally reprograms excitation–contraction coupling in cardiac muscle. Cell. 2005;120:59–72. doi: 10.1016/j.cell.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 34.Meyer T., Ruppert V., Ackermann S., Richter A., Perrot A., Sperling S.R., Posch M.G., Maisch B., Pankuweit S. Novel mutations in the sarcomeric protein myopalladin in patients with dilated cardiomyopathy. Eur. J. Hum. Genet. 2013;21:294–300. doi: 10.1038/ejhg.2012.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.LeWinter M.M., Granzier H.L. Titin is a major human disease gene. Circulation. 2013;127:938–944. doi: 10.1161/CIRCULATIONAHA.112.139717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greaser M.L., Warren C.M., Esbona K., Guo W., Duan Y., Parrish A.M., Krzesinski P.R., Norman H.S., Dunning S., Fitzsimons D.P., et al. Mutation that dramatically alters rat titin isoform expression and cardiomyocyte passive tension. J. Mol. Cell. Cardiol. 2008;44:983–991. doi: 10.1016/j.yjmcc.2008.02.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trahair T., Yeoh T., Cartmill T., Keogh A., Spratt P., Chang V., dos Remedios C.G., Gunning P. Myosin light chain gene expression associated with disease states of the human heart. J. Mol. Cell. Cardiol. 1993;25:577–585. doi: 10.1006/jmcc.1993.1067. [DOI] [PubMed] [Google Scholar]
- 38.Sato Y., Probst H.C., Tatsumi R., Ikeuchi Y., Neuberger M.S., Rada C. Deficiency in APOBEC2 leads to a shift in muscle fiber type, diminished body mass, and myopathy. J. Biol. Chem. 2010;285:7111–7118. doi: 10.1074/jbc.M109.052977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang B., Yan J., Peng Z., Wang J., Liu S., Xie X., Ma X. Teratocarcinoma-derived growth factor 1 (TDGF1) sequence variants in patients with congenital heart defect. Int. J. Cardiol. 2011;146:225–227. doi: 10.1016/j.ijcard.2009.08.046. [DOI] [PubMed] [Google Scholar]
- 40.Chen J., Kubalak S.W., Minamisawa S., Price R.L., Becker K.D., Hickey R., Ross J., Jr., Chien K.R. Selective requirement of myosin light chain 2v in embryonic heart function. J. Biol. Chem. 1998;273:1252–1256. doi: 10.1074/jbc.273.2.1252. [DOI] [PubMed] [Google Scholar]
- 41.Yung C.K., Halperin V.L., Tomaselli G.F., Winslow R.L. Gene expression profiles in end-stage human idiopathic dilated cardiomyopathy: altered expression of apoptotic and cytoskeletal genes. Genomics. 2004;83:281–297. doi: 10.1016/j.ygeno.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 42.Wang Q., Lan Y., Cho E.-S., Maltby K.M., Jiang R. Odd-skipped related 1 (Odd1) is an essential regulator of heart and urogenital development. Dev. Biol. 2005;288:582–594. doi: 10.1016/j.ydbio.2005.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Digilio M.C., Marino B., Giannotti A., Dallapiccola B., Opitz J.M. Specific congenital heart defects in RSH/Smith-Lemli-Opitz syndrome: Postulated involvement of the Sonic Hedgehog pathway in syndromes with postaxial polydactyly or heterotaxia. Birth Defects Res.t A Clin. Mol. Teratol.. 2003;67:149–153. doi: 10.1002/bdra.10010. [DOI] [PubMed] [Google Scholar]
- 44.Jay A., Chikarmane R., Poulik J., Misra V.K. Infantile hypertrophic cardiomyopathy associated with a novel MYL3 mutation. Cardiology. 2013;124:248–251. doi: 10.1159/000347138. [DOI] [PubMed] [Google Scholar]
- 45.Flavigny J., Richard P., Isnard R., Carrier L., Charron P., Bonne G., Forissier J.F., Desnos M., Dubourg O., Komajda M., et al. Identification of two novel mutations in the ventricular regulatory myosin light chain gene (MYL2) associated with familial and classical forms of hypertrophic cardiomyopathy. J. Mol. Med. 1998;76:208–214. doi: 10.1007/s001090050210. [DOI] [PubMed] [Google Scholar]
- 46.Ewen E.P., Snyder C.M., Wilson M., Desjardins D., Naya F.J. The Mef2A transcription factor coordinately regulates a costamere gene program in cardiac muscle. J. Biol. Chem. 2011;286:29644–29653. doi: 10.1074/jbc.M111.268094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martinez-Fernandez A., Li X., Hartjes K.A., Terzic A., Nelson T.J. Natural cardiogenesis-based template predicts cardiogenic potential of induced pluripotent stem cell lines. Circ. Cardiovasc. Genet. 2013;6:462–471. doi: 10.1161/CIRCGENETICS.113.000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith A.J., Nelson N.G., Oommen S., Hartjes K.A., Folmes C.D., Terzic A., Nelson T.J. Apoptotic susceptibility to DNA damage of pluripotent stem cells facilitates pharmacologic purging of teratoma risk. Stem Cells Transl. Med. 2012;1:709–718. doi: 10.5966/sctm.2012-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cazorla O., Freiburg A., Helmes M., Centner T., McNabb M., Wu Y., Trombitás K., Labeit S., Granzier H. Differential expression of cardiac titin isoforms and modulation of cellular stiffness. Circ. Res. 2000;86:59–67. doi: 10.1161/01.res.86.1.59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.