

Abstract
Chromosomal segmental deletion is a frequent cause of human diseases. A familial 1.1 Mb deletion of human chromosome Xq22.1 associates with epilepsy, cleft palate and developmental defects in heterozygous female patients. Here, we describe a mouse mutant with a targeted deletion of the syntenic segment of the mouse X chromosome that phenocopies the human syndrome. Male mice with a deletion of a 1.1 Mb Nxf2–Nxf3 X-chromosomal segment exhibit respiratory failure, neonatal lethality and cleft palate. In female mice, heterozygosity for the deletion manifests cleft palate, early postnatal lethality, postnatal growth delay and spontaneous seizures in surviving animals, apparently due to X-chromosome inactivation. Furthermore, loss of a 0.35 Mb subregion containing Armcx5, Gprasp1, Gprasp2 and Bhlhb9 is sufficient to cause the Xq22.1 syndrome phenotype. Our results support that the 1.1 Mb deletion of human Xq22.1 is the genetic cause of the associated syndrome.
INTRODUCTION
Medical cytogenetics and array comparative genomic hybridization (array-CGH) have identified a number of microdeletions of human chromosomes as common causes of genetic diseases. These regions of segmental deletions, usually a few mega bases, often harbor multiple genes such that their loss could lead to haploinsufficiency syndromes. The 22q11 deletion syndrome (DiGeorge/velocardiofacial syndrome), caused by a 3 Mb deletion, is the most common human microdeletion syndrome, occurring in 1:4000 live births (1, 2). Complementing studies of human patients, the generation of corresponding animal models can greatly facilitate the understanding of the pathogenesis of these complex syndromes, elucidate gene-to-phenotype relationships and identify causative genes. Genetic engineering of large chromosomal segmental deletions remains challenging but has become increasingly feasible.
Loss of a 1.1 Mb region of chromosome Xq22.1 is associated with epilepsy, mental retardation and various developmental defects including cleft palate in heterozygous female patients (3). The severity of symptoms differed between females affected by the deletion, and a male offspring of a heterozygous female died 15 days after birth due to a breathing failure. The 1.1 Mb section of Xq22.1 affected by the deletion contains 13 annotated genes, including NXF5. Mutations of this gene have been previously associated with mental retardation (4–6). A causal relationship between loss of the Xq22.1 region and phenotypic manifestations in human patients remains to be established and the potential role of each of the 13 candidate genes remains to be determined. Here, we report the generation of mice with a 1.1 Mb deletion syntenic to the human Xq22.1 deletion. Mice with this deletion exhibit respiratory failure, cleft palate and epilepsy, phenocopying the human syndrome. Deletion of a 0.35 Mb subregion containing Armcx5, Gprasp1, Gprasp2 and Bhlhb9 causes the same phenotype, identifying these as critical candidate genes.
RESULTS
Respiratory failure and neonatal lethality in DelA/Y males
A 1.1 Mb region (Nxf2–Nxf3) on the mouse X chromosome is syntenic to the 1.1 Mb region deleted in human patients with the Xq22.1 syndrome (Fig. 1A). We previously generated a targeted allele [Del(XNxf2–Nxf3)1Jw] in which the Nxf2–Nxf3 segment is flanked by loxP sites (Fig. 1A) (7). We crossed these mice with ubiquitously expressing Actb-Cre mice (8) to delete the 1.1 Mb Nxf2–Nxf3 segment, here referred to as DelA (Fig. 1A). The DelA allele caused neonatal lethality in all (DelA/Y) male pups. The observation that DelA/Y males develop to term suggests that none of the 20 genes in the DelA region are essential for early embryogenesis (Fig. 1A). Western blot analysis demonstrated absence of GPRASP1 and GPRASP2 proteins (encoded by this region) in DelA/Y male brain and reduced levels in DelA/+ female brain, indicating that the deletion was complete (Fig. 1B). To determine the cause of neonatal lethality in male mutant mice, we examined embryonic Day 18.5 (E18.5) embryos delivered by cesarean section. All wild-type E18.5 pups except one (13 of 14) breathed normally after delivery and were viable. However, all 11 E18.5 DelA/Y male pups examined developed asphyxia and died immediately after delivery, indicating acute respiratory failure (Fig. 2A).
Figure 1.

Deletion mapping analysis of the critical interval on the X chromosome. (A) Schematic representation of the human Xq22.1 segment, the syntenic region on mouse X chromosome after targeted insertion of loxP sites, and the floxed and deletion alleles generated in this study. Dashed lines connect orthologous genes between mouse and human. Deletions on the mouse X chromosome (DelA, DelB and DelC; designated by dashed lines) were obtained by crossing mice carrying the respective floxed allele with Actb-Cre mice that ubiquitously express Cre recombinase (8). All loxP sites are in the same orientation, and in each deletion allele (deletion size indicated in brackets), only one loxP site is left. (B) Western blot analysis of GPRASP1/2 in DelA mutant brain at E16.5. (C) Western blot analysis of GPRASP1/2 in DelC mutant brain at E18.5. ACTB serves as a loading control. Note that the antibody recognizes both GPRASP1 and GPRASP2 (15).
Figure 2.
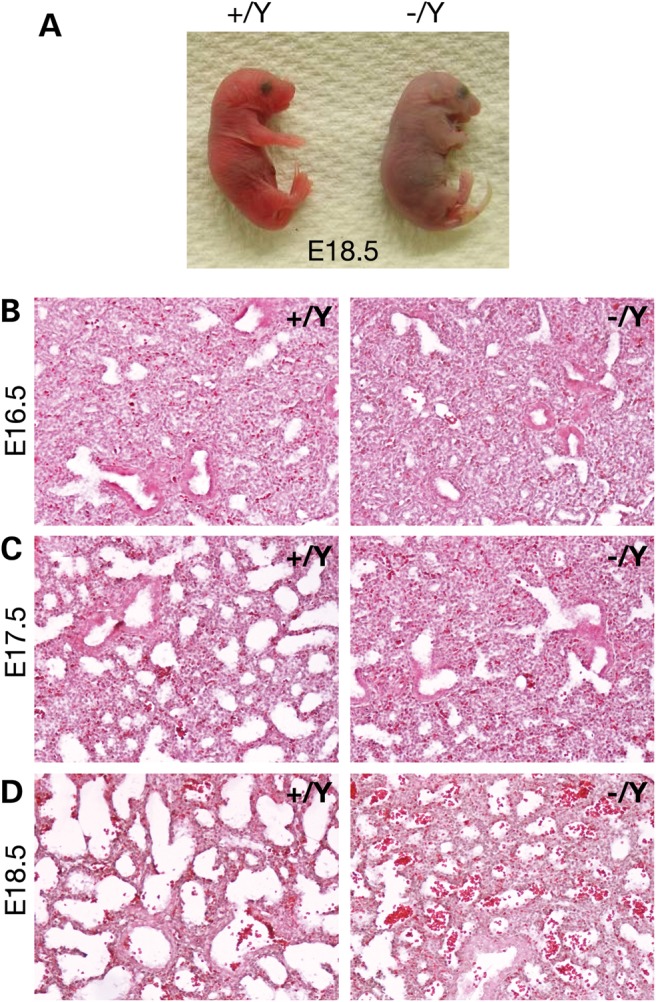
Lung dysplasia in DelA/Y male embryos. (A) Asphyxia in E18.5 DelA/Y male pups. Wild-type pups breathed normally. Pups were delivered at E18.5 by cesarean section. (B–D) Histological analysis of lung from embryos at E16.5 (B), E17.5 (C) and E18.5 (D). Lung sections were stained with hematoxylin and eosin. Genotypes: WT, +/Y; DelA/Y, −/Y.
Alveolarization, the formation of the alveolar gas exchange unit, is one of the most critical steps during lung development, and begins at E16.5 in the mouse (9). Histological analysis of fetal stage (E16.5–E18.5) lungs (Fig. 2B–D) revealed no morphological difference between the lungs of wild-type (XY) and mutant (DelA/Y) mice at E16.5. However, from E17.5 onward, lung development was significantly delayed in mutant compared with wild-type mice. Wild-type lungs exhibited normal progression in the development of sac-like structures and septae. In contrast, mutant lungs developed less septation with thicker interstitial mesenchymal walls. Severe pulmonary hypoplasia may therefore account for the acute respiratory failure at birth in DelA/Y male embryos.
Cleft palate in DelA/Y males and DelA/+ females
The palate separates the oral cavity from the nasal cavity and consists of the primary palate (bony hard plate) and the secondary palate (muscular soft palate) (10). The secondary palate is formed by fusion of two palatal shelves. In the mouse, palatogenesis is initiated at E11.5 and fusion of the palatal shelves normally completed by E15.5. At E18.5, all 20 DelA/Y male pups displayed complete cleft palate (Fig. 3A). Palatal shelves were formed in DelA/Y embryos at E13.5 (Fig. 3B), similar to those present in wild type. At E15.5, the palatal shelves were fused in wild-type embryos but remained separate without any sign of fusion in DelA/Y fetuses (Fig. 3C). Complete cleft palate manifested in 15% (3 of 21) of heterozygous E18.5 DelA/+ females (Table 1). The presence of a cleft palate in a subset of heterozygous females likely reflects phenotypic heterogeneity due to skewed X inactivation.
Figure 3.
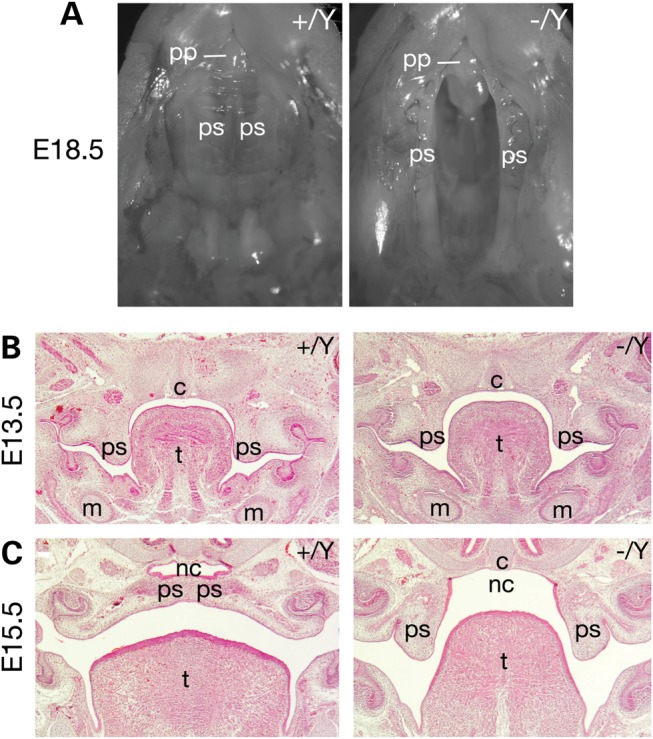
Cleft palate in DelA/Y male embryos. (A) View of the palate from E18.5 wild-type and DelA/Y male embryos. (B and C) Hematoxylin and eosin stained coronal sections through the palate and oral cavity. Genotypes: WT, +/Y; DelA/Y, −/Y. Abbreviations: c, cranial base; m, Meckel's cartilage; nc, nasal cavity; pp, primary palate; ps, palatal shelf; t, tongue.
Table 1.
Comparison of developmental defects among three mouse mutants with deletions in the syntenic region to human Xq22.1
| Deletion allele | Respiratory failure in −/Y neonates | Cleft palate in −/Y males (E18.5) | Cleft palate in +/− females (E18.5) | Epilepsy in +/− females (handling) |
|---|---|---|---|---|
| DelA | 11/11a | 20/20 | 3/21 | 30/35 |
| DelB | None | None | None | None |
| DelC | 8/8 | 37/37 | 1/56 | 9/12 |
aNumber of mice with the defect over the total number of mice examined.
Partial postnatal lethality in juvenile DelA/+ females
All DelA/Y males died immediately after birth, whereas heterozygous DelA/+ females were viable at birth. However, the majority of DelA/+ heterozygous females died postnatally prior to weaning (Fig. 4A). We determined the survival rate of DelA/+ females and wild-type female littermates. None of 31 wild-type females died during a 4-week observation period. In contrast, 45% (22 of 49) of DelA/+ viable newborn pups died within the first 4 days after birth (Fig. 4A), followed by a lower rate of death until Day 24. No substantial lethality was observed in DelA/+ females after 4 weeks. Overall, ∼25% of DelA/+ females survived beyond the age of 4 weeks.
Figure 4.

Postnatal lethality and growth retardation in DelA/+ females. (A) Survival rate of wild-type (n = 31) and DelA/+ females (n = 49). All male littermates were removed immediately after birth. The number of surviving mice was recorded in 4-day intervals. (B) Postnatal growth curve of DelA/+ females that survived to adulthood. Body weight (average ± SD) was measured weekly. All mice were from the breedings described in (C). (C) Body weight of individual DelA/+ females. Each triangle or square represents one mouse. Our breeding strategy for generating offspring was mating of a wild-type male with two DelA/+ females in each cage. All male littermates were removed immediately after birth. Of 14 DelA/+ females monitored starting 2 weeks after birth, four died after 2 weeks, three died after 3 weeks and seven survived to adulthood (corresponding to the +/− animals shown in B). Seven wild-type littermate females were used as controls (+/+) in (B). Genotypes: +/+, wild type; +/−, DelA heterozygous.
Growth delay in juvenile DelA/+ heterozygous females
The body weight of newborn DelA/+ heterozygous females (1.50 ± 0.16 g, n = 18) was similar to that of wild-type female littermates (1.49 ± 0.11 g, n = 23; Student's t-test, P < 0.96), suggesting apparently normal embryonic growth. The suckling behavior of DelA/+ females was normal. However, at 2 weeks after birth, DelA/+ females weighed nearly 50% less than wild-type females, suggesting postnatal developmental delay in heterozygous females (Fig. 4B). After weaning (3 weeks), DelA/+ females grew and eventually caught up with wild-type females in body weight at 6 weeks of age. The DelA/+ pups that died but had no cleft palate tended to have much lower body weight than the DelA/+ pups that survived (Fig. 4C).
Development of epilepsy in DelA/+ heterozygous females
Females heterozygous for the DelA deletion had spontaneous seizures. Thirty of 35 DelA/+ mice examined from 3 weeks to adulthood exhibited handling seizures. In-depth characterization of five DelA/+ adult mice (6 months old) showed that all five mice displayed handling and/or audiogenic seizures; two of them had spontaneous seizures during a prolonged 72-h continuous video electroencephalography (EEG) recording period. The overall seizure frequency was 0.53 ± 0.15 seizures per day (mean ± SE). In contrast, female wild-type littermate controls did not have handling, audiogenic and/or spontaneous seizures. Electrographically, spontaneous seizures in DelA/+ mice were recorded first in hippocampal depth electrodes, and appeared as periods of rhythmic repetitive spike and sharp wave activity (Fig. 5). Semiologically, spontaneous seizures in DelA/+ mice appeared to be of limbic origin, progressing through a typical sequence of behavioral stages, from immobility to facial movements, forelimb and/or tail clonus, repetitive movements, rearing and failing and then tonic–clonic activity (Supplementary Material, Video S1 and Fig. S1 for still images) (11).
Figure 5.

Electrograph of a seizure in DelA/+ females. Representative example of the electrographic changes observed during spontaneous seizures in adult DelA/+ female mice. The animal had a convulsive racine Stage 5 seizure. Electrographic seizure onset is indicated with an arrow. Ctx, cortex; Hip, hippocampus.
Genetic dissection of the critical genomic interval
To narrow down the critical region for the phenotype associated with absence of the DelA genomic region, we generated mutant mice with two subdeletions (DelB, 782 kb and DelC, 350 kb), by insertion of additional loxP sites between the Tmsb15a and Armcx5 genes and subsequent Cre-mediated deletion (Fig. 1A; Supplementary Material, Fig. S2). Among the genes encoded by the DelB region, two (Zmat1 and 3632454L22Rik) are widely expressed, whereas expression of the remaining 15 genes is restricted to testis, brain or placenta (7). DelB/Y null mutant males, DelB/+ heterozygous and DelB/DelB homozygous females were viable and displayed no obvious developmental defects such as lack of respiratory failure, cleft palate or epilepsy (Table 1; Supplementary Material, Fig. S3A). Therefore, none of the 17 genes encoded in the DelB region (Nxf2, Zmat1, 3632454L22Rik, Gm15023, Tceal6, Pramel, Pramel3, Gm6215, Gm5128, Gm6221, Gm7903, Gm7905, Av320801, Nxf7, Prame, Tcp11l3 and Tmsb15a) is essential for development (Fig. 1).
All genes of the DelC region except Nxf3 (Fig. 1) are expressed in a wide range of tissues including lung and brain (7). Western blot analysis confirmed that GPRASP1 and GPRASP2 proteins were absent in the brains of DelC/Y null mutant mice and less abundant in brain tissue from DelC/+ heterozygous females (Fig. 1C). All 15 DelC/Y null mutant males examined exhibited respiratory failure, neonatal lethality and cleft palate (Table 1; Supplementary Material, Fig. S3A). In females, heterozygosity for DelC caused cleft palate, partial postnatal lethality, postnatal developmental delay (Supplementary Material, Fig. S3B) and epilepsy (handling seizure). The phenotypic similarity of DelC and DelA mutant mice strongly implies a role for genes encoded by the DelC region in developmental defects such as respiratory failure, cleft palate and epilepsy (Table 1).
Four genes in the mouse DelC region (Arxes2, Arxes1, Bex2 and Nxf3) have no corresponding orthologs in the human Xq22.1 deletion interval and are therefore likely not relevant for a phenotype observed in both human and mouse (Fig. 1). The remaining four genes (Armcx5, Gprasp1, Gprasp2 and Bhlhb9) are orthologous genes between the mouse DelC region and the human Xq22.1 deletion interval (Fig. 1A); loss of these four genes appears to be causative for the developmental defects of DelA mice. All four proteins (ARMCX5, GPRASP1, GPRASP2 and BHLHB9) contain an Arm2 (armadillo-like repeats) domain and belong to the GPRASP (GPCR-associated sorting protein) family, members of which interact with G-protein-coupled receptors (GPCRs) (12).
DISCUSSION
We have generated a mouse model for the newly described human Xq22.1 deletion syndrome. In female patients, a familial 1.1 Mb deletion in Xq22.1 is associated with epilepsy, mental retardation and behavioral disorders (3). DelA mutant mice carrying a deletion of the syntenic region of the mouse X chromosome exhibit all major developmental defects of human patients, including cleft palate and epilepsy. These findings strongly imply the 1.1 Mb deletion in Xq22.1 as the underlying genetic cause of developmental defects in humans. In addition, DelA/Y mutant males die perinatally from respiratory failure due to pulmonary hypoplasia, suggesting that boys with the Xq22.1 deletion may be similarly affected. The initial description of the Xq22.1 deletion in a family notes that a son of the heterozygous mother died 15 days after birth due to respiratory failure (3).
Interestingly, in both mice and humans, females heterozygous for the deletion exhibit variable phenotypes. Some heterozygous females have cleft palate, whereas in other females palatogenesis is normal. A majority of heterozygous female mice die postnatally, but a quarter of them survive to adulthood. This variation in observed phenotypes is likely due to X-chromosome inactivation (XCI), where one of the two X chromosomes is inactivated in females during early development and silencing is maintained into adulthood (13). Due to random X inactivation, females are mosaics in terms of the parental origin of the active X chromosome, resulting in two distinct populations of cells in females (14). Some X-linked mutations may adversely affect cell proliferation. X-inactivation skewing has dramatic effects on the clinical phenotypes of a number of X-linked disorders such as Rett syndrome (14). The variability of phenotypes in individual DelA/+ and DelC/+ heterozygous females is likely attributed to skewed X inactivation. DelA/+ heterozygous females that preferentially inactivate the wild-type X chromosome are more likely to develop cleft palate or die postnatally, whereas females with a skewed inactivation of the DelA mutant X chromosome are less likely to exhibit such a phenotype. The developmental delay in DelA heterozygous females may also be attributed to skewed XCI. The body weight of DelA/+ females is significantly lower during the first 5 weeks but is comparable with wild type at the age of 6 weeks. This phenomenon is not likely due to competition from wild-type littermates for maternal nutrients, because DelC/+ females exhibit a similar postnatal growth pattern in the absence of wild-type pups (Supplementary Material, Fig. S3B). One possible explanation is that cells with an inactivated mutant X chromosome (functionally equivalent to wild-type cells) have a growth advantage over cells with an inactivated wild-type X chromosome (functionally null for the DelA deletion) and thus can compensate over time. Consistent with such a hypothesis, phenotypic severity in reported human cases correlated with X-inactivation patterns: in the daughter, who exhibited more severe defects than the mother, both X chromosomes were equally (50:50) inactivated in the peripheral blood, whereas the mother had highly skewed preferential inactivation of the X chromosome with the Xq22.1 deletion (90:10) (3).
Our genetic studies have identified the 350 kb DelC region as the critical interval for the phenotypes of DelA mice. Four orthologous genes (Armcx5, Gprasp1, Gprasp2 and Bhlhb9) are located in the mouse DelC region and the 1.1 Mb human Xq22.1 deletion region and thus remain candidate genes. Gprasp1 knockout mice lack cleft palate, epilepsy or respiratory failure, showing that deletion of Grasp1 alone is not responsible for the developmental defects in DelC mutants (15–17). The question remains as to which gene is responsible for these phenotypes. There are three possibilities. First, the deletion of a single gene (Armcx5, or Gprasp2, or Bhlhb9) could be responsible for all defects. Second, deletion of any combination of these genes (Armcx5, Gprasp1, Gprasp2 and Bhlhb9) could be responsible for the multiple defects; and finally, deletion of a single gene (Armcx5, or Gprasp2, or Bhlhb9) or any combination of these genes (Armcx5, Gprasp1, Gprasp2 and Bhlhb9) could be responsible for a subset of phenotypes, respectively. Further genetic (knockout and transgenic rescue) studies in mice are needed to distinguish between these possibilities and identify the gene or genes responsible for respiratory failure, cleft palate and epilepsy in the human Xq22.1 deletion syndrome. These studies will set the stage for functionally characterizing the role of these novel factors in the regulation of lung development, palatogenesis and brain development.
MATERIALS AND METHODS
Generation of DelA mice
DelA mice were generated as previously described (7). Nxf2fl and Nxf3fl mice were of mixed genetic background (129xC57BL/6) (18,19). Both Nxf2 and Nxf3 are X linked. Intercrossing of Nxf2fl and Nxf3fl mice eventually led to the production of Nxf2fl Nxf3fl/Y mice. Nxf2fl Nxf3fl/+ female mice were crossed with Actb-Cre males to generate the Del(XNxf2–Nxf3)1Jw allele (MGI ID: 5297602), referred to as DelA (Fig. 1A). The Cre recombinase is ubiquitously expressed in Actb-Cre mice (8).
Generation of DelB and DelC mice
To generate the DelB and DelC mutants, we generated a targeting construct in which loxP sites were inserted between Tmsb15a and Armcx5 (Supplementary Material, Fig. S2A), flanked by two homologous arms amplified from BAC DNA (BAC clone RP23-250F8) by PCR using high-fidelity HF2 Taq DNA polymerase (Clontech). The floxed PGK-neo selection cassette replaced a 5.6 kb intergenic region between Tmsb15a and Armcx5; a HyTK negative selection cassette was subcloned adjacent to the left arm. The targeting vector is referred to as pUP108. The approved symbol (MGI) for the targeted intergenic insertion site allele is Iis8tm1Jw.
To generate the DelB mutant allele, Nxf2fl/Y ES cells, previously generated (18), were electroporated with the linearized pUP108 vector. ES cells were cultured in the presence of 350 μg/ml G418 and 2 μm ganciclovir. Double-resistant ES cell clones (n = 192) were screened for homologous recombination events by long-distance PCR with primers P1 and P2, producing a 2.6 kb product from the left arm, and primers P3 and P4 producing a 2.6 kb product from the right arm (Supplementary Material, Fig. S2A). All PCR primer sequences are provided in Supplementary Material, Table S1. Twenty-four homologously targeted ES cell clones were identified, resulting in a targeting frequency of 13%. The targeted conditional allele with four loxP sites is referred to as Del(XNxf2-Tmsb15a)Jw1 (abbreviated DelB; Fig. 1A; Supplementary Material, Fig. S2B). One ES cell clone was injected into blastocysts and germline transmission of the conditional allele from male chimeras confirmed by PCR genotyping of the floxed PGKNeo allele (wild type, 571 bp, primers P5 and P6; and mutant, 346 bp, primers P3 and P7; Supplementary Material, Fig. S2A). To delete the DelB region, mice carrying the floxed allele were crossed with Actb-Cre mice, and the resulting allele confirmed by PCR genotyping with primers P7 and P8 (427 bp; Supplementary Material, Fig. S2B).
To generate DelC mice, Nxf3fl/Y ES cells (19) were electroporated with the linearized pUP108 vector, producing a targeted conditional allele with four loxP sites referred to as Del(XArmcx5–Nxf3)Jw1 (abbreviated DelC; Fig. 1A; Supplementary Material, Fig. S2C). Injection of one ES cell clone into blastocysts resulted in germline transmission of the targeted allele from male chimeras. Mice carrying the floxed DelC allele were crossed with Actb-Cre mice, and the resulting DelC deletion allele was confirmed by PCR genotyping (558 bp) with primers P9 and P10 (Supplementary Material, Fig. S2C). All mice were maintained and used in accordance with standards approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Histological analysis
Lungs from embryos between E16.5 and E18.5 and heads from embryos at E13.5 and E15.5 were fixed in 4% paraformaldehyde in phosphate buffered saline overnight at 4°C, dehydrated in ethanol and embedded in paraffin. Sections of 8 μm in thickness were cut and stained with hematoxylin and eosin.
Western blot analysis
Brains dissected from mouse embryos of E16.5 and E18.5 were frozen on dry ice and stored at −80°C. After confirmation of the genotype, tissues were homogenized in SDS–PAGE sample buffer and heated at 95°C for 10 min. Protein lysates (20 μg) were separated on 10% SDS–PAGE gel and electro-blotted on nitrocellulose membranes using iBlot (Invitrogen). Primary antibodies used for western blotting were: GPRASP1/GASP-1 (1:2500; a gift from F. Simonin) (15) and ACTB (1:10 000, cat. no. A5441, clone AC-15, Sigma).
EEG recordings
All EEG recordings were performed on 6-month-old DelA/+ heterozygous females using a Stellate-Harmonie (16-bit, 24 channel digital EEG machine Stellate Inc., Montreal, Canada). Two surface frontal cortical electrodes, bilateral hippocampal depth electrodes and ground and reference electrodes were placed. Animals were recorded for 72 h. EEG tracings were visually analyzed for the presence of electrographic seizures and video was reviewed for the presence of behavioral seizure activity. Electrographic seizures were defined as the presence of rhythmic repetitive spike and sharp activity lasting 5 s or more.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by NIH grants R01GM089893 and R01GM076327 to P.J.W. and NINDS K12 NS049453 to E.M.G.
Supplementary Material
ACKNOWLEDGEMENTS
We thank F. Simonin for the GPRASP1 antibody, T. Bale and L. Kubin for discussion on the studies. We thank M. Anguera and S. Kasowitz for comments on the manuscript, S. Eckardt for help with manuscript preparation.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Saitta S.C., Harris S.E., Gaeth A.P., Driscoll D.A., McDonald-McGinn D.M., Maisenbacher M.K., Yersak J.M., Chakraborty P.K., Hacker A.M., Zackai E.H., et al. Aberrant interchromosomal exchanges are the predominant cause of the 22q11.2 deletion. Hum. Mol. Genet. 2004;13:417–428. doi: 10.1093/hmg/ddh041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Devriendt K., Fryns J.P., Mortier G., van Thienen M.N., Keymolen K. The annual incidence of DiGeorge/velocardiofacial syndrome. J. Med. Genet. 1998;35:789–790. doi: 10.1136/jmg.35.9.789-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grillo L., Reitano S., Belfiore G., Spalletta A., Amata S., Bottitta M., Barone C., Falco M., Fichera M., Romano C. Familial 1.1 mb deletion in chromosome Xq22.1 associated with mental retardation and behavioural disorders in female patients. Eur. J. Med. Genet. 2010;53:113–116. doi: 10.1016/j.ejmg.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Jun L., Frints S., Duhamel H., Herold A., Abad-Rodrigues J., Dotti C., Izaurralde E., Marynen P., Froyen G. NXF5, a novel member of the nuclear RNA export factor family, is lost in a male patient with a syndromic form of mental retardation. Curr. Biol. 2001;11:1381–1391. doi: 10.1016/s0960-9822(01)00419-5. [DOI] [PubMed] [Google Scholar]
- 5.Frints S.G., Jun L., Fryns J.P., Devriendt K., Teulingkx R., Van den Berghe L., De Vos B., Borghgraef M., Chelly J., Des Portes V., et al. Inv(X)(p21.1;q22.1) in a man with mental retardation, short stature, general muscle wasting, and facial dysmorphism: Clinical study and mutation analysis of the NXF5 gene. Am. J. Med. Genet. A. 2003;119A:367–374. doi: 10.1002/ajmg.a.20195. [DOI] [PubMed] [Google Scholar]
- 6.Vanmarsenille L., Verbeeck J., Belet S., Roebroek A.J., Van de Putte T., Nevelsteen J., Callaerts-Vegh Z., D'Hooge R., Marynen P., Froyen G. Generation and characterization of an Nxf7 knockout mouse to study NXF5 deficiency in a patient with intellectual disability. PLoS ONE. 2013;8:e64144. doi: 10.1371/journal.pone.0064144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou J., McCarrey J.R., Wang P.J. A 1.1-mb segmental deletion on the X chromosome causes meiotic failure in male mice. Biol. Reprod. 2013;88:159. doi: 10.1095/biolreprod.112.106963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewandoski M., Meyers E.N., Martin G.R. Analysis of Fgf8 gene function in vertebrate development. Cold Spring Harb. Symp. Quant. Biol. 1997;62:159–168. [PubMed] [Google Scholar]
- 9.Morrisey E.E., Hogan B.L. Preparing for the first breath: Genetic and cellular mechanisms in lung development. Dev. Cell. 2010;18:8–23. doi: 10.1016/j.devcel.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bush J.O., Jiang R. Palatogenesis: Morphogenetic and molecular mechanisms of secondary palate development. Development. 2012;139:231–243. doi: 10.1242/dev.067082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Racine R.J. Modification of seizure activity by electrical stimulation. II. motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 12.Heese K. G proteins, p60TRP, and neurodegenerative diseases. Mol. Neurobiol. 2013;47:1103–1111. doi: 10.1007/s12035-013-8410-1. [DOI] [PubMed] [Google Scholar]
- 13.Lee J.T., Bartolomei M.S. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell. 2013;152:1308–1323. doi: 10.1016/j.cell.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 14.Migeon B.R. The role of X inactivation and cellular mosaicism in women's health and sex-specific diseases. JAMA. 2006;295:1428–1433. doi: 10.1001/jama.295.12.1428. [DOI] [PubMed] [Google Scholar]
- 15.Boeuf J., Trigo J.M., Moreau P.H., Lecourtier L., Vogel E., Cassel J.C., Mathis C., Klosen P., Maldonado R., Simonin F. Attenuated behavioural responses to acute and chronic cocaine in GASP-1-deficient mice. Eur. J. Neurosci. 2009;30:860–868. doi: 10.1111/j.1460-9568.2009.06865.x. [DOI] [PubMed] [Google Scholar]
- 16.Thompson D., Martini L., Whistler J.L. Altered ratio of D1 and D2 dopamine receptors in mouse striatum is associated with behavioral sensitization to cocaine. PLoS ONE. 2010;5:e11038. doi: 10.1371/journal.pone.0011038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathis C., Bott J.B., Candusso M.P., Simonin F., Cassel J.C. Impaired striatum-dependent behavior in GASP-1-knock-out mice. Genes Brain Behav. 2011;10:299–308. doi: 10.1111/j.1601-183X.2010.00666.x. [DOI] [PubMed] [Google Scholar]
- 18.Pan J., Eckardt S., Leu N.A., Buffone M.G., Zhou J., Gerton G.L., McLaughlin K.J., Wang P.J. Inactivation of Nxf2 causes defects in male meiosis and age-dependent depletion of spermatogonia. Dev. Biol. 2009;330:167–174. doi: 10.1016/j.ydbio.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou J., Pan J., Eckardt S., Leu N.A., McLaughlin K.J., Wang P.J. Nxf3 is expressed in sertoli cells, but is dispensable for spermatogenesis. Mol. Reprod. Dev. 2011;78:241–249. doi: 10.1002/mrd.21291. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.