

Abstract
Measurements of specific interactions between proteins are challenging. In redox systems, interactions involve surfaces near the attachment sites of cofactors engaged in interprotein electron transfer (ET). Here we analyzed binding of cytochrome c2 to cytochrome bc1 by measuring paramagnetic relaxation enhancement (PRE) of spin label (SL) attached to cytochrome c2. PRE was exclusively induced by the iron atom of heme c1 of cytochrome bc1, which guaranteed that only the configurations with SL to heme c1 distances up to ∼30 Å were detected. Changes in PRE were used to qualitatively and quantitatively characterize the binding. Our data suggest that at low ionic strength and under an excess of cytochrome c2 over cytochrome bc1, several cytochrome c2 molecules gather near the binding domain forming a “cloud” of molecules. When the cytochrome bc1 concentration increases, the cloud disperses to populate additional available binding domains. An increase in ionic strength weakens the attractive forces and the average distance between cytochrome c2 and cytochrome bc1 increases. The spatial arrangement of the protein complex at various ionic strengths is different. Above 150 mM NaCl the lifetime of the complexes becomes so short that they are undetectable. All together the results indicate that cytochrome c2 molecules, over the range of salt concentration encompassing physiological ionic strength, do not form stable, long-lived complexes but rather constantly collide with the surface of cytochrome bc1 and ET takes place coincidentally with one of these collisions.
Introduction
Cytochrome bc1 is a central enzyme of many bioenergetic pathways. It catalyzes electron transfer (ET) from hydroquinone to the water-soluble cytochrome c and uses the energy released during this process to transport protons across the membrane. One of the requirements for efficient catalysis is the presence of interprotein ET between the hemes of cytochrome c1 subunit and cytochrome c within a millisecond time scale of enzymatic turnover. This should be viewed in two major terms: structural, describing a spatial orientation of the interacting proteins, and dynamic, describing association and dissociation processes.
It has been generally accepted that protein–protein interactions involve long-range electrostatic interactions that facilitate formation of an encounter complex whereas short-range interactions lead to the stabilization of a tight complex in a proper spatial configuration.1−4 Electrostatic forces are usually considered to be a dominant factor that contributes to the binding of cytochrome c to cytochrome bc1. This is inferred from many experiments showing a significant salt dependence of ET between heme c and c1.5−8 In the yeast system, an exceptional importance of hydrophobic interactions between the surfaces of the cytochrome c1 subunit and cytochrome c was proposed on the basis of X-ray structure analysis of cytochrome bc1 cocrystallized with its redox partner.9 However, further analysis of these structures with molecular dynamics simulations suggested that salt bridges and H-bonds may be of greater importance than expected from the crystallographic data alone.10
A convenient model of the mitochondrial system is a bacterial counterpart consisting of cytochrome c2 and cytochrome bc1. Extensive chemical modifications and mutagenetic studies revealed the presence of crucial amino acid residues responsible for salt bridge formation that stabilize the complex. The existence of a complex between cytochrome c and cytochrome bc1 was shown by measurements of ET between heme c2 and heme c1(11,12) as well as by techniques that are independent of ET, such as plasmon waveguide resonance spectroscopy13 or electron paramagnetic resonance (EPR).14,15 Under low ionic strength conditions, the presence of a relatively long-lived complex was inferred from the intramolecular ET between the proteins or by changes in paramagnetic properties of spin-labeled cytochrome c2 after binding to cytochrome bc1. Increasing salt concentration or mutations of some charged residues resulted in the progressive disappearance of the complexes observed by EPR14 or in a change of the ET kinetics from intermolecular to bimolecular.11 These observations were interpreted in terms of a collisional model of ET between cytochrome c2 and cytochrome bc1.14
It can be rationally envisaged that isolated cytochrome c1 makes a 1:1 complex with cytochrome c. However, cytochrome bc1 is a homodimer containing two subunits of cytochrome c1 thus prediction of how many cytochrome c molecules can be bound simultaneously to the cytochrome bc1 dimer is not straightforward. This question is relevant to catalysis given that cytochrome bc1 has been shown to exchange electrons between monomers on a catalytic time scale.16,17 Intriguingly, the X-ray crystal structures of yeast cytochrome bc1 revealed the presence of only one cytochrome c bound to the cytochrome bc1 dimer.9 On the other hand, experiments on the bacterial system with the use of plasmon waveguide resonance suggested that oxidized cytochrome c2 binds to two separate sites on oxidized cytochrome bc1 with different affinities. The observed biphasic binding was interpreted as an influence of the mobile iron–sulfur head domain on the association and dissociation of cytochrome c2.13
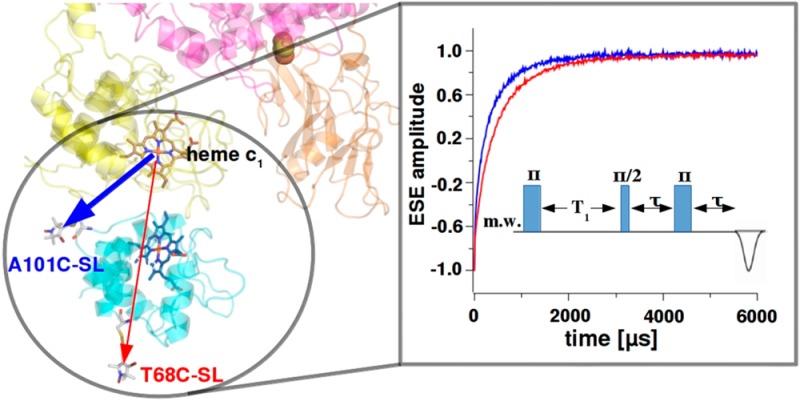
In light of all these studies, the question of how the structure and dynamics of the complex between cytochrome bc1 and cytochrome c is related to the interprotein ET remains open. In this work we addressed this issue by analyzing the binding of Rhodobacter (Rba.) capsulatus cytochrome c2 to cytochrome bc1 using an inversion recovery (IR) EPR technique intended to detect relatively short-range of distances between interacting proteins in a highly specific manner. The approach was based on measurements of magnetic interactions between spin label (SL) on cytochrome c2 and a fast-relaxing iron ion of heme c1 in cytochrome c1. These interactions were detected as paramagnetic relaxation enhancement (PRE),18 a phenomenon that has been exploited to study protein–protein interactions by both EPR and NMR spectroscopy, in systems such as cytochrome c–cytochrome c oxidase,19 cytochrome c–cytochrome c peroxidase,20,21 plastocyanin–cytochrome f,22 and Rieske protein–cytochrome b.23 Our experiments further support the collisional mechanism in which cytochrome bc1 does not form long-lived complexes with cytochrome c2 and ET between the proteins is a product of several collisions between proteins. In addition, they provide insight into how cytochrome c2 molecules are arranged around cytochrome bc1 under different ionic strength conditions at different molar ratios of the proteins.
Experimental Methods
Biochemical Procedures
A101C and T68C mutants of cytochrome c2 were isolated from Rba. capsulatus strains and spin-labeled with BrMTSL24 as described in ref (14). Cytochrome bc1 mutants c1:M183K and c1:M183K/FeS:S158A were also isolated from Rba. capsulatus strains according to the procedure described in ref (25). Before sample preparation, cytochrome c2 and cytochrome bc1 solutions were dialyzed against 5 mM bicine buffer, pH 8.0, containing 25% v/v of glycerol and 100 mg/L n-dodecyl-β-d-maltopyranoside (DDM). Samples with different ionic strengths were prepared by addition of appropriate amounts of NaCl. Concentrations of cytochrome c2 and cytochrome bc1 were determined spectrophotometrically as described in ref (14). The cytochrome bc1 concentration refers to the concentration of monomers throughout the whole text. Uncertainty in determined protein concentration was around 10–15%. Oxidized cytochrome c2 was obtained by addition of potassium ferricyanide to a final concentration of 300 μM, which allowed full oxidation of cytochrome c2 but did not influence the relaxation of SL directly. Before measurements all samples were frozen by immersing the EPR tubes into liquid nitrogen.
EPR Measurements
All EPR measurements were performed on a Bruker Elexsys E580 spectrometer equipped with an Oxford Instrument temperature control system. Pulse EPR experiments were conducted at the Q-band using a Bruker ER5107D2 resonator inserted into a CF935 cryostat. Spin–lattice relaxation times were measured using an IR sequence: π–T–π/2−τ–π with constant τ = 200 ns. The initial T = 300 ns delay was incremented to cover the whole recovery curve after the inverting π pulse. A four-step phase cycling was applied and microwave power was adjusted to achieve a 40 ns π pulse. The effect of spectral diffusion on the measured relaxation times was found to be negligible, as inferred from measurements where the first π pulse of the IR sequence was replaced with a picket fence of ten π pulses26 (Figure S1 in Supporting Information). Each IR curve was recorded at the spectral position where the maximum amplitude of the echo is observed (g = 2.0082 for SL at Q-band). Application of the 40 ns π pulse at this spectral position minimized the effect of orientation selection. The measured IR curves were found to be highly reproducible and estimated uncertainties of fitted parameters were less than 1%. X-band continuous wave EPR spectra of FeS were measured at 20 K with the use of an SHQ4122 resonator equipped with an ESR900 cryostat.
Results
Spin–Lattice PRE in Spin-Labeled Cytochrome c2
As a starting point to measure magnetic interactions that influence the spin–lattice relaxation rates of SL attached to cytochrome c2, we examined the IR curves over a temperature range of 20–120 K for the two single mutants specifically labeled at positions A101C or T68C (A101C-SL and T68C-SL, respectively; Figure 1A). Without addition of any oxidants, the cytochrome c2 iron ion is reduced (Fe2+), which is diamagnetic (S = 0). Therefore, it has no influence on longitudinal relaxation of the attached SL (Figure 1B open squares and open circles). Thus, the relaxation of SL was dominated by the two-phonon Raman process and the temperature profiles followed that of the nitroxides in frozen water:glycerol mixtures and the relaxation rates were similar to those previously reported.27,28
Figure 1.
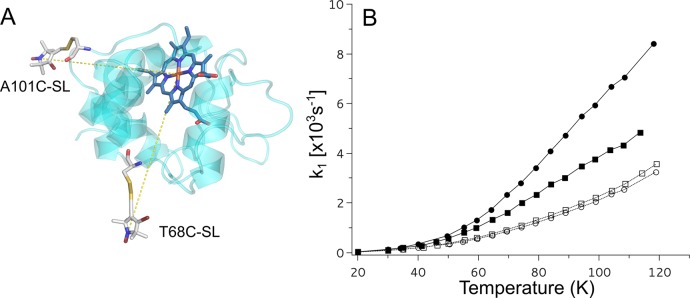
Positions of SL in cytochrome c2 and spin–lattice relaxation rates of SL attached to cytochrome c2. (A) Model of the crystal structure of Rba. capsulatus cytochrome c2 with SL attached at positions A101C or T68C. (B) Temperature dependence of spin–lattice relaxation rates of A101C-SL (circles) and T68C-SL (squares) for 25 μM cytochrome c2 (0 mM NaCl) in reduced or oxidized state (open and solid symbols, respectively).
Ferricyanide oxidizes the ion in cytochrome c2 to Fe3+, which is a low spin (S = 1/2) paramagnetic center and a source of rapidly fluctuating local magnetic field that impacts relaxation of SL attached to the protein. This process manifested itself in an enhancement of the spin–lattice relaxation observed for both of the attached labels in available temperature range for IR experiments. As shown in Figure 1B, the increase in the observed relaxation rates depends on the labeling position (stronger for A101C-SL). The difference in PRE of A101C-SL and T68C-SL is predominantly related to a difference in average distance between each of the SLs and heme iron: 16.7 and 19.3 Å for A101C-SL and T68C-SL, respectively (Figure 1A), as calculated using a rotamer library approach (RLA) method in the MMM software package29,30 (Figure S4, Supporting Information). As the spin–lattice PRE decreases with the sixth power of the interspin distance, the determined difference in distance corresponds to about 2.4 times larger spin–lattice PRE in A101C-SL compared to T68C-SL. This is in agreement with measured values (for example, at 95 K, values of spin–lattice PRE for A101C-SL and T68C-SL are 3.5 × 103 and 1.5 × 103 s–1, respectively).
Spin–Lattice PRE in Cytochrome c2 in the Presence of Cytochrome bc1
The next series of experiments were based on the assumption that upon binding of cytochrome c2 to cytochrome bc1, cytochrome c2 approaches cytochrome c1 close enough that the paramagnetic iron of heme c1 can influence the longitudinal relaxation rate of SL. Initially, we examined relaxation rates of A101C-SL and T68C-SL in cytochrome c2 mixed with an excess of cytochrome bc1 to ensure that almost all cytochrome c2 molecules are bound in the complex. In these measurements we used the M183K mutant of cytochrome bc1 to fully control the redox states of heme c1. M183K refers to a point mutation in the cytochrome c1 subunit, that changes the heme c1 ligation pattern resulting in a large decrease of redox potential of this heme31 (more than 200 mV vs wild type). Consequently, M183K was used as a form in which all molecules of cytochrome bc1 contained oxidized (Fe3+), therefore paramagnetic (S = 1/2), heme c1 without the necessity to use any external oxidants. Heme c1 remained oxidized after mixing cytochrome bc1 with reduced cytochrome c2. We note that this is not possible with native cytochrome bc1, where the obligatory presence of an oxidant to keep heme c1 fully oxidized results in unwanted oxidation of heme c2.
Parts A and B of Figure 2 compare the temperature dependence of the spin–lattice relaxation rates of A101C-SL and T68C-SL in mixtures containing M183K and respective cytochromes c2. The profiles obtained for high ionic strength conditions were almost the same, irrespective of labeled position, and overlapped with respective profiles obtained for A101C-SL and T68C-SL in 0 mM NaCl without cytochrome bc1 (Figure 2A,B). This is consistent with the lack of detectable interaction between cytochrome c2 and cytochrome bc1 at high ionic strength. In addition, these overlaps infer that NaCl has no effect on spin–lattice relaxation rate of SL attached to cytochrome c2 (and SL alone), as has been confirmed by separate control experiments (not shown). The profile obtained for A101C-SL at low ionic strength (Figure 2A) showed a significant PRE. In case of T68C-SL PRE was much weaker (Figure 2B). Nevertheless, in both cases PRE disappears at high ionic strength, which is expected if one considers the electrostatic nature of binding of cytochrome c2 to cytochrome bc1.
Figure 2.

Temperature dependence of the spin–lattice relaxation of spin-labeled cytochrome c2 in the presence of cytochrome bc1. (A) Relaxation rate (k1) for mixtures of 25 μM A101C-SL and 100 μM cytochrome bc1 M183K in buffer with 0 and 200 mM NaCl (solid circles and open squares, respectively). (B) Similar experiment as in (A) but with the use of 25 μM T68C-SL (symbols are as in (A)). Open circles in (A) and (B) show the relaxation measured in the presence of 100 μM cytochrome bc1 M183K/S158A. Solid triangles in (A) and (B) are the relaxation rates measured in the absence of cytochrome bc1 (replotted from Figure 1B).
The site-specificity of the observed PRE is generally consistent with the structural model of binding. Figure 3 shows spatial orientation of cytochrome c2 interacting with Rba. capsulatus cytochrome bc1 derived from the model created on the basis of the crystal structure of iso-1 cytochrome c bound to yeast cytochrome bc1(9) as described in ref (14). In this orientation A101C-SL is at shorter distance from the iron of heme c1 than T68C-SL. Because the spin–lattice PRE is effective over relatively short distances (in this case up to ∼30 Å), only A101C-SL lies within the range of the strong magnetic dipolar field modulated by relaxation of heme c1 iron. This is reflected in the faster relaxation of A101C-SL than T68C-SL. This also implies that the spin–lattice PRE of A101C-SL specifically monitors the close contact of cytochrome c2 and cytochrome c1 that occurs just at the binding interface so that A101C-SL can sense the presence of heme c1.
Figure 3.
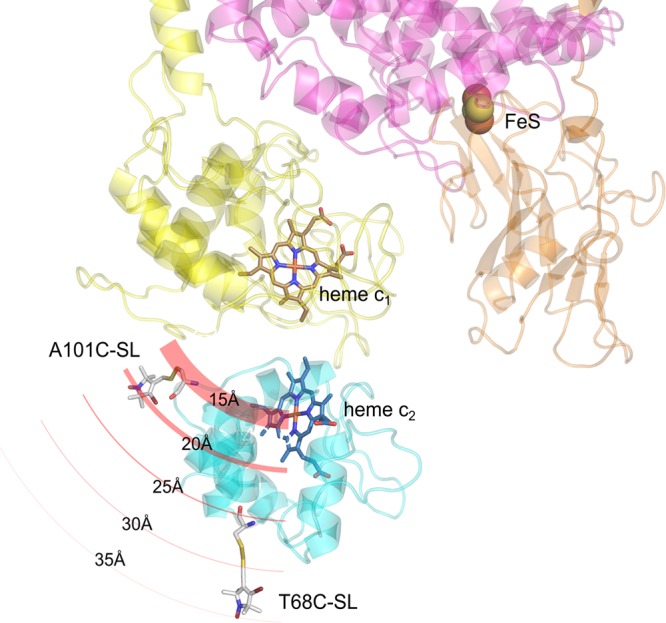
Structural model of the complex of cytochrome c2 with cytochrome bc1. The cytochrome c2 molecule (turquoise) with attached SL at positions A101C or T68C binds to the binding domain of cytochrome c1 subunit (yellow). The FeS subunit and part of cytochrome b are shown in orange and pink, respectively. Circular red lines depict PRE on SL, induced by the iron atom of heme c1. The thickness of the lines represents the effective strength of PRE, which decreases radially as the distance from the heme c1 iron increases. A101C-SL and T68C-SL sense the different strength of PRE. For simplicity, only one monomer of cytochrome bc1 is shown.
As shown in Figure 3, besides oxidized heme c1, reduced Rieske cluster (FeS) is another paramagnetic metal center of cytochrome bc1 that potentially could influence relaxation of A101C-SL or T68C-SL. To assess its possible contribution to PRE, we used a double mutant M183K/S158A. M183K/S158A, in addition to M183K in cytochrome c1, contained a point mutation S158A in the FeS subunit in the vicinity of the coordinating shell of FeS. The S158A mutation (similarly to the corresponding S154A in Rba. sphaeroides(32)) lowers the midpoint potential of FeS by 130 mV (unpublished data); thus M183K/S158A under our experimental conditions maintains paramagnetic heme c1 and diamagnetic FeS (both fully oxidized). This was verified by X-band CW EPR (not shown). The overlay of open and solid circles in Figure 2A,B showed that the temperature profiles of the relaxation rates of A101C-SL or T68C-SL did not depend on the redox state of the FeS (were identical for samples containing either M183K or M183K/S158A). This indicates that the reduced FeS does not contribute to PRE within the observed temperature region. The lack of detectable effect of FeS on SL spin–lattice relaxation rate can be explained, first, by the larger separation between the centers and, second, by the fact that the spin–lattice relaxation rate of the FeS (manuscript in preparation) is an order of magnitude slower than the low spin heme iron.33,34 In this case the effect of the heme iron dominates any possible contribution from the FeS to observed PRE. Remaining experiments were performed using M183K/S158A.
PRE Dependence on Ionic Strength
Figure 4 shows relaxation rates for samples containing M183K/S158A and spin labeled cytochrome c2 (A101C-SL or T68C-SL) mixed at a 6:1 molar ratio in buffers with varying concentrations of NaCl. All measurements were performed at 95 K, at which PRE effect was sufficiently large (Figure 2A) and the signal-to-noise ratio was optimal.
Figure 4.

Ionic strength dependence of the spin–lattice relaxation rate (k1) of spin-labeled cytochrome c2 in the presence of cytochrome bc1. Cytochrome c2 A101C-SL (solid circles) and T68C-SL (solid squares) were mixed with cytochrome bc1 mutant M183K/S158A in a 1:6 molar ratio and measured at 95 K. The total concentrations of cytochrome c2 and cytochrome bc1 were 25 and 150 μM, respectively. The ionic strength was varied by changing the NaCl concentration.
For A101C-SL (Figure 4, circles) the salt dependence was steep, in particular within the range up to 50 mM NaCl. At around 30 mM NaCl the strength of PRE fell to 50% of its maximum value. A further increase in NaCl concentration caused the relaxation rate to decrease less profoundly, with values approaching the 1.9 × 103 s–1 limit at NaCl concentrations above 150 mM. This limit corresponds to the rate in the absence of PRE; the measured relaxation rate of A101C-SL becomes equal to the rate obtained in buffer without cytochrome bc1.
For T68C-SL (Figure 4, squares) the salt dependence was much less pronounced than for A101C-SL. In the whole tested range of ionic strength, the relaxation rate of T68C-SL mixed with cytochrome bc1 did not deviate much from the value obtained for T68C-SL in buffer without cytochrome bc1.
In general, the increase in the ionic strength causes weakening of the electrostatic forces that stabilize the complex. It can be expected that, as a result of this weakening, the average distance between proteins increases and/or the spatial orientation of the two proteins in the complex changes. Both effects will cause an increase in the average distance between heme c1 and SL, thereby a decrease in the observed PRE. Such a decrease is clearly visible in the case of A101C-SL, where even small changes in the distance result in significant changes in PRE. The changes for T68C-SL are much less visible because the maximal PRE, at the lowest ionic strength, is much weaker (consistent with the larger interspin distance in the modeled complex, Figure 3). We note that a larger sensitivity of PRE to ionic strength seen in A101C-SL is consistent with the observation that the temperature dependence profile of PRE in A101C-SL also shows larger ionic strength sensitivity when compared to that in T68C-SL (Figure 2).
PRE Dependence on the Cytochrome c2/bc1 Ratio
In general, as stated above, the change in the ionic strength may influence both the average distance between proteins (ratio between bound and unbound cytochrome c2) and their spatial orientation. To estimate the contribution of these two factors to the binding process, we performed measurements of the spin–lattice relaxation times of A101C-SL upon titration with increasing amounts of cytochrome bc1. We analyzed the data in terms of the relaxation times as they are linearly correlated with the changes in the fractions of bound and unbound A101C-SL (Supporting Information). The titration experiments were conducted under three different ionic strength conditions in attempt to determine both the stoichiometry and the affinity of binding.
When 25 μM A101C-SL was titrated with increasing amounts of M183K/S158A in buffer containing 0 mM NaCl, we observed a progressive decrease in the relaxation time of the SL for concentrations of M183K/S158A up to approximately 30 μM (Figure 5, circles). The relaxation time approached a limit of 370 μs and did not change significantly upon further addition of M183K/S158A. The titrations performed at 10 and 25 mM NaCl yielded similar profiles, but the slopes and limit levels were different. For 10 mM NaCl a limit of 400 μs was obtained at about 50 μM cytochrome bc1 (Figure 5, solid squares) whereas for 25 mM NaCl a limit of 465 μs was obtained at about 70 μM cytochrome bc1 (Figure 5, open squares).
Figure 5.

Titration of 25 μM cytochrome c2 A101C-SL with cytochrome bc1 at different ionic strengths. Binding of cytochrome c2 to cytochrome bc1 mutant M183K/S158A was monitored as changes in average spin–lattice relaxation time. Measurements were performed for solutions containing 0 mM NaCl (circles), 10 mM NaCl (solid squares), and 25 mM NaCl (open squares) at 95 K. Dashed lines represent fits of eq 1 in which na was substituted with eq 2.
The data shown in Figure 5 revealed that the shortest measured relaxation time of A101C-SL at saturating concentrations of cytochrome bc1 (greater than the dissociation constant; Kd) for each curve is different. This result is interesting in light of the standard approach used for analysis of ligand binding, where weakening of the affinity is usually reflected in a rising of Kd value while the amount of bound ligand (which is proportional to the number of binding sites) under saturating conditions remains unchanged. If this were the case, the curves should converge to the same value of the relaxation time. It follows that the observed lack of convergence could, in principle, be interpreted as an indication that the number of the specific binding sites for cytochrome c2 on cytochrome bc1 changes upon increasing the salt concentration. However, this explanation seems unlikely. Rather, it can be suggested that the salt influences the spatial orientation of cytochrome c2 bound to cytochrome bc1 thereby decreasing PRE.
We attempted to fit the data in Figure 5 with an equation that relates the amount of bound ligand Lb (A101C-SL in our case) to the concentration of binding sites P (i.e. cytochrome bc1):35,36
| 1 |
where Kd is a dissociation constant, L0 is a total A101C-SL concentration, and na is the number of binding sites. We further assumed that the final structure of the complex changes upon increasing salt concentration and the minimum relaxation time observed for each ionic strength for A101C-SL mixed with excess of cytochrome bc1 corresponds to the state in which a whole population of cytochrome c2 molecules is bound to cytochrome bc1.
We performed a fitting the titration data with eq 1, in which the number of binding sites was set to either na = 1, 2, or 3. For na = 1 only the data obtained for 25 mM NaCl were fitted well, whereas in two lower ionic strengths this model did not reproduce the titration curves and the greatest discrepancy was observed for the lowest ionic strength (0 mM NaCl). The model assuming na = 2 yielded a relatively good fit for each titration curve but still was not perfect in the case of lowest ionic strength (0 mM NaCl). A further increase of the number of binding sites (na = 3) gave the best fit only for the lowest ionic strength but at the same time could not reproduce the titration curve for the highest ionic strength (25 mM NaCl; for details see Figure S2, Supporting Information). The observation that an increase in na improves fits for lower ionic strengths whereas a decrease in na leads to better fits at high ionic strengths suggests that the apparent number of cytochrome c2 molecules gathered near the cytochrome c1 changes with the ionic strength—the lower the salt concentration, the larger the number of interacting molecules (SLs interacting with heme c1). At the same time it can be anticipated that the average number of the cytochrome c2 molecules gathered near the single cytochrome bc1 binding domain depends also on the molar ratio between the two proteins. Therefore, we considered a model in which apparent number of cytochrome c2 molecules na depends on both the ionic strength and the cytochrome bc1–cytochrome c2 molar ratio according to the following formula:
| 2 |
where R is the cytochrome bc1/A101C-SL ratio, n is the number of specific binding sites, and α is the scaling parameter that defines how strongly the negative charges on cytochrome bc1 attract the cytochrome c2 molecules to the proximity of the specific binding site. In this formula, na should be considered as the parameter proportional to an average number of A101C-SL molecules per cytochrome bc1, gathered close enough that the relaxation rate of SL is influenced by the heme c1 iron atom (details in the Supporting Information). In our analysis we assumed the number of specific binding sites (n in eq 2) is one per cytochrome bc1 monomer (in this type of experiment it is impossible to determine the n value because n is strongly correlated with Kd) (Figure S3, Supporting Information).
The fits based on the ligand binding eq 1 with constant parameter na substituted by eq 2 neatly reproduced all experimental data points (Figure 5), yielding the parameters shown in Table 1. This correction allowed us to describe the progressive changes in the number of cytochrome c2 molecules located near the cytochrome bc1 binding domain from at least 3 for the lowest ionic strength and low R and to 1 for the highest ionic strength in which it becomes independent of R.
Table 1. Cytochrome c2–Cytochrome bc1 Interaction Parameters.
The confidence intervals were calculated with significance level of 5%
Estimation of Average Interspin Distances in the Cytochrome c2–Cytochrome bc1 System Using a “Dipolar Ruler”
On the basis of our data and the available structural information, we plotted the extent of PRE (kdip) as a function of distance between SL and the fast-relaxing heme iron (Figure 6). The kdip values were calculated as the difference between the relaxation rates measured in the presence of PRE and the relaxation rate in its absence. The first two points in Figure 6 included A101C-SL and T68C-SL interacting with the iron of heme c2. The average interspin distances for these samples were calculated in MMM software using the structure of spin-labeled cytochrome c2, as shown in Figure 1A (also Figure S4, Supporting Information, for rotamer visualization). The shortest distance of 16.7 Å between A101C-SL and the iron of heme c2 resulted in the largest kdip (Figure 6). The corresponding distance for T68C-SL was larger (19.0 Å) and kdip was 2.5 times smaller in comparison to those for A101C-SL. The two additional points included A101C-SL and T68C-SL interacting with the iron of heme c1 on cytochrome bc1. In this case, distances between SL and the iron of heme c1 (20.5 and 35.0 Å, respectively) were calculated assuming that the spatial orientation of cytochrome c2 and cytochrome bc1 was as in the modeled structure of the complex shown in Figure 3 (Figure S3, Supporting Information, for rotamer visualization). The values of kdip were calculated from the data obtained for samples of A101C-SL or T68C-SL mixed with an excess of cytochrome bc1 in buffer without NaCl (we assumed that under those conditions a dominant fraction of cytochrome c2 interacting with cytochrome bc1 was in a spatial orientation as shown in the model in Figure 3).
Figure 6.

Spin–lattice PRE dependence on the distance between SL and fast-relaxing heme iron. Solid symbols represent the PRE obtained for A101C-SL interacting with heme c2 (circle), T68C-SL interacting with heme c2 (square), A101C-SL interacting with heme c1 of cytochrome bc1:M183K/S158A in buffer without NaCl (diamond), and T68C-SL interacting with heme c1 of cytochrome bc1:M183K/S158A in buffer without NaCl (triangle). For these samples the average distances between interacting paramagnetic centers were determined using MMM software on the basis of the structural data shown in Figures 1A and 3. These four points were used to fit the r–6 distance dependence of PRE (for details, see the Supporting Information). Open symbols represent samples for which the measured PRE was used to determine the average interspin distances in samples containing a mixture of A101C-SL and cytochrome bc1:M183K/S158A in buffer containing 10 mM and 25 mM NaCl (square and circle, respectively).
Using these four points (Figure 6, solid symbols) as a reference, we fit the distance dependence of kdip with the r–6 decay function (for details, see the Supporting Information). The quality of the fit (Figure 6, dotted line) was good enough to conclude that the interspin distance must be a dominant factor in determining the value of kdip. If this is the case, the different levels of the relaxation time approached by the titrations curves of Figure 5 under saturating conditions (cytochrome bc1 ≫ Kd) reflect salt-dependent changes in the average distance between A101C-SL and the iron of heme c1. This prompted us to use the fitted curve as a “dipolar ruler” to estimate the average interspin distance between A101C-SL and the iron of heme c1 for cytochrome c2 interacting with cytochrome bc1 in buffer containing NaCl. Plotting kdip values calculated from the ending points of the titrations of Figure 5 yielded distances of 22.0 and 26.0 Å for cytochrome c2–cytochrome bc1 mixtures at 10 and 25 mM NaCl, respectively.
Discussion
When studying protein–protein interactions, it is desirable to obtain information about not only the affinity but also the spatial orientation of the interacting proteins. Ideally, the method should distinguish specific from nonspecific sites, where the term specific means a type of interaction that occurs with engagement of specific regions of the proteins (binding domains) and leads to a catalytically active complex. Toward this purpose we used the measurements of intermolecular interactions between two paramagnetic centers located on two different proteins. One of these centers is the naturally present iron atom of heme c1 of cytochrome bc1, whereas the second is an SL attached to cytochrome c2. As the paramagnetic iron atom is a fast relaxing species, it may influence the relaxation properties of the SL on the second molecule as long as the distance between the centers is close enough to be detected by electron paramagnetic resonance spectroscopy.
There are many different techniques allowing a distance determination by EPR spectroscopy such as DEER, RIDME, ESEEM, or relaxation based measurement.37−39 In our case we exploited the spin–lattice PRE of the SL induced by the heme iron as it is relatively short-ranged,34 thus providing high specificity. This means that any detectable dipolar interaction must take place very close to the specific binding domain where interprotein ET occurs. At the same time this method is “blind” to spurious, nonspecific binding of cytochrome c2 to cytochrome bc1 occurring at regions remote enough from heme c1 that its impact on spin–lattice relaxation of SL vanishes (Figure 3).
The SL was attached to the molecule containing a metal center that, when oxidized, strongly impacts the relaxation of SL (Figure 1B). Therefore, in measurements of the interactions between SL and the iron atom of heme c1, the fully reduced form of cytochrome c2 was used for which the intraprotein dipolar coupling did not exist. On the other hand, heme c1 was kept oxidized and unable to exchange electrons with cytochrome c2 due to the M183K mutation that drastically lowered its midpoint potential. The redox reactions between the two hemes were intentionally turned off to make sure that the measurements monitored just the structural association of two macromolecules (otherwise, the rather uncontrolled changes in redox states of those hemes would affect measurements and obscure the interpretation of the results). A set of experiments described in Figure 2 established that only heme c1 influenced relaxation of SL and no contribution from the reduced FeS was observed.
A “Cloud” of Cytochrome c2 Molecules near the Binding Domain of Cytochrome bc1 at Low Ionic Strength
The first interesting observation that comes from the titration experiments shown in Figure 5 is that the initial decrease of the titration curves is different for each ionic strength: it is the steepest for 0 mM NaCl, intermediate for 10 mM, and the least steep for 25 mM NaCl. Because this trend cannot be simply explained by changes in Kd value (Supporting Information), the fitting of these data to the binding equation required a modification of the term describing the number of binding sites. This modification involved introducing additional contribution to the determined number of bound cytochrome c2 molecules (eq 2) that depends on the ionic strength (α parameter) and cytochrome bc1/cytochrome c2 ratio (R).
The fitted α values (Table 1) reduced from 2.1 for 0 mM NaCl, through 1.5 at 10 mM, to 0 at 25 mM NaCl. When α = 0, the modified equation turns to its classical form, where a single molecule of cytochrome c2 can interact with a single binding domain of cytochrome bc1 irrespective of the molar ratio between the two proteins. An alteration of α to a larger value indicates that, in some cases (R < 1, cytochrome c2 in excess over cytochrome bc1), more than one molecule of cytochrome c2 can approach the binding domain close enough that the relaxation time of SL is influenced by heme c1 iron. This could be described as a “cloud” of several cytochrome c2 molecules gathered in close proximity to the binding domain. As the concentration of cytochrome bc1 increases (R becomes larger than 1), the cloud of cytochrome c2 molecules disperses to populate the additional available binding sites. We note that determination of the exact number of gathered molecules is not possible, because α should only be considered as a parameter that is proportional to the number of SLs attached to cytochrome c2 that are in the range of detectable magnetic interaction with heme c1. In other words, an increase of the α value by 1 would correspond to one additional cytochrome molecule c2 only if the SLs in all bound cytochrome molecules c2 interacted magnetically with heme c1 with the same strength, which is unlikely. If one cytochrome c2 molecule binds in orientation similar to that shown in Figure 3, additional cytochrome c2 molecules are expected to experience magnetic interaction of different strength.
Short-Lived Complexes between Cytochrome c2 and Cytochrome bc1 at High Ionic Strength
Electrostatic forces have been proposed to be a major factor responsible for formation and stabilization of the cytochrome c2 complex with cytochrome bc1, which is essential to support effective ET between the proteins. Generally, two models of interprotein ET can be proposed. A sequential model, in which a long-lived complex is formed, then ET occurs, and then finally the complex dissociates.40 Alternatively, two proteins may collide several times before ET occurs and the lifetime of the complex is relatively short.41 In our case, the complexes between cytochrome c2 and cytochrome bc1 at ionic strength corresponding to <150 mM of NaCl must be relatively long-lived; therefore, they can be detected by EPR for A101C-SL (Figure 4). However, at higher salt concentrations (>150 mM) typical of physiological ionic strength the lifetime of the complexes is so short that their concentration falls below the detection level of this method (Figure 4). This is consistent with the results of our previous experiments in which the binding was monitored by changes in the shape of the CW EPR spectrum of SL attached to cytochrome c2.14
Salt Dependence of the Average Distance between Heme c1 and SL in Cytochrome c2
When the dependence of PRE on cytochrome bc1/c2 ratio for various ionic strengths was examined, the titration curves approached different ending values of the average relaxation time under saturating conditions (excess of cytochrome bc1 over cytochrome c2) (Figure 5). Considering that under saturating conditions (R > 1) all molecules of cytochrome c2 should be bound to cytochrome bc1, the measured relaxation time originates from cytochrome c2 interacting with cytochrome bc1. Thus, the different value obtained for each ionic strength can be interpreted as differences in the spatial orientation of the two proteins and/or changes in the distance between the proteins.
On the basis of our dipolar ruler curve (Figure 6), we found a linear increase in the average distance between the interacting paramagnetic centers upon the salt concentration increase (raise of ∼0.2 Å in distance on every 1 mM increase of NaCl). Extrapolation of this trend to 100 mM NaCl gives an average distance ∼40 Å, which is beyond the limiting value for the magnetic interactions that can be detected by spin–lattice PRE measurements.42 However, at this ionic strength we can still detect the residual effect of interaction with heme c1 (Figure 4), which indicates that some fraction of cytochrome c2 is much closer to cytochrome bc1 than the average 40 Å.
By virtue of the use of SL attached at two different positions on the surface of cytochrome c2, we were able to obtain information on the spatial orientation of these two proteins forming the complex. Under all investigated conditions PRE was weaker for T68C-SL than for A101C-SL. Such a result indicates that the electric dipole moment of cytochrome c2 fosters the binding to cytochrome bc1 in a configuration that facilitates ET between the proteins15 (in the orientation shown in Figure 3). This orientation seems to be maintained even at higher ionic strengths where PRE is still detected. This means that only a small fraction of SL in T68C-SL, if any, approaches heme c1 closer than shown in Figure 3 as a result of constrained rotation of cytochrome c2 in the vicinity of cytochrome bc1. Rather, a dominant fraction of cytochrome c2 molecules approaching the binding domain are already oriented in the configuration where heme c2 faces heme c1 (Figure 3).
The fact that the titration curves do not converge to the same ending point (Figure 5) introduces additional degrees of freedom to the mathematical analysis of binding. In particular, a definition of the number of binding sites for cytochrome c2 on cytochrome bc1 dimer (discrimination between 1:1, 1:2, etc.) becomes ambiguous within the frame of the models describing ligand binding isotherms (Figure S3, Supporting Information). This is because there is no possibility to determine the universal value of this parameter independently of the conditions of salt concentration and/or the ratio between the proteins. In all cases the parameters α, n, and Kd remain strongly correlated, which means that they cannot be obtained simultaneously by the fitting procedure (many sets of these parameters reproduce the same curve).
Interprotein ET as a Product of Several Collisions
The general view that emerges from our analysis of PRE between SL attached to cytochrome c2 and cytochrome bc1 is summarized in the picture that schematically depicts how cytochrome c2 molecules might be arranged around cytochrome bc1 (Figure 7). This ordering depends not only on the ionic strength but also on the reciprocal amounts of the interacting proteins expressed as the ratio R. At low ionic strength and with R < 1 (excess of cytochrome c2 over cytochrome bc1), the charged residues on the surface of the cytochrome bc1 binding domain attract cytochrome c2 molecules very effectively such that they gather forming a cloud of molecules near the binding domain. As we add more cytochrome bc1, the cytochrome c2 molecules occupy the new binding sites and the cloud of cytochrome c2 molecules disperses. At saturating conditions (R > 1, ionic strength still low) each molecule of cytochrome c2 occupies a separate binding domain in proximity to heme c1 (each SL experiences the strongest PRE). This process is illustrated by passing through the top panels of Figure 7 horizontally from left to right. The increase in ionic strength brings about the weakening of attractive electrostatic forces, which also results in dispersing the cloud at R < 1 or increase in the average distance of the complexes at R > 1 (passing through the panels in Figure 7 vertically from top to the bottom). If the salt concentration is sufficiently high (>100 mM), only a few cytochrome c2 molecules (that are below the detection limit) locate near the binding domain even when R ≫ 1. At the same time the ET process is not stopped at high salt concentration which indicates that the proteins must still interact with each other. However, under these conditions the interaction does not lead to any long-lived stabilized complex, rather the cytochrome c2 molecules constantly collide with cytochrome bc1 and the transfer of one electron between hemes is a product of several collisions. This result is an additional support for the simple model of diffusion-coupled and not diffusion-limited mechanism of ET between cytochrome c2 and cytochrome bc1 under conditions of physiological ionic strength.41,43
Figure 7.
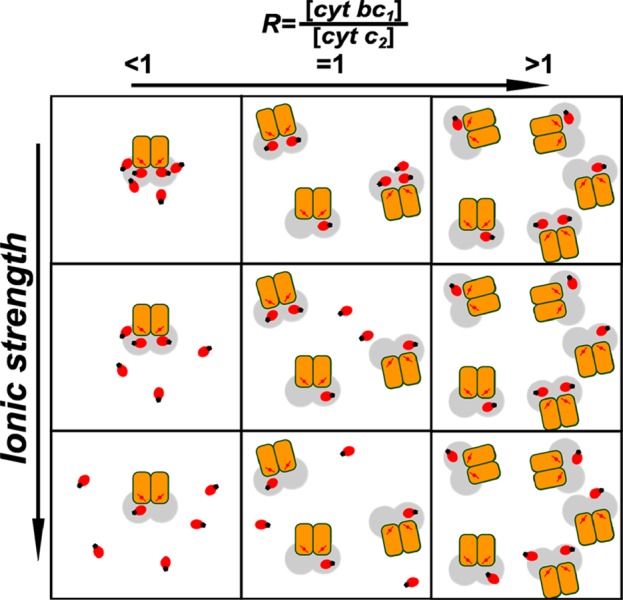
Model of the arrangement of cytochrome c2 molecules in the presence of cytochrome bc1. In this scheme red ovals represent cytochrome c2 molecules with attached SL (black dot). Cytochrome bc1 monomers are depicted as orange rectangles. The ranges of distances from heme c1 (red line with dot) where PRE can be detected are shown in gray. Each square represents different conditions of ionic strength (increase in salt concentration from top to bottom) and cytochrome bc1/cytochrome c2 ratio (increase of cytochrome bc1 concentration from left to right).
The dynamic process of changes in macromolecular organization of cytochrome c2 near the binding domain of cytochrome bc1, described here, emerged from the analysis of changes in spin–lattice PRE which, by its physical nature, was very specific to the studied system. We anticipate that this property makes the method an attractive tool to study specific protein–protein interactions in more complicated systems, for example, when isolating the interactions of interest from a variety of other interactions in the complex molecular-crowded environment.44,45
Acknowledgments
This work was supported by The Wellcome Trust International Senior Research Fellowship (to A.O.). We thank the reviewers for insightful comments and suggestions.
Glossary
Abbreviations
- ET
electron transfer
- PRE
paramagnetic relaxation enhancement
- SL
spin label
- EPR
electron paramagnetic resonance
- FeS
2Fe-2S Rieske cluster
- IR
inversion recovery
- CW EPR
continuous wave EPR
- RLA
rotamer library approach
- heme c1
heme C in cytochrome c1
- heme c2
heme C in cytochrome c2
Supporting Information Available
Comparison of IR and picket fence curves (Figure S1), processing and analysis of IR curves, comparison of ligand binding models (Figure S2), evidence of the ambiguity in determination of binding stoichiometry (Figure S3 and Table S1), visualization of SL rotamers (Figure S4), description of dipolar ruler, and references. This material is available free of charge via the Internet at http://pubs.acs.org
The authors declare no competing financial interest.
Supplementary Material
References
- Northrup S. H.; Erickson H. P. Kinetics of Protein-Protein Association Explained by Brownian Dynamics Computer Simulation. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 3338–3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selzer T.; Schreiber G. New Insights into the Mechanism of Protein–protein Association. Proteins Struct. Funct. Bioinf. 2001, 45, 190–198. [DOI] [PubMed] [Google Scholar]
- Schreiber G. Kinetic Studies of Protein-Protein Interactions. Curr. Opin. Struct. Biol. 2002, 12, 41–47. [DOI] [PubMed] [Google Scholar]
- Ubbink M. The Courtship of Proteins: Understanding the Encounter Complex. FEBS Lett. 2009, 583, 1060–1066. [DOI] [PubMed] [Google Scholar]
- Ahmed A. J.; Smith H. T.; Smith M. B.; Millett F. S. Effect of Specific Lysine Modification on the Reduction of Cytochrome c by Succinate-Cytochrome c Reductase. Biochemistry 1978, 17, 2479–2483. [DOI] [PubMed] [Google Scholar]
- Smith H. T.; Ahmed A. J.; Millett F. Electrostatic Interaction of Cytochrome c with Cytochrome c1 and Cytochrome Oxidase. J. Biol. Chem. 1981, 256, 4984–4990. [PubMed] [Google Scholar]
- Speck S. H.; Ferguson-Miller S.; Osheroff N.; Margoliash E. Definition of Cytochrome c Binding Domains by Chemical Modification: Kinetics of Reaction with Beef Mitochondrial Reductase and Functional Organization of the Respiratory Chain. Proc. Natl. Acad. Sci. U. S. A. 1979, 76, 155–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J.; Kriaucionas A.; Knaff D.; Millett F. The Reaction Domain on Rhodospirillum Rubrum Cytochrome c2 and Horse Cytochrome c for the Rhodospirillum Rubrum Cytochrome bc1 Complex. J. Biol. Chem. 1987, 262, 14005–14009. [PubMed] [Google Scholar]
- Lange C.; Hunte C. Crystal Structure of the Yeast Cytochrome bc1 Complex with Its Bound Substrate Cytochrome C. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 2800–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokhan O.; Wraight C. A.; Tajkhorshid E. The Binding Interface of Cytochrome c and Cytochrome c1 in the bc1 Complex: Rationalizing the Role of Key Residues. Biophys. J. 2010, 99, 2647–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engstrom G.; Rajagukguk R.; Saunders A. J.; Patel C. N.; Rajagukguk S.; Merbitz-Zahradnik T.; Xiao K.; Pielak G. J.; Trumpower B.; Yu C.-A.; et al. Design of a Ruthenium-Labeled Cytochrome c Derivative to Study Electron Transfer with the Cytochrome bc1 Complex. Biochemistry 2003, 42, 2816–2824. [DOI] [PubMed] [Google Scholar]
- Millett F.; Havens J.; Rajagukguk S.; Durham B. Design and Use of Photoactive Ruthenium Complexes to Study Electron Transfer within Cytochrome bc1 and from Cytochrome bc1 to Cytochrome C. Biochim. Biophys. Acta 2013, 1827, 1309–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devanathan S.; Salamon Z.; Tollin G.; Fitch J. C.; Meyer T. E.; Berry E. a; Cusanovich M. a. Plasmon Waveguide Resonance Spectroscopic Evidence for Differential Binding of Oxidized and Reduced Rhodobacter Capsulatus Cytochrome c2 to the Cytochrome bc1 Complex Mediated by the Conformation of the Rieske Iron-Sulfur Protein. Biochemistry 2007, 46, 7138–7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarewicz M.; Borek A.; Daldal F.; Froncisz W.; Osyczka A. Demonstration of Short-Lived Complexes of Cytochrome c with Cytochrome bc1 by EPR Spectroscopy. J. Biol. Chem. 2008, 283, 24826–24836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarewicz M.; Pietras R.; Froncisz W.; Osyczka A. Reorientation of Cytochrome c2 upon Interaction with Oppositely Charged Macromolecules Probed by SR EPR: Implications for the Role of Dipole Moment to Facilitate Collisions in Proper Configuration for Electron Transfer. Metallomics 2011, 3, 404–409. [DOI] [PubMed] [Google Scholar]
- Świerczek M.; Cieluch E.; Sarewicz M.; Borek A.; Moser C. C.; Dutton P. L.; Osyczka A. An Electronic Bus Bar Lies in the Core of Cytochrome bc1. Science 2010, 329, 451–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanciano P.; Lee D.-W.; Yang H.; Darrouzet E.; Daldal F. Intermonomer Electron Transfer between the Low-Potential B Hemes of Cytochrome bc1. Biochemistry 2011, 50, 1651–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsh D. D. J.; Beck W. W. F.; Innes J. J. B.; Brudvig G. W. Using Saturation-Recovery EPR to Measure Distances in Proteins: Applications to Photosystem II. Biochemistry 1992, 31, 532–541. [DOI] [PubMed] [Google Scholar]
- Lyubenova S.; Siddiqui M. K.; Vries M. J. M. P. De; Ludwig B.; Prisner T. F. Protein-Protein Interactions Studied by EPR Relaxation Measurements: Cytochrome c and Cytochrome c Oxidase. J. Phys. Chem. B 2007, 111, 3839–3846. [DOI] [PubMed] [Google Scholar]
- Volkov A. N.; Worrall J. a R.; Holtzmann E.; Ubbink M. Solution Structure and Dynamics of the Complex between Cytochrome c and Cytochrome c Peroxidase Determined by Paramagnetic NMR. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 18945–18950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir Q.; Volkov A. N.; Ullmann G. M.; Ubbink M. Visualization of the Encounter Ensemble of the Transient Electron Transfer Complex of Cytochrome c and Cytochrome c Peroxidase. J. Am. Chem. Soc. 2010, 132, 241–247. [DOI] [PubMed] [Google Scholar]
- Scanu S.; Foerster J.; Timmer M. Loss of Electrostatic Interactions Causes Increase of Dynamics within the Plastocyanin–Cytochrome F Complex. Biochemistry 2013, 52, 6615–6626. [DOI] [PubMed] [Google Scholar]
- Sarewicz M.; Dutka M.; Froncisz W.; Osyczka A. Magnetic Interactions Sense Changes in Distance between Heme b(L) and the Iron-Sulfur Cluster in Cytochrome bc(1). Biochemistry 2009, 48, 5708–5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenbach C.; Oh K. K.; Hubbell W. L.; Trabanino R.; Hideg K. Estimation of Inter-Residue Distances in Spin Labeled Proteins at Physiological Temperatures: Experimental Strategies and Practical Limitations. Biochemistry 2001, 40, 15471–15482. [DOI] [PubMed] [Google Scholar]
- Valkova-Valchanova M. B.; Saribas A. S.; Gibney B. R.; Dutton P. L.; Daldal F. Isolation and Characterization of a Two-Subunit Cytochrome B-c1 Subcomplex from Rhodobacter Capsulatus and Reconstitution of Its Ubihydroquinone Oxidation (Qo) Site with Purified Fe-S Protein Subunit. Biochemistry 1998, 16242–16251. [DOI] [PubMed] [Google Scholar]
- Hung S.; Grant C. V.; Peloquin J. M.; Waldeck A. R.; Britt R. D.; Chan S. I. Electron Spin-Lattice Relaxation Measurement of the 3Fe-4S (S-3) Cluster in Succinate:Ubiquinone Reductase from Paracoccus Denitrificans. A Detailed Analysis Based on a Dipole-Dipole Interaction Model. J. Phys. Chem. A 2000, 104, 4402–4412. [Google Scholar]
- Du J.; Eaton G.; Eaton S. Temperature, Orientation, and Solvent Dependence of Electron Spin-Lattice Relaxation Rates for Nitroxyl Radicals in Glassy Solvents and Doped Solids. J. Magn. Reson. Ser. A 1995, 115, 213–221. [Google Scholar]
- Sato H.; Bottle S. E.; Blinco J. P.; Micallef A. S.; Eaton G. R.; Eaton S. S. Electron Spin-Lattice Relaxation of Nitroxyl Radicals in Temperature Ranges That Span Glassy Solutions to Low-Viscosity Liquids. J. Magn. Reson. 2008, 191, 66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyhach Y.; Jeschke G. Prediction of Favourable Sites for Spin Labelling of Proteins. Spectroscopy 2010, 24, 651–659. [Google Scholar]
- Polyhach Y.; Bordignon E.; Jeschke G. Rotamer Libraries of Spin Labelled Cysteines for Protein Studies. Phys. Chem. Chem. Phys. 2011, 2356–2366. [DOI] [PubMed] [Google Scholar]
- Darrouzet E.; Mandaci S.; Li J.; Qin H.; Knaff D. B.; Daldal F. Substitution of the Sixth Axial Ligand of Rhodobacter Capsulatus Cytochrome c1 Heme Yields Novel Cytochrome c1 Variants with Unusual Properties. Biochemistry 1999, 38, 7908–7917. [DOI] [PubMed] [Google Scholar]
- Kolling D. J.; Brunzelle J. S.; Lhee S.; Crofts A. R.; Nair S. K. Atomic Resolution Structures of Rieske Iron-Sulfur Protein: Role of Hydrogen Bonds in Tuning the Redox Potential of Iron-Sulfur Clusters. Structure 2007, 15, 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Bowler B. E.; Eaton G. R.; Eaton S. S. Electron Spin Lattice Relaxation Rates for S = 1/2 Molecular Species in Glassy Matrices or Magnetically Dilute Solids at Temperatures between 10 and 300 K. J. Magn. Reson. 1999, 139, 165–174. [DOI] [PubMed] [Google Scholar]
- Ulyanov D.; Bowler B.; Eaton G. Electron-Electron Distances in Spin-Labeled Low-Spin Metmyoglobin Variants by Relaxation Enhancement. Biophys. J. 2008, 95, 5306–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. X.; Kumar N. R.; Srivastava D. K. A Novel Spectroscopic Titration Method for Determining the Dissociation Constant and Stoichiometry of Protein-Ligand Complex. Anal. Biochem. 1992, 206, 376–381. [DOI] [PubMed] [Google Scholar]
- Sarewicz M.; Szytuła S.; Dutka M.; Osyczka A.; Froncisz W. Estimation of Binding Parameters for the Protein-Protein Interaction Using a Site-Directed Spin Labeling and EPR Spectroscopy. Eur. Biophys. J. 2008, 37, 483–493. [DOI] [PubMed] [Google Scholar]
- Schiemann O.; Prisner T. F. Long-Range Distance Determinations in Biomacromolecules by EPR Spectroscopy. Q. Rev. Biophys. 2007, 40, 1–53. [DOI] [PubMed] [Google Scholar]
- Sahu I.; McCarrick R.; Lorigan G. Use of Electron Paramagnetic Resonance To Solve Biochemical Problems. Biochemistry 2013, 52, 5967–5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astashkin A. V.; Elmore B. O.; Fan W.; Guillemette J. G.; Feng C. Pulsed EPR Determination of the Distance between Heme Iron and FMN Centers in a Human Inducible Nitric Oxide Synthase. J. Am. Chem. Soc. 2010, 132, 12059–12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLendon G.; Hake R. Interprotein Electron Transfer. Chem. Rev. 1992, 92, 481–490. [Google Scholar]
- Hackenbrock C. R.; Chazotte B.; Gupte S. S. The Random Collision Model and a Critical Assessment of Diffusion and Collision in Mitochondrial Electron Transport. J. Bioenerg. Biomembr. 1986, 18, 331–368. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Bowler B. E.; Lynch K.; Eaton S. S.; Eaton G. R. Interspin Distances in Spin-Labeled Metmyoglobin Variants Determined by Saturation Recovery EPR. Biophys. J. 2000, 79, 1039–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupte S.; Wu E. S.; Hoechli L.; Hoechli M.; Jacobson K.; Sowers A. E.; Hackenbrock C. R. Relationship between Lateral Diffusion, Collision Frequency, and Electron Transfer of Mitochondrial Inner Membrane Oxidation-Reduction Components. Proc. Natl. Acad. Sci. U. S. A. 1984, 81, 2606–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeds E. J.; Ashenberg O.; Gerardin J.; Shakhnovich E. I. Robust Protein Protein Interactions in Crowded Cellular Environments. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 14952–14957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A.; Kim Y. C.; Mittal J. Protein–protein Interactions in a Crowded Environment. Biophys. Rev. 2013, 5, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.