

Abstract
The MYC proto-oncogene is an essential regulator of many normal biological programmes. MYC, when activated as an oncogene, has been implicated in the pathogenesis of most types of human cancers. MYC overexpression in normal cells is restrained from causing cancer through multiple genetically and epigenetically controlled checkpoint mechanisms, including proliferative arrest, apoptosis and cellular senescence. When pathologically activated in the correct epigenetic and genetic contexts, MYC bypasses these mechanisms and drives many of the ‘hallmark’ features of cancer, including uncontrolled tumour growth associated with DNA replication and transcription, cellular proliferation and growth, protein synthesis and altered cellular metabolism. MYC also dictates tumour cell fate by enforcing self-renewal and by abrogating cellular senescence and differentiation programmes. Moreover, MYC influences the tumour microenvironment, including activating angiogenesis and suppressing the host immune response. Provocatively, brief or even partial suppression of MYC back to its physiological levels of activation can lead to the restoration of intrinsic checkpoint mechanisms, resulting in acute and sustained tumour regression associated with tumour cells undergoing proliferative arrest, differentiation, senescence and apoptosis, as well as remodelling of the tumour microenvironment, recruitment of an immune response and shutdown of angiogenesis. Hence, tumours appear to be addicted to the MYC oncogene because of both tumour cell intrinsic and host-dependent mechanisms. MYC is important for the regulation of both the initiation and maintenance of tumorigenesis.
Keywords: Oncogene Addiction, Targeted Therapeutics, MYC Oncogene, Transgenic Mouse Models
Introduction
The MYC proto-oncogene was identified as the aetiological agent of leukemogenesis induced by avian myelocytomatosis retrovirus (MC29). Later, MYC was shown to be activated through genomic events including chromosomal translocation in Burkitt’s lymphoma as well as gene amplifications [1–3]. More recently, MYC was found to be overexpressed in human tumours [4–6]; indeed, MYC is thought to be causally involved in more than half of all human cancers [4, 7–11]. Thus, MYC appears to be one of the most important oncogenic events in human tumorigenesis.
MYC largely functions as a transcription factor that coordinates many biological processes [12]. MYC activation can usurp these programmes resulting in the characteristic features of cancer. Thus, MYC activation contributes to autonomous proliferation and growth, persistent DNA replication, increased ribosomal biogenesis and protein synthesis, global changes in cellular metabolism, activation of the angiogenic switch and suppression of host immune responses [5, 13–16]. Hence, MYC activation appears to be a molecular hallmark of cancer.
In this review, we will examine the notion that MYC activation is one of the necessary events for the initiation of tumorigenesis and frequently results in the dependence of tumour survival on high levels of MYC, herein referred to as MYC addiction.
MYC and the initiation of cancer
The expression of MYC is tightly regulated by transcriptional control, mRNA turnover and protein expression and degradation. Through insertional mutagenesis, chromosomal translocations and genomic amplifications, MYC can be activated to drive cell transformation [17, 18]. However, generally MYC activation alone cannot induce tumorigenesis. MYC causes transformation only in specific cell lines presumed to have already acquired other oncogenic events that rendered them permissive [19, 20]. Surprisingly, MYC overexpression alone is incapable of inducing neoplastic transformation of most cells. Instead, MYC overexpression in normal human and mouse cells induces proliferative arrest, senescence and/or apoptosis [21–26] (Fig. 1). Thus, there appear to be cellular programmes that inherently prevent MYC activation from initiating tumorigenesis.
Fig. 1.
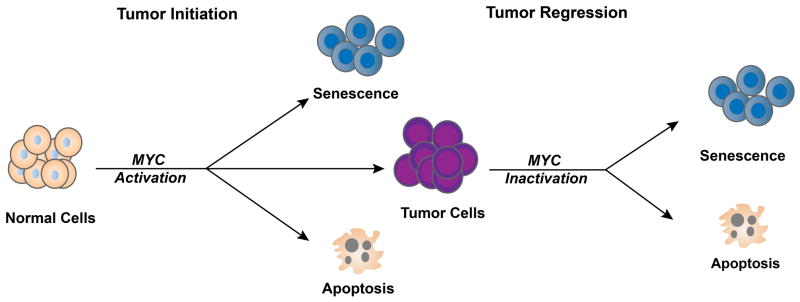
MYC-induced cancer initiation and maintenance. MYC induces tumorigenesis by evading multiple tumour suppressing checkpoint mechanisms including proliferative arrest, apoptosis and/or senescence. Upon MYC suppression, these barriers are restored, enabling sustained tumour regression.
MYC overexpression has also been found to induce DNA replication and entry into S phase [27–30]. MYC is part of the replication complex [28, 31]. It is interesting that MYC alone blocks mitotic cellular division [29]. Normal cells grow and replicate in response to MYC but they cannot divide; rather, these cells become polyploid [19, 29, 30, 32, 33]. Indeed, this occurs in part because MYC overexpression can enforce replication that results in DNA breaks [34]. This appears to be the consequence of MYC directly blocking double-strand DNA repair, but it could also be related to the dysregulation of oxidative stress [34–36]. The dosage of MYC overexpression may dictate whether cells undergo proliferative arrest [29], cellular senescence [24] or apoptosis. Thus, MYC deregulation alone cannot force complete transit through the cell division cycle.
The precise consequences of MYC overexpression in a normal cell are dependent on both the epigenetic and genetic setting. MYC overexpression in the embryonic liver induces cellular proliferation, whereas it promotes cellular growth without mitotic division associated with polyploidy in the adult liver [37]. Similarly, MYC overexpression induces proliferation in embryonic heart, but cellular hypertrophy in adults (Felsher and Bishop, unpublished data). Circumstances in an adult host that promote proliferation may favour MYC-induced cellular proliferation. For example, in the liver, a partial hepatectomy or exposure to toxins that cause liver damage can enable MYC to induce cellular proliferation [37, 38]. Similarly, the loss of the p53 tumour suppressor cooperates with MYC to induce cellular proliferation and tumorigenesis in adult hepatocytes [37]. Thus, cellular context and specific genetic defects can enable MYC to more readily induce tumorigenesis.
The gene dosage of MYC strongly influences the consequences of its activation. Highly robust activation of MYC is more commonly associated with DNA damage and apoptosis; conversely, less robust MYC activation appears to be associated with proliferative arrest and cellular senescence [24, 29]. Similarly, the MYC gene dosage appears to strongly influence the effect on cellular proliferation versus apoptosis [39]. Thus, the level and context of MYC dictate the consequences of its activation.
MYC cooperates with other oncogenes
MYC cooperates with other oncogenes [40–44]; many oncogenes, such as Bmi1 and Pim1, were first identified in genetic screens to establish events that cooperate with MYC to induce lymphomagenesis [45–49]. Oncogenes or tumour suppressor genes that regulate apoptosis are often dysregulated in MYC-induced tumorigenesis, including expression of BCL2 or loss of p53 or p19ARF [50–54]. Hence, there are ‘intrinsic’ mechanisms of tumour suppression that prevent MYC-induced malignant transformation [55].
The use of in vivo mouse models has illustrated that host-dependent mechanisms also influence the ability of MYC to initiate tumorigenesis. Examples of such host-dependent mechanisms include environmental toxins or carcinogens [56], cytokines such as transforming growth factor alpha [57, 58], innate immunity [59] and autocrine factors [60].
The particular stage of differentiation of a cellular lineage may also have an influence on the consequences of MYC activation. As described above, MYC activation in embryonic hepatocytes induces robust cellular proliferation; by contrast MYC activation in adult cells induces DNA replication associated with mitotic arrest and hyperdiploid cells [37]. Thus, the ability of MYC expression to initiate tumorigenesis is a consequence of the constellation of other oncogene activating or tumor suppressor inactivating genetic events as well as likely through nongenetic or even epigenetic mechanisms.
MYC initiates tumorigenesis only in a permissive epigenetic and genetic context which overcomes cell intrinsic mechanisms that mitigate proliferation, induce apoptosis and activate innate and adaptive immunity. Genetic events may be required to bypass these mechanisms. Changes in the microenvironment can create a setting that is permissive for tumorigenesis. Thus, changes both inside tumour cells and outside in the tumour microenvironment are causally involved in the mechanism of MYC-induced tumorigenesis.
MYC and the maintenance of cancer
Because cancers are caused by oncogenes, suppression of oncogenes should in theory reverse cancer [61, 62]. Yet, several questions remain unanswered: which and how many events must be targeted? Will cancers acquire compensatory mutations? Does an oncogene need to harbour mutations to be essential for maintenance of a neoplastic state? Could therapeutically targeting oncogenes be toxic to the host because their inactivation in normal cells could disrupt their required normal physiologic function?
The notion that a neoplastic state is reversible was first illustrated using conditional temperature-sensitive oncogene mutants [63, 64]. Further, anti-sense oligonucleotides that targeted oncogenes could reverse neoplasia [65–69].
To determine experimentally whether an autochthonously arising cancer is reversible, we employed transgenic mouse models utilizing a conditional oncogene. Use of mouse models with the tetracycline-regulated system and/or chimeric gene products that could be activated in an on/off fashion are the most common approaches [70–72].
The suppression of MYC was shown to reverse tumorigenesis. Similar results were seen in a wide variety of tumours, including haematopoietic (T cell and B cell lymphoma and leukaemia), epithelial (hepatocellular, breast and squamous cell carcinomas) and mesenchymal tumours (osteogenic sarcoma) [73–76]. Of note, in some cases it was confirmed that these tumours were clonal and genetically complex [77].
MYC-induced tumorigenesis is not always reversible. The introduction of additional genetic features, such as a mutant RAS, can impede the reversibility of MYC-induced breast adenocarcinoma [78]. Absence of p53 prevents MYC-induced lymphoma from being reversible [79]. However, when examined, all tumours that recurred after MYC suppression had reactivated MYC expression [80]. Thus, tumours do not appear to be able to completely escape MYC addiction.
Tumorigenesis may be reversible even when MYC is not the initiating oncogenic lesion. Utilizing a dominant negative MYC, termed ‘omoMYC’, it was shown that conditional suppression of MYC appears to reverse RAS-induced tumorigenesis [81, 82]. However, it is likely that RAS is activating endogenous MYC, which may explain why these tumours appear to be addicted to MYC. Moreover, recent observations suggest that omoMYC blocks some of the interactions between MYC and other partners [83]. Thus, addiction to MYC appears to be a feature of cancers that do not necessarily require its genetic activation.
MYC-associated oncogene addiction
Cancer appears to be addicted to MYC [84–87]. MYC inactivation reverses cancer, restoring normal cellular checkpoint mechanisms and resulting in proliferative arrest, differentiation, apoptosis and/or cellular senescence. In addition, MYC inactivation remodels the microenvironment, restoring normal tissue architecture [73] and shutdown of angiogenesis [79]. Thus, oncogene addiction to MYC restores physiological programmes both within the tumour cell and in the host.
The specific outcome of MYC suppression is influenced by the type of cancer (Fig. 2). Haematopoietic tumours appear to undergo proliferative arrest, differentiation and senescence, followed by robust apoptosis [73]. Osteosarcoma undergoes proliferative arrest, differentiation and senescence with minimal if any evidence of apoptosis [74]. Hepatocellular or breast adenocarcinoma follow two main courses; most tumour cells undergo proliferative arrest, senescence and apoptosis, but a subpopulation of cells exhibits tumour dormancy [88, 89].
Fig. 2. MYC inactivation leads to cancer-specific consequences.

MYC inactivation elicits oncogene addiction by multiple mechanisms that differ depending on the tumour type. MYC inactivation in lymphoma induces proliferative arrest, differentiation/senescence and apoptosis. In an osteosarcoma model, MYC inactivation induces proliferative arrest and differentiation/senescence but not apoptosis. Finally, in liver cancer, MYC inactivation induces proliferative arrest, differentiation/senescence and apoptosis. MYC reactivation restores the tumour in liver cancer but not in the osteosarcoma model.
Predicting oncogene addiction
The stereotypical changes associated with oncogene addiction suggest that it may be possible to predict when tumours will regress shortly after therapeutic measures. Oncogene addiction can be modelled as a consequence of differential changes in survival and death signals [90]. Tumours regress because both survival and death signals dissipate upon oncogene suppression, but the latter signals dissipate more slowly. This could be related to differential regulation of either the actual effectors or of survival and death signalling [90, 91], differential levels of metabolites that regulate or are required for survival or death [92, 93] and/or non-cell autonomous mechanisms such as autocrine or paracrine host signalling [6].
Oncogene addiction and the immune system
MYC can influence immune mechanisms that may contribute to tumorigenesis [94–96]; MYC inactivation could contribute to tumour regression through the restoration of immune mechanisms. Indeed, MYC inactivation in a RAG1−/− (lacking both B and T cells) or a CD4−/− mouse host (lacking CD4+ T helper cells) demonstrated reduced kinetics of tumour regression, increased minimal residual disease and inevitable tumour recurrence [97]. It was also shown that CD4+ T helper cells were required for MYC or BCR-ABL inactivation to induce cellular senescence of tumour cells and the shutdown of angiogenesis in the tumour microenvironment [97].
In situ analysis showed that the absence of host immune effectors had little impact on proliferative arrest or apoptosis in the tumour, but the absence of immune effectors largely abrogated cellular senescence and the shutdown of angiogenesis. Thrombospondins were implicated as critical effectors. Similarly, suppression of MYC through omoMYC induces changes in the tumour microenvironment associated with tumour regression [6, 98]. The suppression of MYC mediates its effect on the tumour both through direct effects on cancer cells as well as through specific immune effectors and chemokines [99].
Thus, oncogene addiction may occur via mechanisms that operate on multiple levels: (i) tumour cell intrinsic induction of proliferative arrest, senescence and apoptosis; (ii) recruitment of immune effectors that is probably heralded by a non-canonical CD4+ T cell-specific mechanism; and (iii) remodelling of the tumour microenvironment. The initial regression of a tumour is cell autonomous, but complete regression requires host-dependent mechanisms (Fig. 3).
Fig. 3. MYC inactivation elicits tumour regression through both tumour-intrinsic and host-dependent mechanisms.

MYC activation leads to tumorigenesis through suppression of critical safeguards such as apoptosis, proliferative arrest, differentiation and senescence. Activation of MYC also facilitates engagement of the hallmarks of tumour growth, as well as cell-extrinsic phenomena such as host immunity.
The rational combination of immune therapy with oncogene-targeted therapy could cooperate to induce optimal treatment of human cancers [100, 101]. Hence, oncogene suppression may result in tumour regression via the cell autonomously killing the tumour and then through host cell-dependent immune activation that eliminates the residual tumour cells.
Brief or partial suppression of MYC can reverse tumorigenesis
Brief suppression of MYC is associated with an irreversible change in the cellular programme; in some settings, tumours cannot be restored upon MYC reactivation [74]. Similarly, a two-fold decrease in oncogenic levels of MYC was sufficient to induce tumour regression [15]. This effect is tumour-type specific, as evidenced by the fact that lymphoma and osteosarcoma exhibit this phenotype [74, 79], in contrast to epithelial tumours such as hepatocellular or breast carcinoma [88, 89].
In osteosarcoma, MYC suppression results in terminal cellular differentiation from osteoblasts into differentiated osteocytes that are associated with bone formation in vivo [74]. The reactivation of MYC not only fails to restore the cancer, but either has no effect or is associated with apoptosis. Gene expression analysis showed that there were irreversible changes in gene expression involving ribosome biosynthesis and protein synthesis [16, 102]. Changes in protein biogenesis may be important mechanisms of oncogene addiction.
Partial suppression of MYC can also result in sustained tumour regression when the levels of MYC are below those of human tumour-derived cell lines and above those of proliferating normal human cells or Epstein barr virus-transformed lymphocytes [103]. Thus, there is a specific threshold level of MYC required to sustain a malignant phenotype [103]. Protein and gene expression analysis identified many specific changes but, of note, ribosomal gene products were suppressed. Collectively, these results suggest that a global shift in protein biogenesis is an important part of how MYC suppression results in tumour regression, as has been described by others [104].
An important implication of these results is that it may be sufficient to partially and/or briefly suppress MYC expression in at least some tumour types in order to induce a sustained clinical effect on human cancer. The transient inactivation of MYC may be effective as a result of the dependence of MYC-associated oncogene addiction on molecular features that are determined shortly after oncogene inactivation [90].
MYC activation is also associated with global changes in the energy metabolism of cancer cells [19, 105]. Hence, MYC addiction observed in many cancer cells could at least in part relate to acute changes in metabolism. Alternatively, suppression of MYC may induce tumour regression by acutely disrupting the means by which tumour cells maintain survival or suppress death signalling.
Summary: MYC as an important mediator of cancer initiation and maintenance
MYC initiates and thereby maintains tumorigenesis through the regulation of multiple programmes. These include programmes within cells (e.g. DNA replication, survival, death, self-renewal and energy metabolism), within the tumour microenvironment (e.g. regulation of autocrine factors and angiogenesis) and within the host (i.e. through effects on the immune response) (see Fig. 3). MYC suppression elicits addiction and tumour regression precisely because of the reversal of these cellular, microenvironmental and immune-regulated programmes.
MYC, like most transcription factors, is generally considered to be ‘undruggable’ [106–109]. Nevertheless, siRNA/shRNA/anti-sense oligos could provide potential strategies to target MYC [67, 110, 111]. Alternatively, synthetic lethal screens have provided some strategies to target MYC-addicted tumours [112–114]. Therapies that suppress MYC indirectly may be efficacious as illustrated by inhibition via statins or the bromodomain containing 4 gene [108, 115, 116]. The central role of MYC in the initiation and maintenance of tumorigenesis suggests that efforts to identify therapies that can target this oncogene are of paramount importance.
Acknowledgments
The authors would like to acknowledge all current and past colleagues for their contributions in characterizing various models of oncogene addiction. Our research has been generously funded by grants from the National Institutes of Health (R01CA170378, U54CA149145, 5T32AI07290, 1F32CA177139 and P50CA114747) and the Leukemia and Lymphoma Society (R6223-07), and by a Burroughs Welcome Fund Career Award.
Footnotes
Conflict of interest statement
No conflict of interest was declared.
References
- 1.Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci U S A. 1982;79:7824–7. doi: 10.1073/pnas.79.24.7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neel BG, Jhanwar SC, Chaganti RS, Hayward WS. Two human c-onc genes are located on the long arm of chromosome 8. Proc Natl Acad Sci U S A. 1982;79:7842–6. doi: 10.1073/pnas.79.24.7842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taub R, Kirsch I, Morton C, et al. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci U S A. 1982;79:7837–41. doi: 10.1073/pnas.79.24.7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boxer LM, Dang CV. Translocations involving c-myc and c-myc function. Oncogene. 2001;20:5595–610. doi: 10.1038/sj.onc.1204595. [DOI] [PubMed] [Google Scholar]
- 5.Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eilers M, Eisenman RN. Myc’s broad reach. Genes Dev. 2008;22:2755–66. doi: 10.1101/gad.1712408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Escot C, Theillet C, Lidereau R, et al. Genetic alteration of the c-myc protooncogene (MYC) in human primary breast carcinomas. Proc Natl Acad Sci U S A. 1986;83:4834–8. doi: 10.1073/pnas.83.13.4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gamberi G, Benassi MS, Bohling T, et al. C-myc and c-fos in human osteosarcoma: prognostic value of mRNA and protein expression. Oncology. 1998;55:556–63. doi: 10.1159/000011912. [DOI] [PubMed] [Google Scholar]
- 9.Kawate S, Fukusato T, Ohwada S, Watanuki A, Morishita Y. Amplification of c-myc in hepatocellular carcinoma: correlation with clinicopathologic features, proliferative activity and p53 overexpression. Oncology. 1999;57:157–63. doi: 10.1159/000012024. [DOI] [PubMed] [Google Scholar]
- 10.Ladanyi M, Park CK, Lewis R, Jhanwar SC, Healey JH, Huvos AG. Sporadic amplification of the MYC gene in human osteosarcomas. Diagn Mol Pathol. 1993;2:163–7. [PubMed] [Google Scholar]
- 11.Stock C, Kager L, Fink FM, Gadner H, Ambros PF. Chromosomal regions involved in the pathogenesis of osteosarcomas. Genes Chromosomes Cancer. 2000;28:329–36. doi: 10.1002/1098-2264(200007)28:3<329::aid-gcc11>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 12.Dang CV, Resar LM, Emison E, et al. Function of the c-Myc oncogenic transcription factor. Exp Cell Res. 1999;253:63–77. doi: 10.1006/excr.1999.4686. [DOI] [PubMed] [Google Scholar]
- 13.Bachireddy P, Rakhra K, Felsher DW. Immunology in the clinic review series; focus on cancer: multiple roles for the immune system in oncogene addiction. Clin Exp Immunol. 2012;167:188–94. doi: 10.1111/j.1365-2249.2011.04514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Felsher DW. Cancer revoked: oncogenes as therapeutic targets. Nat Rev Cancer. 2003;3:375–80. doi: 10.1038/nrc1070. [DOI] [PubMed] [Google Scholar]
- 15.Shachaf CM, Felsher DW. Tumor dormancy and MYC inactivation: pushing cancer to the brink of normalcy. Cancer Res. 2005;65:4471–4. doi: 10.1158/0008-5472.CAN-05-1172. [DOI] [PubMed] [Google Scholar]
- 16.van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer. 2010;10:301–9. doi: 10.1038/nrc2819. [DOI] [PubMed] [Google Scholar]
- 17.Alitalo K, Bishop JM, Smith DH, Chen EY, Colby WW, Levinson AD. Nucleotide sequence to the v-myc oncogene of avian retrovirus MC29. Proc Natl Acad Sci U S A. 1983;80:100–4. doi: 10.1073/pnas.80.1.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sheiness D, Fanshier L, Bishop JM. Identification of nucleotide sequences which may encode the oncogenic capacity of avian retrovirus MC29. J Virol. 1978;28:600–10. doi: 10.1128/jvi.28.2.600-610.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spencer CA, Groudine M. Control of c-myc regulation in normal and neoplastic cells. Adv Cancer Res. 1991;56:1–48. doi: 10.1016/s0065-230x(08)60476-5. [DOI] [PubMed] [Google Scholar]
- 21.Evan GI, Wyllie AH, Gilbert CS, et al. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–28. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 22.Felsher DW, Bishop JM. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc Natl Acad Sci U S A. 1999;96:3940–4. doi: 10.1073/pnas.96.7.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gibson AW, Cheng T, Johnston RN. Apoptosis induced by c-myc overexpression is dependent on growth conditions. Exp Cell Res. 1995;218:351–8. doi: 10.1006/excr.1995.1166. [DOI] [PubMed] [Google Scholar]
- 24.Grandori C, Wu KJ, Fernandez P, et al. Werner syndrome protein limits MYC-induced cellular senescence. Genes Dev. 2003;17:1569–74. doi: 10.1101/gad.1100303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoffman B, Liebermann DA. Apoptotic signaling by c-MYC. Oncogene. 2008;27:6462–72. doi: 10.1038/onc.2008.312. [DOI] [PubMed] [Google Scholar]
- 26.Nilsson JA, Cleveland JL. Myc pathways provoking cell suicide and cancer. Oncogene. 2003;22:9007–21. doi: 10.1038/sj.onc.1207261. [DOI] [PubMed] [Google Scholar]
- 27.Cerni C, Mougneau E, Zerlin M, Julius M, Marcu KB, Cuzin F. c-myc and functionally related oncogenes induce both high rates of sister chromatid exchange and abnormal karyotypes in rat fibroblasts. Curr Top Microbiol Immunol. 1986;132:193–201. doi: 10.1007/978-3-642-71562-4_28. [DOI] [PubMed] [Google Scholar]
- 28.Dominguez-Sola D, Ying CY, Grandori C, et al. Non-transcriptional control of DNA replication by c-Myc. Nature. 2007;448:445–51. doi: 10.1038/nature05953. [DOI] [PubMed] [Google Scholar]
- 29.Felsher DW, Zetterberg A, Zhu J, Tlsty T, Bishop JM. Overexpression of MYC causes p53-dependent G2 arrest of normal fibroblasts. Proc Natl Acad Sci U S A. 2000;97:10544–8. doi: 10.1073/pnas.190327097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mai S, Hanley-Hyde J, Fluri M. c-Myc overexpression associated DHFR gene amplification in hamster, rat, mouse and human cell lines. Oncogene. 1996;12:277–88. [PubMed] [Google Scholar]
- 31.Srinivasan SV, Dominguez-Sola D, Wang LC, Hyrien O, Gautier J. Cdc45 is a critical effector of myc-dependent DNA replication stress. Cell Rep. 2013;3:1629–39. doi: 10.1016/j.celrep.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnston LA, Prober DA, Edgar BA, Eisenman RN, Gallant P. Drosophila myc regulates cellular growth during development. Cell. 1999;98:779–90. doi: 10.1016/s0092-8674(00)81512-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neto-Silva RM, de Beco S, Johnston LA. Evidence for a growth-stabilizing regulatory feedback mechanism between Myc and Yorkie, the Drosophila homolog of Yap. Dev Cell. 2010;19:507–20. doi: 10.1016/j.devcel.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karlsson A, Deb-Basu D, Cherry A, Turner S, Ford J, Felsher DW. Defective double-strand DNA break repair and chromosomal translocations by MYC overexpression. Proc Natl Acad Sci U S A. 2003;100:9974–9. doi: 10.1073/pnas.1732638100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ray S, Atkuri KR, Deb-Basu D, et al. MYC can induce DNA breaks in vivo and in vitro independent of reactive oxygen species. Cancer Res. 2006;66:6598–605. doi: 10.1158/0008-5472.CAN-05-3115. [DOI] [PubMed] [Google Scholar]
- 36.Vafa O, Wade M, Kern S, et al. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell. 2002;9:1031–44. doi: 10.1016/s1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- 37.Beer S, Zetterberg A, Ihrie RA, et al. Developmental context determines latency of MYC-induced tumorigenesis. PLoS Biol. 2004;2:e332. doi: 10.1371/journal.pbio.0020332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Makino R, Hayashi K, Sugimura T. C-myc transcript is induced in rat liver at a very early stage of regeneration or by cycloheximide treatment. Nature. 1984;310:697–8. doi: 10.1038/310697a0. [DOI] [PubMed] [Google Scholar]
- 39.Murphy DJ, Junttila MR, Pouyet L, et al. Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell. 2008;14:447–57. doi: 10.1016/j.ccr.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murray MJ, Cunningham JM, Parada LF, Dautry F, Lebowitz P, Weinberg RA. The HL-60 transforming sequence: a ras oncogene coexisting with altered myc genes in hematopoietic tumors. Cell. 1983;33:749–57. doi: 10.1016/0092-8674(83)90017-x. [DOI] [PubMed] [Google Scholar]
- 41.Clegg NJ, Couto SS, Wongvipat J, et al. MYC cooperates with AKT in prostate tumorigenesis and alters sensitivity to mTOR inhibitors. PLoS One. 2011;6:e17449. doi: 10.1371/journal.pone.0017449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Compere SJ, Baldacci P, Sharpe AH, Thompson T, Land H, Jaenisch R. The ras and myc oncogenes cooperate in tumor induction in many tissues when introduced into midgestation mouse embryos by retroviral vectors. Proc Natl Acad Sci U S A. 1989;86:2224–8. doi: 10.1073/pnas.86.7.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DeoCampo ND, Wilson MR, Trosko JE. Cooperation of bcl-2 and myc in the neoplastic transformation of normal rat liver epithelial cells is related to the down-regulation of gap junction-mediated intercellular communication. Carcinogenesis. 2000;21:1501–6. [PubMed] [Google Scholar]
- 44.Welm AL, Kim S, Welm BE, Bishop JM. MET and MYC cooperate in mammary tumorigenesis. Proc Natl Acad Sci U S A. 2005;102:4324–9. doi: 10.1073/pnas.0500470102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jonkers J, Berns A. Retroviral insertional mutagenesis as a strategy to identify cancer genes. Biochim Biophys Acta. 1996;1287:29–57. doi: 10.1016/0304-419x(95)00020-g. [DOI] [PubMed] [Google Scholar]
- 46.Kool J, Berns A. High-throughput insertional mutagenesis screens in mice to identify oncogenic networks. Nat Rev Cancer. 2009;9:389–99. doi: 10.1038/nrc2647. [DOI] [PubMed] [Google Scholar]
- 47.Mikkers H, Allen J, Knipscheer P, et al. High-throughput retroviral tagging to identify components of specific signaling pathways in cancer. Nat Genet. 2002;32:153–9. doi: 10.1038/ng950. [DOI] [PubMed] [Google Scholar]
- 48.Mikkers H, Berns A. Retroviral insertional mutagenesis: tagging cancer pathways. Adv Cancer Res. 2003;88:53–99. doi: 10.1016/s0065-230x(03)88304-5. [DOI] [PubMed] [Google Scholar]
- 49.Mendrysa SM, Akagi K, Roayaei J, et al. An Integrated Genetic-Genomic Approach for the Identification of Novel Cancer Loci in Mice Sensitized to c-Myc-Induced Apoptosis. Genes Cancer. 2010;1:465–79. doi: 10.1177/1947601910374875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Green DR. A Myc-induced apoptosis pathway surfaces. Science. 1997;278:1246–7. doi: 10.1126/science.278.5341.1246. [DOI] [PubMed] [Google Scholar]
- 51.Schmitt CA, Lowe SW. Bcl-2 mediates chemoresistance in matched pairs of primary E(mu)-myc lymphomas in vivo. Blood Cells Mol Dis. 2001;27:206–16. doi: 10.1006/bcmd.2000.0372. [DOI] [PubMed] [Google Scholar]
- 52.Schmitt CA, McCurrach ME, de Stanchina E, Wallace-Brodeur RR, Lowe SW. INK4a/ARF mutations accelerate lymphomagenesis and promote chemoresistance by disabling p53. Genes Dev. 1999;13:2670–7. doi: 10.1101/gad.13.20.2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jacobs JJ, Scheijen B, Voncken JW, Kieboom K, Berns A, van Lohuizen M. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. 1999;13:2678–90. doi: 10.1101/gad.13.20.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zindy F, Eischen CM, Randle DH, et al. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998;12:2424–33. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–15. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- 56.Beer S, Komatsubara K, Bellovin DI, Kurobe M, Sylvester K, Felsher DW. Hepatotoxin-induced changes in the adult murine liver promote MYC-induced tumorigenesis. PLoS One. 2008;3:e2493. doi: 10.1371/journal.pone.0002493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Calvisi DF, Thorgeirsson SS. Molecular mechanisms of hepatocarcinogenesis in transgenic mouse models of liver cancer. Toxicol Pathol. 2005;33:181–4. doi: 10.1080/01926230590522095. [DOI] [PubMed] [Google Scholar]
- 58.Cavin LG, Wang F, Factor VM, et al. Transforming growth factor-alpha inhibits the intrinsic pathway of c-Myc-induced apoptosis through activation of nuclear factor-kappaB in murine hepatocellular carcinomas. Mol Cancer Res. 2005;3:403–12. doi: 10.1158/1541-7786.MCR-04-0186. [DOI] [PubMed] [Google Scholar]
- 59.Reimann M, Lee S, Loddenkemper C, et al. Tumor stroma-derived TGF-beta limits myc-driven lymphomagenesis via Suv39h1-dependent senescence. Cancer Cell. 2010;17:262–72. doi: 10.1016/j.ccr.2009.12.043. [DOI] [PubMed] [Google Scholar]
- 60.van Riggelen J, Muller J, Otto T, et al. The interaction between Myc and Miz1 is required to antagonize TGFbeta-dependent autocrine signaling during lymphoma formation and maintenance. Genes Dev. 2010;24:1281–94. doi: 10.1101/gad.585710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bowden GT, Schneider B, Domann R, Kulesz-Martin M. Oncogene activation and tumor suppressor gene inactivation during multistage mouse skin carcinogenesis. Cancer Res. 1994;54:1882s–5s. [PubMed] [Google Scholar]
- 62.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moss PS, Honeycutt N, Pawson T, Martin GS. Viral transformation of chick myogenic cells. The relationship between differentiation and the expression of the SRC gene. Exp Cell Res. 1979;123:95–105. doi: 10.1016/0014-4827(79)90425-7. [DOI] [PubMed] [Google Scholar]
- 64.Boettiger D, Soltesz R, Holtzer H, Pacifici M. Infection of chick limb bud presumptive chondroblasts by a temperature-sensitive mutant of Rous sarcoma virus and the reversible inhibition of their terminal differentiation in culture. Mol Cell Biol. 1983;3:1518–26. doi: 10.1128/mcb.3.8.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dias N, Stein CA. Antisense oligonucleotides: basic concepts and mechanisms. Mol Cancer Ther. 2002;1:347–55. [PubMed] [Google Scholar]
- 66.Maksimenko A, Malvy C. Oncogene-targeted antisense oligonucleotides for the treatment of Ewing sarcoma. Expert Opin Ther Targets. 2005;9:825–30. doi: 10.1517/14728222.9.4.825. [DOI] [PubMed] [Google Scholar]
- 67.Pastorino F, Brignole C, Marimpietri D, et al. Targeted delivery of oncogene-selective antisense oligonucleotides in neuroectodermal tumors: therapeutic implications. Ann N Y Acad Sci. 2004;1028:90–103. doi: 10.1196/annals.1322.010. [DOI] [PubMed] [Google Scholar]
- 68.Stein CA, Benimetskaya L, Mani S. Antisense strategies for oncogene inactivation. Semin Oncol. 2005;32:563–72. doi: 10.1053/j.seminoncol.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 69.Tamm I, Dorken B, Hartmann G. Antisense therapy in oncology: new hope for an old idea? Lancet. 2001;358:489–97. doi: 10.1016/S0140-6736(01)05629-X. [DOI] [PubMed] [Google Scholar]
- 70.Arvanitis C, Felsher DW. Conditionally MYC: insights from novel transgenic models. Cancer Lett. 2005;226:95–9. doi: 10.1016/j.canlet.2004.10.043. [DOI] [PubMed] [Google Scholar]
- 71.Felsher DW. Tumor dormancy: death and resurrection of cancer as seen through transgenic mouse models. Cell Cycle. 2006;5:1808–11. doi: 10.4161/cc.5.16.3111. [DOI] [PubMed] [Google Scholar]
- 72.Giuriato S, Rabin K, Fan AC, Shachaf CM, Felsher DW. Conditional animal models: a strategy to define when oncogenes will be effective targets to treat cancer. Semin Cancer Biol. 2004;14:3–11. doi: 10.1016/j.semcancer.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 73.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4:199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- 74.Jain M, Arvanitis C, Chu K, et al. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science. 2002;297:102–4. doi: 10.1126/science.1071489. [DOI] [PubMed] [Google Scholar]
- 75.Marinkovic D, Marinkovic T, Mahr B, Hess J, Wirth T. Reversible lymphomagenesis in conditionally c-MYC expressing mice. Int J Cancer. 2004;110:336–42. doi: 10.1002/ijc.20099. [DOI] [PubMed] [Google Scholar]
- 76.Pelengaris S, Khan M, Evan GI. Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell. 2002;109:321–34. doi: 10.1016/s0092-8674(02)00738-9. [DOI] [PubMed] [Google Scholar]
- 77.Karlsson A, Giuriato S, Tang F, Fung-Weier J, Levan G, Felsher DW. Genomically complex lymphomas undergo sustained tumor regression upon MYC inactivation unless they acquire novel chromosomal translocations. Blood. 2003;101:2797–803. doi: 10.1182/blood-2002-10-3091. [DOI] [PubMed] [Google Scholar]
- 78.D’Cruz CM, Gunther EJ, Boxer RB, et al. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat Med. 2001;7:235–9. doi: 10.1038/84691. [DOI] [PubMed] [Google Scholar]
- 79.Giuriato S, Ryeom S, Fan AC, et al. Sustained regression of tumors upon MYC inactivation requires p53 or thrombospondin-1 to reverse the angiogenic switch. Proc Natl Acad Sci U S A. 2006;103:16266–71. doi: 10.1073/pnas.0608017103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Choi PS, van Riggelen J, Gentles AJ, et al. Lymphomas that recur after MYC suppression continue to exhibit oncogene addiction. Proc Natl Acad Sci U S A. 2011;108:17432–7. doi: 10.1073/pnas.1107303108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Soucek L, Whitfield J, Martins CP, et al. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455:679–83. doi: 10.1038/nature07260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Soucek L, Whitfield JR, Sodir NM, et al. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013;27:504–13. doi: 10.1101/gad.205542.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Savino M, Annibali D, Carucci N, et al. The action mechanism of the Myc inhibitor termed Omomyc may give clues on how to target Myc for cancer therapy. PLoS One. 2011;6:e22284. doi: 10.1371/journal.pone.0022284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Felsher DW. Oncogene addiction versus oncogene amnesia: perhaps more than just a bad habit? Cancer Res. 2008;68:3081–6. doi: 10.1158/0008-5472.CAN-07-5832. discussion 6. [DOI] [PubMed] [Google Scholar]
- 85.Sharma SV, Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007;21:3214–31. doi: 10.1101/gad.1609907. [DOI] [PubMed] [Google Scholar]
- 86.Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science. 2002;297:63–4. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 87.Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68:3077–80. doi: 10.1158/0008-5472.CAN-07-3293. discussion 80. [DOI] [PubMed] [Google Scholar]
- 88.Boxer RB, Jang JW, Sintasath L, Chodosh LA. Lack of sustained regression of c-MYC-induced mammary adenocarcinomas following brief or prolonged MYC inactivation. Cancer Cell. 2004;6:577–86. doi: 10.1016/j.ccr.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 89.Shachaf CM, Kopelman AM, Arvanitis C, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431:1112–7. doi: 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- 90.Tran PT, Bendapudi PK, Lin HJ, et al. Survival and death signals can predict tumor response to therapy after oncogene inactivation. Sci Transl Med. 2011;3:103ra99. doi: 10.1126/scitranslmed.3002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sharma SV, Gajowniczek P, Way IP, et al. A common signaling cascade may underlie “addiction” to the Src, BCR-ABL, and EGF receptor oncogenes. Cancer Cell. 2006;10:425–35. doi: 10.1016/j.ccr.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gao P, Tchernyshyov I, Chang TC, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–5. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wise DR, DeBerardinis RJ, Mancuso A, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105:18782–7. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rooney CM, Rowe M, Wallace LE, Rickinson AB. Epstein-Barr virus-positive Burkitt’s lymphoma cells not recognized by virus-specific T-cell surveillance. Nature. 1985;317:629–31. doi: 10.1038/317629a0. [DOI] [PubMed] [Google Scholar]
- 96.Soucek L, Lawlor ER, Soto D, Shchors K, Swigart LB, Evan GI. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat Med. 2007;13:1211–8. doi: 10.1038/nm1649. [DOI] [PubMed] [Google Scholar]
- 97.Rakhra K, Bachireddy P, Zabuawala T, et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell. 2010;18:485–98. doi: 10.1016/j.ccr.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sodir NM, Swigart LB, Karnezis AN, Hanahan D, Evan GI, Soucek L. Endogenous Myc maintains the tumor microenvironment. Genes Dev. 2011;25:907–16. doi: 10.1101/gad.2038411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Restifo NP. Can antitumor immunity help to explain “oncogene addiction”? Cancer Cell. 2010;18:403–5. doi: 10.1016/j.ccr.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368:1365–6. doi: 10.1056/NEJMc1302338. [DOI] [PubMed] [Google Scholar]
- 101.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12:237–51. doi: 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wu CH, Sahoo D, Arvanitis C, Bradon N, Dill DL, Felsher DW. Combined analysis of murine and human microarrays and ChIP analysis reveals genes associated with the ability of MYC to maintain tumorigenesis. PLoS Genet. 2008;4:e1000090. doi: 10.1371/journal.pgen.1000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shachaf CM, Gentles AJ, Elchuri S, et al. Genomic and proteomic analysis reveals a threshold level of MYC required for tumor maintenance. Cancer Res. 2008;68:5132–42. doi: 10.1158/0008-5472.CAN-07-6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer. 2003;3:179–92. doi: 10.1038/nrc1015. [DOI] [PubMed] [Google Scholar]
- 105.Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 2009;15:6479–83. doi: 10.1158/1078-0432.CCR-09-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Verdine GL, Walensky LD. The challenge of drugging undruggable targets in cancer: lessons learned from targeting BCL-2 family members. Clin Cancer Res. 2007;13:7264–70. doi: 10.1158/1078-0432.CCR-07-2184. [DOI] [PubMed] [Google Scholar]
- 107.Yan C, Higgins PJ. Drugging the undruggable: transcription therapy for cancer. Biochim Biophys Acta. 2013;1835:76–85. doi: 10.1016/j.bbcan.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–17. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hermeking H. The MYC oncogene as a cancer drug target. Curr Cancer Drug Targets. 2003;3:163–75. doi: 10.2174/1568009033481949. [DOI] [PubMed] [Google Scholar]
- 111.Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Semin Cancer Biol. 2006;16:318–30. doi: 10.1016/j.semcancer.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 112.Yang D, Liu H, Goga A, Kim S, Yuneva M, Bishop JM. Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proc Natl Acad Sci U S A. 2010;107:13836–41. doi: 10.1073/pnas.1008366107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Toyoshima M, Howie HL, Imakura M, et al. Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc Natl Acad Sci U S A. 2012;109:9545–50. doi: 10.1073/pnas.1121119109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kessler JD, Kahle KT, Sun T, et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. 2012;335:348–53. doi: 10.1126/science.1212728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shachaf CM, Perez OD, Youssef S, et al. Inhibition of HMGcoA reductase by atorvastatin prevents and reverses MYC-induced lymphomagenesis. Blood. 2007;110:2674–84. doi: 10.1182/blood-2006-09-048033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cao Z, Fan-Minogue H, Bellovin DI, et al. MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG-CoA reductase. Cancer Res. 2011;71:2286–97. doi: 10.1158/0008-5472.CAN-10-3367. [DOI] [PMC free article] [PubMed] [Google Scholar]