

Abstract
Pseudomonas aeruginosa use N-(3-oxododecanoyl)-homoserine lactone (C12) as a quorum-sensing molecule to regulate gene expression in the bacteria. It is expected that in patients with chronic infections with P. aeruginosa, especially as biofilms, local [C12] will be high and, since C12 is lipid soluble, diffuse from the airways into the epithelium and underlying fibroblasts, capillary endothelia and white blood cells. Previous work showed that C12 has multiple effects in human host cells, including activation of apoptosis. The present work tested the involvement of Bak and Bax in C12-triggered apoptosis in mouse embryo fibroblasts (MEF) by comparing MEF isolated from embryos of wild type (WT) and Bax−/−/Bak−/− (DKO) mice. In WT MEF C12 rapidly triggered (mins to 2 hr): activation of caspases 3/7 and 8, depolarization of mitochondrial membrane potential (Δφmito) release of cytochrome C from mitochondria into the cytosol, blebbing of plasma membranes, shrinkage/condensation of cells and nuclei and, subsequently, cell killing. A DKO MEF line that was relatively unaffected by the Bak/Bax-dependent proapoptotic stimulants staurosporine and etoposide responded to C12 similarly to WT MEF: activation of caspase 3/7, depolarization of Δφmito and release of cytochrome C and cell death. Re-expression of Bax or Bak in DKO MEF did not alter the WT-like responses to C12 in DKO MEF. These data showed that C12 triggers novel, rapid proapoptotic Bak/Bax-independent responses that include events commonly associated with activation of both the intrinsic pathway (depolarization of Δφmito and release of cytochrome C from mitochondria into the cytosol) and the extrinsic pathway (activation of caspase 8). Unlike the proapoptotic agonists staurosporine and etoposide that release cytochrome C from mitochondria, C12’s effects do not require participation of either Bak or Bax.
INTRODUCTION
Pseudomonas aeruginosa are opportunistic bacteria that accumulate and form biofilms in the lungs of cystic fibrosis (CF) patients. P. aeruginosa uses N-(3-oxododecanoyl)-homoserine lactone (C12) as a quorum-sensing molecule to control bacterial gene expression, including production of biofilms and virulence factors (Schuster and Greenberg, 2006; Rumbaugh, 2007; Irie and Parsek, 2008). C12 elicits both pro- and anti-inflammatory effects and also proapoptotic effects in host cells that are likely to be exposed in CF lungs, including epithelial cells, leukocytes, fibroblasts and endothelial cells (Smith et al, 2001, 2002; Tateda et al, 2003; Li et al, 2004, Kravchenko et al, 2006, Jahoor et al, 2008, Jacobi et al, 2009, Li et al, 2009, Schwarzer et al, 2012). Inflammatory responses may vary depending on whether C12 was added in the absence (DiMango et al, 1995; Smith et al, 2001, 2002; Jahoor et al, 2008) or presence (Kravchenko et al, 2008) of other agonists.
The goal of the present study was to test whether Bak and Bax were involved in mediating apoptotic responses of mouse embryo fibroblasts (MEF) to C12. We used MEF for these studies for two reasons. First, fibroblasts are relevant biologically since it is expected that in patients with chronic infections with P. aeruginosa biofilms local [C12] will be high and, since C12 is lipid soluble, it will diffuse from the airways across and into the epithelium and underlying fibroblasts. Second, MEF are advantageous experimentally since MEF from wild type (WT) and Bak−/−/Bax−/− (DKO) mice are readily available, and complete knock out of these genes in airway epithelia using siRNA or similar method is extremely difficult to achieve.
Bak/Bax are commonly involved in apoptosis, particularly in the release of cytochrome C from mitochondria to the cytosol to trigger downstream activation of caspases 3/7 and 9 (Degenhardt et al, 2002; Scorrano et al, 2003; Mikhailov et al, 2003; Oakes et al, 2003; Wang et al, 2011). Schwarzer et al (2012) previously showed that C12 rapidly triggered events commonly associated with activation of apoptosis (membrane blebbing, activation of caspases 3/7, 8 and 9; depolarization of mitochondria and release of cytochrome C). Recent experiments (Valentine et al, 2013a) that were reported while work for this paper was in progress showed that C12 caused ZVAD-blocked cleavage and increased activity of caspase 3/7 and killing in MEF over 4-8 hrs. These experiments showed that these C12-triggered proapoptotic events were totally dependent on IRE1α and XBP-1 in MEF. The present experiments performed direct tests of the roles of Bak and Bax in C12-triggered apoptosis in MEF. We measured/assayed cellular and nuclear morphology, activities of caspases 3/7 and 8, cell killing and three different aspects of mitochondrial function/morphology in MEF from WT and DKO mice and also in DKO MEF that had been transduced to re-express Bak and/or Bax. The results indicated that C12 triggers multiple proapoptotic events in MEF, including release of cytochrome C from mitochondria into the cytosol, and Bak or Bax were not required for any of these effects. As far as we can tell, C12 is a unique bacterial product in that it is a membrane-permeant small molecule that triggers cytochrome C release from mitochondria without the involvement of Bak or Bax.
RESULTS
Effects of C12 on caspase 3/7 and cell morphology in WT MEF
We tested for C12-induced apoptosis in WT MEF by performing assays of caspases, membrane blebbing and cellular and nuclear shrinkage. In dose-response experiments conducted after 1 hr exposure, C12 increased caspase 3/7 activity at 10 and 50 μM in WT MEF, but not at 1 μM (Fig. 1). Control, untreated cells showed typical, flattened morphology with oval nuclei and BCECF distributed throughout the cytosol and nuclei. 50 μM C12 caused WT cells and nuclei to shrink/condense, and plasma membrane had multiple blebs containing BCECF, indicating continuity with the cytosol. These effects were very prominent by 2 hr of treatment (Fig. 2).
Fig. 1. Dose-dependent effects of C12 on caspase 3/7 in WT MEF.
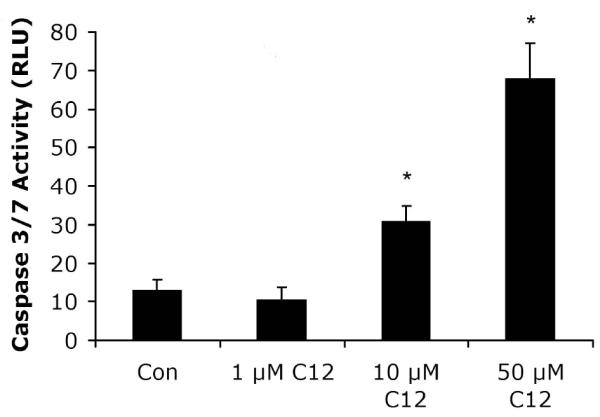
MEF were grown on 12 well plates for two days, and growth medium was changed to Ringer’s solution. C12 was added at the concentrations shown for 1 hr, followed by preparation for measurement of caspase 3/7 activities in RLU. The threshold concentration for C12-triggered activation of caspase was between 1 and 10 μM. Avg +/− SD, n = 3 experiments. * p< 0.05 for comparisons of control vs. C12.
Fig. 2. C12 causes cell and nuclear shrinkage in WT MEF.

WT MEF were observed under DIC (A, C) or fluorescence (B, D) optics at 2 hr. Fluorescence images of blue Hoechst-stained nuclei and green BCECF-stained cytosol were overlain using Adobe Photoshop. Control, untreated cells showed typical, flattened morphology with oval nuclei and BCECF distributed throughout the cytosol and nuclei. Compared to control conditions, C12-treated WT cells and nuclei were obviously shrunken, and plasma membrane had multiple blebs (arrows) containing BCECF. Images typical of >15 each in three different coverglasses each.
Effects of C12 on caspases 3/7 and 8 and cell killing in WT, DKO and Bak or Bax-corrected MEF
The roles of Bax and Bak in apoptotic responses were tested using MEF from WT and DKO mice (Wang et al, 2011). DKO MEF were also transduced with retrovirus expressing Bak or Bax as a further test of the role of Bak and Bax in the MEF responses to C12. We first tested for Bak and Bax expression in the cell lines. Western blots showed that WT MEF and the GFP-transduced MEF (used as control for transductions) and Bak- and/or Bax-corrected DKO MEF expressed Bak and Bax as predicted (Figs 3). C12 caused activation of caspases 8 and 3/7 after 2 hrs in both WT and DKO MEF (Fig. 4). Further, re-expression of Bak or Bax in DKO MEF did not change caspase 3/7 responses to C12 (Fig. 5). Staurosporine, a commonly used proapoptotic agonist, was also tested to assure that the DKO and Bak/Bax-corrected DKO MEF were behaving as predicted (Oakes et al, 2003). Staurosporine had no effect on caspase 3/7 in DKO MEF, but did activate caspase 3/7 activity in DKO+Bak and DKO+Bax MEF. We also tested whether the anti-apoptotic Bcl-2 affected C12-induced apoptosis. WT MEF were transduced with Bcl-2 using a retroviral vector (Supplementary Data Fig. S1A). However, C12-induced activation of caspase 3/7 occurred similarly in WT MEF and in WT MEF expressing either GFP (as control for transduction) or BCl-2 (Supplementary Data Fig. S2B).
Fig. 3. Expression of Bax and Bak in WT and DKO MEF.
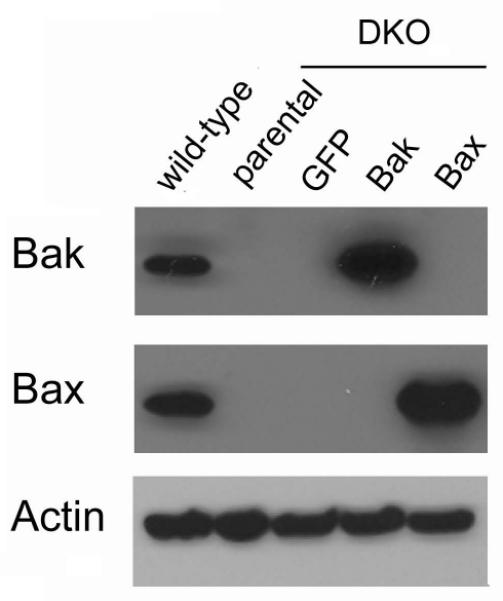
Western blots to identify Bak and Bax were performed on WT and DKO MEF transduced with GFP (served as control for transductions), Bak or Bax. Results typical of two similar.
Fig. 4. Effects of C12 on caspases 3/7 and 8 in WT and DKO MEF.

WT and DKO MEF were exposed to 50 μM C12 for 2 hrs, then prepared for assays of caspase 8 (A) and caspase 3/7 (B). C12 activated caspases 3/7 and 8 in WT and in DKO MEF. Avg +/− SD, n = 3 expts each. * p< 0.05 for comparisons between C12-treated vs. control.
Fig. 5. Effects of C12 on caspase 3/7 in WT and DKO MEF, and the (lack of) effect of Bak and Bax expression.
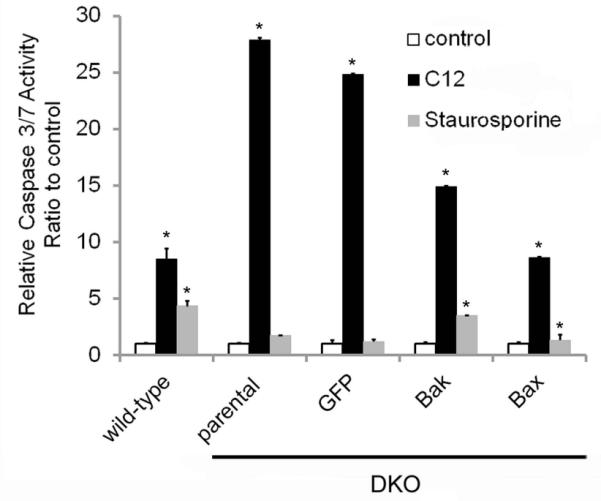
WT and DKO MEF stably expressing GFP, Bak or Bax were treated with either 50 μM C12 or 10 μM staurosporine for 2 hrs, followed by measurement of caspase 3/7 activity. C12 activated caspase 3/7 in WT and in DKO MEF, whether or not they expressed GFP or Bak or Bax. Staurosporine activated caspase 3/7 in WT and in DKO MEF that expressed Bak or Bax, but not in DKO (parental) or DKO+GFP MEF. Caspase activities in RLU were normalized in each comparison to values observed in the absence of C12 (control). Data are avg +/− SD (n = 3 expts). * p< 0.05 for comparisons between C12-treated or staurosporine-treated vs. control.
Measurements of cell death (uptake of propidium iodide, PI) after 24 hr treatment with C12 were also used to test the roles of Bak/Bax. PI responses to the common proapoptotic agonist etoposide were used to assure WT and DKO and Bak/Bax-corrected DKO MEF were behaving as predicted. Preliminary dose-response experiments showed that 50 μM C12 caused roughly 20% killing, and 100 and 150 μM C12 caused equivalent 70-75% killing of WT MEF (Zhao and Li, data not shown). We used 100 μM C12 for cell killing experiments. Consistent with results measuring caspase 3/7 activity, C12 caused PI uptake (killing) of both WT and DKO MEF, and re-expression of Bak and/or Bax in DKO MEF did not change killing responses (Fig. 6A). Etoposide caused predictable cell killing in WT MEF and in DKO MEF that expressed Bak or Bax, but much less in DKO and DKO+GFP MEF (Fig. 6B). C12 also caused roughly equivalent, dose-dependent killing of WT and Bax−/−/Bak−/− cells in another cell line, NIH3T3 MEF (Fig. S2A, B). Overall, these data showed that C12 caused activation of caspase 3/7 (Fig. 5) and cell killing (Fig. 6) in both WT and DKO MEF, and C12-triggered responses in DKO MEF’s did not increase in DKO MEF in which Bak/Bax had been re-expressed.
Fig. 6. Effects of C12 and etoposide on killing (PI uptake) in WT and DKO MEF and effects of Bak and Bax expression.

WT and DKO MEF and DKO MEF transduced with GFP, Bak, or Bax were treated with 100 μM C12 or 10 μM etoposide for 24 hrs, followed by treatment with PI and FACS analysis of PI uptake. A. C12 elicited PI uptake (compared to untreated controls) in WT and in DKO or DKO+GFP, DKO+Bak, DKO+Bax MEF. B. Etoposide increased PI uptake in WT and in DKO MEF that expressed Bak or Bax, but much less in DKO or DKO+GFP. Data in A and B are avg +/− SD (n = 3 expts). * p < 0.05 for comparisons between C12- or etoposide-treated and controls.
Testing roles of Bax and Bak in C12-induced depolarization of Δφmito and release of cytochrome C from mitochondria in MEF
Imaging of the voltage-sensitive dye JC-1 was used to test for Bak/Bax-dependent effects of C12 in mitochondria of MEF. JC1 accumulated in WT MEF in punctate patterns throughout the cytosol consistent with mitochondrial localization. Depolarization of Δφmito was assessed from the increase in fluorescence as JC1 was released from mitochondria into the cytosol and nucleus. In WT MEF C12 caused a slow increase of JC1 fluorescence over the course of 15 mins (Fig. 7A), similar to effects in airway epithelial cells (Schwarzer et al, 2012). Complete depolarization of Δφmito was triggered by FCCP (10 μM) treatment at the end of each experiment (Fig. 7A). C12 also caused depolarization of Δφmito in DKO MEF (Fig. 7B) that was very similar to that in WT MEF (Fig. 7A). Averages from these time course experiments are summarized in Fig. 8: C12 caused 70% depolarization of Δφmito (i.,e., steady state values in response to C12 compared to effects of FCCP 100%) in WT and DKO MEF.
Fig. 7. Effects of C12 on Δφmito in WT and DKO MEF.

MEF were loaded with JC1 then mounted on the imaging microscope for measurement of fluorescence under control conditions and during treatment with C12 (50 μM) and FCCP (10 μM). JC1 fluorescence showed steady value in control conditions and slow increase (equivalent to depolarization) in response to C12 in both WT (A) and DKO (B) MEF. FCCP caused maximal depolarization of Δφmito at the end of the experiments. Results typical of 3-5 experiments each.
Fig. 8. Summary: C12 on Δφmito in WT and DKO MEF.
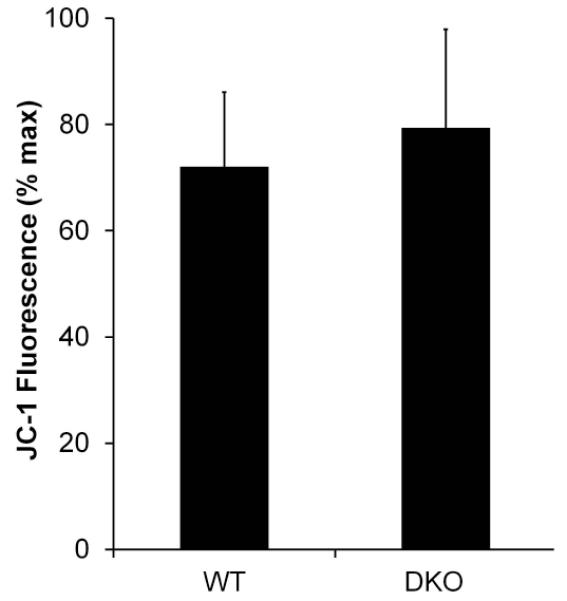
Average effects of C12 on magnitude of steady state depolarization of Δφmito (increase in fluorescence) in WT and DKO MEF. C12 caused roughly equivalent 70% depolarization of Δφmito (compared to FCCP = 100%) in both WT and DKO MEF. Averages +/− SD for 3-5 experiments. Responses of WT and DKO MEF were insignificantly different from each other (p>0.5).
We performed both immunofluorescence and ELISA assays to test whether C12’s proapoptotic effects included Bak/Bax-dependent release of cytochrome C from mitochondria into the cyotosol (Goldstein et al, 2005). Typical images of control and C12-treated MEF are shown in Fig. 9. Under control conditions WT MEF exhibited characteristic cytochrome C staining throughout the cytosol and typical oval nuclei with smooth boundaries that excluded cytochrome C. In control conditions DKO MEF exhibited less distinct and more punctate staining of cytochrome C-labeled mitochondria, consistent with the mitochondrial morphology described in previous work (Karbowski et al, 2006). Cytochrome C was largely excluded from the nuclei of both WT and DKO MEF in control conditions. In WT and DKO MEF C12 caused cytochrome C staining to become diffuse in both the cytosol and nuclei (see arrows). Nuclei of C12-treated WT and DKO MEF were often shrunken and fragmented (arrowheads). These results indicated cytochrome C had been released from mitochondria into the cytosol and had diffused into the nuclei of WT and DKO MEF.
Fig. 9. Effects of C12 on cytochrome C and nuclei in WT and DKO MEF.

WT and DKO MEF grown on coverslips were incubated with Ringer’s (control) or Ringer’s + 50 μM C12 (C12) for 1 hr followed by fixation, staining (Hoechst 33342 for nuclei and anti-cytochrome C) and confocal imaging of cytochrome C and Hoechst 33342. Arrows point to cytochrome C staining in the nuclei in C12-treated cells. Arrowheads point to nuclei that were shrunken and fragmented in C12-treated cells. Images are typical of >100 cells each in three similar experiments.
Release of cytochrome C from mitochondria into cytosol and nucleus of MEF was quantitated from these and other immunofluorescence images of cytochrome C staining. We used images like those shown in Fig. 9 to measure the ratio of intensity of cytochrome C staining in the nucleus compared to that in the cytosol of WT and DKO MEF, i.e., [(nuclear intensity – background)/cytosol intensity – background)], in control conditions and during C12 treatment. As summarized in Fig. 10, C12 caused large and roughly equivalent increases in the nuclear/cytosol distribution of cytochrome C in WT and DKO MEF. These data indicated that cytochrome C was being prominently released from mitochondria into the cytosol (and diffusion into the nuclei) of WT and DKO MEF.
Fig. 10. Summary: Quantitation of cytochrome C release from mitochondria of WT and DKO MEF during C12 treatment.
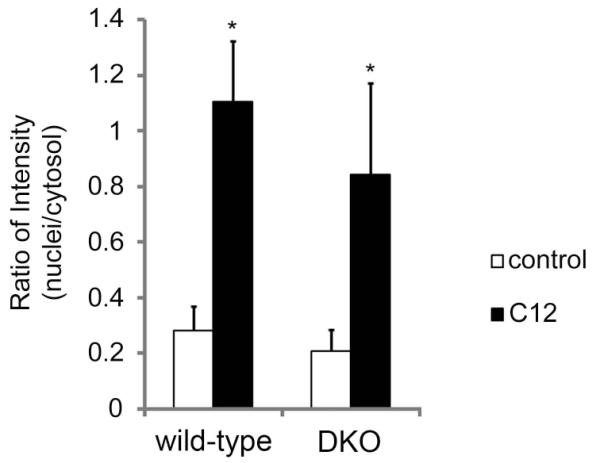
Average of nuclear/cytosol cytochrome C in control vs. C12-treated WT and DKO MEF. Compared to controls, there were C12-induced increases in nucleus/cytosol ratio for cytochrome C in WT and DKO MEF. Data are averages-/+SD for n = 10 cells from one field on one slide; *p<0.05 for comparison C12 vs. control. Results are typical for cells in 3 experiments.
ELISA assays for cytochrome C were performed on cytosolic and mitochondrial fractions from WT and DKO MEF exposed to 50 μM C12 for 1 hr. The same fractions were also analyzed for the presence of mitochondria by performing western blots with antibodies to the mitochondrial marker VDAC (voltage-dependent anion channel; Huang et al., 2013) and actin, as a loading control. C12 increased cytosol/mitochondria ratio of cytochrome C in WT and DKO MEF (Fig. 11A). Western blots showed that VDAC was present in the mitochondrial fractions but not in the cytosolic fractions of all cells under all conditions (Fig. 11B). Actin staining indicated that equal amounts of protein were loaded to each lane. Thus, western blots were consistent with the idea that the C12-induced increase in cytosol/mitochondria cytochrome C ratio in WT and DKO MEF resulted from release of cytochrome C into the cytosol and not from an artifact resulting from ineffective cell fractionation or homogenization of mitochondria. Immunomicroscopy and ELISA data were therefore consistent in showing that C12 caused cytochrome C release from mitochondria into the cytosol in WT and DKO cells.
Fig. 11. Effects of C12 on cytosol/mitochondria cytochrome C in WT and DKO MEF: ELISA assays.
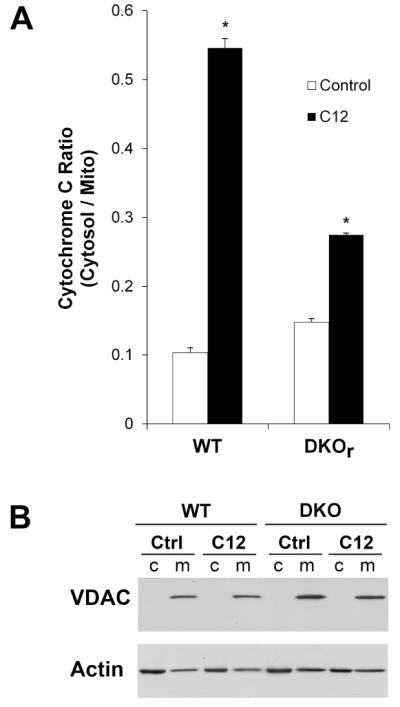
WT and DKO MEF grown on plastic dishes were incubated with Ringer’s (control) or Ringer’s + 50 μM C12 (C12) for 1 hr followed by preparation of cytosolic and mitochondrial fractions and assays for cytochrome C (by ELISA) and VDAC and actin (by western blot). A. C12 increased cytosolic/mitochondrial cytochrome C ratio for WT and DKO MEF. Average+/−SD, n = 3 separate samples. *p<0.05 for C12 vs. control. B. Western blots showed VDAC in mitochondrial but not cytosolic fractions; actin was present prominently in cytosolic and less in mitochondrial fractions. Results typical of two similar experiments.
DISCUSSION
An important conclusion from this study was that C12 rapidly triggered multiple events associated with apoptosis in MEF. C12 caused apoptosis-characteristic activation of caspases 3/7 and 8, release of cytochrome C from mitochondria into the cytosol, shrinkage of cells and nuclei and fragmentation of nuclei and blebbing of plasma membranes. A striking aspect of these effects of C12 was the rapidity of the proapoptotic responses: C12-induced depolarization of mitochondrial membrane potential began within 5 mins and was complete within 20 mins; release of cytochrome C from mitochondria into the cytosol and activation of executioner caspase 3/7 occurred within 60 mins. . In contrast, many other proapoptotic agonists require hrs or even days for apoptosis to become manifest (Goldstein et al., 2000, 2005). C12 appears also to trigger similar apoptosis in other cell types, including mast cells, macrophages, neutrophils, airway epithelial cells and mammary epithelial cells (Tateda et al, 2003; Li et al, 2004; Shiner et al, 2006; Kravchenko et al, 2006; Schwarzer et al, 2012).
C12 induced killing of WT and DKO MEF after 24 hrs. Although PI uptake could have resulted from either necrosis or apoptosis, PI uptake also occurred in response to the proapoptotic agonist etoposide, a result consistent with the idea that C12-induced PI uptake may have similarly resulted from apoptosis. Whilc C12-induced cell killing varied between 1.5-8-fold over control levels (Fig. 6), caspase activations varied between 8-25-fold activation over controls. This apparent quantitative discrepancy may result from differences in the methods. Caspase measurements were performed after 2 hrs on one set of cells while cell killing was performed after 24 hrs on cells that had different passage number and cell density, both of which could affect responses. Further tests of the quantitative relationship between C12-induced caspase activation and cell killing will require performing experiments over similar time courses on cells in which growth patterns were controlled. Despite this quantitative inconsistency in killing vs. caspase assays, the data were clear in showing that C12 induced very rapid activation of proapoptotic responses.
A second major conclusion was that C12’s proapoptotic effects occurred without the involvement of Bak or Bax. WT and DKO MEF exhibited equivalent depolarization of mitochondrial membrane potential and release of cytochrome C into the cytosol. C12 also induced equivalent activation of executioner caspase 3/7 and cell killing in WT and DKO MEF. Further, expressing Bak or Bax in DKO MEF did not increase C12’s ability to activate caspase 3/7 or to kill MEF. In contrast, effects of staurosporine and etoposide, known activators of the intrinsic pathway to apoptosis (Degenhardt et al, 2002; Mikailov et al, 2002) were dependent on Bak or Bax. Thus, staurosporine activated caspase 3/7 in WT and DKO MEF expressing Bak or Bax, but not in DKO MEF. And etoposide caused cell killing in WT and DKO MEF expressing Bak or Bax, but not in DKO MEF. Experiments on WT and DKO NIH3T3 cells (Suppl. Fig. S2) confirmed dose-dependent cell killing by C12 in WT and DKO MEF used for all the other experiments (Fig. 6A).
These results showing no involvement of proapoptotic Bak/Bax in C12-triggered apoptosis were consistent with the finding that overexpression of anti-apoptotic Bcl-2 in WT MEF also did not alter C12’s ability to activate caspase 3/7. A previous study showed that C12-triggered depolarization of Δφmito and activation of casapses 3 and 8 and cell killing was reduced in Jurkat cells that overexpressed Bcl-2 (Tateda et al, 2003), consistent with the idea that Bcl-2 and related proteins including Bak and Bax were intimately involved in C12-triggered apoptosis. Different cell lines or methods or even genes in apparently matched cell lines may explain such apparent discrepancies. Recent work (Valentine et al, 2013a) showed that C12-induced apoptosis in MEF required IRE1α and XBP1, also consistent with the idea that something besides Bak and Bax was mediating C12-induced apoptosis.
The most striking and important results of this study were that C12 caused cytochrome C release from mitochondria into the cytosol equally well in WT and DKO MEF. It is generally thought that following activation by upstream BH3-only proteins and inactivation of Bcl-2 and Bcl-xl, Bak and Bax oligomerize in the outer membrane of mitochondria and facilitate release of cytochrome C into the cytosol (Bender and Martinou, 2013). C12’s ability to release cytochrome C from mitochondria into the cytosol without the involvement of Bak and Bax is, as far as we can tell, novel for bacteria-derived products. The triterpenoid plant derivative betulinic acid causes similar release of cytochrome C from mitochondria and activation of apoptosis, an effect that appears to be mediated by the opening of the mitochondrial permeability transition pore or PTP (Mullauer et al 2009, 2010). The role of the PTP in C12-induced apoptosis should be tested.
Although it is clear that C12’s proapoptotic effects are Bak/Bax-independent, the specific molecules or pathways triggered by C12 remain unknown. C12 clearly affects mitochondria and downstream caspase 3/7, so it seems likely that C12 activates molecule(s) in the intrinsic pathway to apoptosis in MEF. In addition, C12 appears also to activate the extrinsic pathway (via caspase 8 activation), implicating a role for C12 activating a receptor like the TNF receptor or TNFR-like receptor (Spaekeert et al, 2012; de Wilt et al, 2013). Previous studies showed that C12 triggers events often associated with receptor-activation: changes in activities of Stat 3 (Li et al, 2003), PPARγ (Jahoor et al, 2008), Ca2+ and cAMP signaling (Schwarzer et al, 2010, 2012), Ca2+-triggered phosphorylation (Vikstrom et al, 2009, 2010) and activation of IRE1α, splicing of XBP1 mRNA and production of XBP1s protein (Valentine et al, 2013a). However, the roles of and connections among these potential receptors, pathways and molecules to activation of the wide array of C12-induced proapoptotic responses remain unknown. A recent study showed that C12’s effects to inhibit NF-κB activation were blocked by several triazolo[4,3-a]quinolones with nM affinities, indicating that C12 elicits at least some of its effects selectively (Valentine et al, 2013b). Further tests of these compounds on C12’s activation of proapoptotic and inflammatory responses may provide insights into whether C12’s multiple effects result from one or multiple activation steps. Although the molecular mechanisms remain to be determined, it seems possible that C12, with its high lipid solubility, activates a pattern associated molecular pattern (PAMP) receptor or damage-associated molecular pattern receptor (DAMP) (Kumar et al, 2011) in the cytosol. In lungs or other tissues infected with P. aeruginosa biofilms, where local [C12] >10 μM, epithelia, dendritic cells, white cells, fibroblasts, neurons, smooth muscle and endothelial cells, may all exhibit increased proapoptotic signaling. Such effects may contribute to the persistence of P. aeruginosa infections in CF lungs.
EXPERIMENTAL PROCEDURES
Reagents
Unless otherwise specified, reagents and chemicals were obtained from Sigma. C12 was dissolved in DMSO and frozen in separate vials and then thawed for single experiments. Preliminary experiments showed that C12 lost potency with repeated thaw-freeze-thaw cycles.
Generation and culture of WT and DKO and Bax/Bak-corrected MEF
Uncloned population of wild-type and Bak−/−Bax−/− (DKO) MEF immortalized by SV40 large T antigen expression have been described previously (Wang et al, 2011). DKO MEF were resistant to the apoptosis stimuli etoposide. Bax/Bak-corrections of DKO MEF were performed. The cDNA of murine Bak or murine Bax was subcloned into the retroviral expression vector pBABE-IRES-EGFP with the marker protein enhanced green fluorescent protein (EGFP) expressing from an internal ribosomal entry site (IRES). To generate retrovirus, the package cell line human embryonic kidney (HEK)-293T was transfected with the plasmid carrying the respective gene of interest or the empty vector along with the retroviral helper plasmids pUVMC and pMDG2.0 using the jetPRIME® transfection reagent (Polyplus-transfection; New York, NY). Medium containing retrovirus was collected 48-72 hours following transfection and supplemented with 10 μg/ml polybrene (Sigma; St. Louis, MO) to enhance infection efficiency. Bak, Bax, or the control GFP alone was expressed in DKO MEF by retroviral infection. Infected cells were again subcloned by limited dilution to identify cells that expressed high levels of GFP.
Wild-type and Bak−/−Bax−/− murine fibroblasts immortalized by the NIH3T3 spontaneous immortalization method were a gift from Dr. Wei-xing Zong (Zong et al, 2003).
All cells were cultured in DMEM/high glucose medium (Mediatech; Manassas, VA) with 10% (v/v) fetal bovine serum (Gemini; West Sacramento, CA), 100 U/ml penicillin and 100 μg/ml streptomycin (Mediatech). Cells were grown in a humidified 95/5% air/CO2 incubator at 37°C. The cells were passaged at 1:5-1:10 dilutions and the remaining cell suspension was seeded directly onto a 96-well, 24-well or 12-well tissue culture plate (BD Falcon, Bedford, MA) or onto coverglasses for imaging.
Western blot analysis of Bax and Bak
Whole cell lysates with equal number of cells (5 × 104) were loaded on a 4-12% Bis-Tris gel (Bio-Rad; Hercules, CA) and transferred onto PVDF membrane (Millipore; Billerica, MA). The membrane was incubated with appropriate primary or secondary antibodies either at 4°C overnight or at room temperature for 3 hours in blotting buffer (1 × PBS + 0.2% tween-20) with10% (w/v) nonfat dry milk (Bio-Rad). Protein levels were detected using the enhanced chemiluminescent detection system (Pierce; Rockford, IL). Antibodies used for western blot analysis were anti-β-actin mAb (Sigma), anti-Bak pAb (Enzo; Farmingdale, NY), and anti-Bax pAb (Santa Cruz; Santa Cruz, CA).
Assay for caspases 3/7 and 8
Caspase 3/7 and caspase 8 activities were measured by cell-based homogeneous luminescent assays (Caspase-Glo, Promega, Madison, WI), in which a specific substrate that contains the tetrapeptide DEVD (specific for caspase 3/7) and LETD (specific for caspase 8) was cleaved by the activated caspases from the cells to release aminoluciferin reacting with the luciferase and resulting in the production of light. WT and DKO MEF were plated on a clear-bottom, white 96-well plate in 100 μl media per well for 4-5 days until they were confluent. During the experiment, cells were treated with drugs in the 37°C incubator for 60-120 mins, depending on the experiment, or were left untreated as controls. On the same plate, some wells without cells but 100 μl of the same media served as blanks. After treatment, 100 μl reagent was added to each well with cells (treated or controls) and their media, or blank (media only). The plate was incubated at room temperature for 1 hour on a shaker, and the end-point luminescence was measured in a plate-reading luminometer (LmaxII 384, Molecular Devices, Sunnyvale, CA). Data were background (blank) subtracted and averaged.
Cell viability assay
The indicated cell lines were plated (2 × 104) in each well of a 48-well tissue culture plate and grown for 24 hours. The different cell lines were treated with various concentrations of C12 or with 10 μM etoposide for a further 24 hrs, then incubated with Trypsin-EDTA (Mediatech) followed by harvesting in the presence of 1.0 μg/ml propidium iodide (PI). Cell viability was determined by PI exclusion method using flow cytometry analysis (FACScalibur, Beckon Dickinson; San Jose, CA). Briefly, on a dot plot of forward scatter (FSC) vs FL3, the events with normal cell size (high FSC values) and low PI staining (small FL3 values) were defined as viable cells. The viability was determined as the percentage of viable events in total events, and the percentage of cell death represented 100 minus the cell viability percentage.
Cell morphology in live MEF
MEF grown on coverslips were treated in media in the incubator for one hr with the nuclear stain Hoechst 33342 (5 μM) and BCECF/AM (2 μM), which becomes cleaved and retained in the cytosol. MEF were then rinsed with Ringer’s solution and mounted in a chamber that allowed observation of cells on the stage of an inverted fluorescence microscope (Nikon T2000). Cells were then incubated with Ringer’s or Ringer’s + 50 μM C12 for 2 hrs and observed using DIC bright field and green and blue fluorescence. Blue and green images were overlain using Adobe Photoshop to permit observation of cytosol and nuclei to compare to DIC images.
Measuring Δφmito using imaging microscopy of JC1
For imaging experiments to measure mitochondrial membrane potential (Δφmito), cells were incubated with growth media containing the Δφmito probe JC-1 (10 μM) for 10 mins at room temperature, and then washed three times with Ringer’s solution to remove the extra dye. Dye-loaded cells were mounted onto a chamber on the stage of either a wide field imaging microscope or a spinning disk confocal imaging microscope. Cells were maintained at room temperature during the experiment. Treatments were made by diluting stock solutions into Ringer’s solution at the concentrations stated in the text. Fluorescence imaging measurements of Δφmito were performed using equipment and methods that have been reported previously (Fu et al., 2007; Hybiske et al., 2007; Schwarzer et al., 2008). Briefly, a Nikon Diaphot inverted microscope was used with a 40x Neofluar objective (1.4 NA). A CCD camera collected emission images (em: >510 nm for JC-1) during excitation [490± 5 nm for JC-1] using a filter wheel (Lambda-10, Sutter Instruments, Novato, CA). Axon Imaging Workbench 5.0 (Axon Instruments, Foster City, CA) controlled both light exposure and collection of data. Images were corrected for background (region without cells). Data have been reported as relative ratios. Maximum JC1 ratios were measured at the end of experiments by treating cells with 10 μM FCCP to completely depolarize Δφmito. Minimum JC1 fluorescence was defined as the starting value, and maximal depolarization was defined as fluorescence measured in presence of FCCP.
Cytochrome C, nuclear and cellular morphology in fixed and stained MEF
MEF grown on coverglasses were left untreated or incubated with C12 (50 μM) for times mentioned in the text; cells were then rinsed with PBS, fixed for 30 min with 2% paraformaldehyde in PBS at 4°C, rinsed with PBS and permeabilized with 0.2% Triton X-100 (in PBS) for 15 min. After blocking with 2% BSAPBS for 60 min, cells were incubated for 1 hr at room temperature with an anti-cytochrome C (Invitrogen, Grand Island, NY) followed with thorough rinses with PBS (+0.05% Tween-20). Finally, cells were incubated for 1 hr with an Alexa Fluor 546 anti-rabbit - or Alexa Fluor 488 anti-mouse secondary antibodies (Invitrogen, Grand Island, NY) followed by 10 min incubation with 1 μM Hoechst 33342. Fluorescence images were obtained using an inverted confocal microscope with a 63x objective (Zeiss 710). Quantitation of resulting cytochrome C distribution was performed on these images using NIH Image. Using Adobe Photoshop, we outlined images of nuclei in blue images. These outlines were then overlaid on the cytochrome C (green images). Equal-sized areas of the outlined nuclei and cytosol (area outside the nuclei) were corrected for background (area without cells) and the nuclear/cytosol ratio was calculated for each cell: (nucleus – background)/(cytosol – background). At least 10 cells in three fields from each slide were measured and averaged for quantitation of nuclear/cytosol ratio for cytochrome C.
ELISA assays of cytosolic and mitochondrial cytochrome C
ELISA assays of cytosolic cytochrome C were performed to test whether C12 altered cytosol/mitochondrial distribution of WT and DKO MEF, which were grown on plastic plates, rinsed with Ringer’s and exposed for one hr to Ringer’s alone or Ringer’s+50 μM C12. Cells were then homogenized and processed using differential centrifugation to isolate cytosolic and mitochondrial fractions as previously described (Frezza et al., 2007). Protein content of fractions was measured by Bradford and equal amounts of protein/well were assayed by Quantikine ELISA (R&D Systems,Minneapolis, MN) to quantify rat/mouse cytochrome C content. Aliquots of the same fractions were also analyzed in western blots using anti-VDAC (voltage-dependent anion channel, Huang et al, 2013) and anti-actin antibodies (Cell Signaling Technology, Danvers, MA, and MP Biomedicals, Solon, OH) to determine whether the cytosolic fractions had contamination from mitochondria that occurred during preparation of the cells and to assure equivalent protein loading into the lanes of the gel.
Statistics
Quantitative data were presented as averages +/− SD, where n = number of different biological samples or experiments. Statistical comparisons between C12-treatment and control (untreated) were determined using t-tests for paired or unpaired samples as appropriate; p<0.05 was considered statistically significant.
Supplementary Material
Fig. S1. Bcl-2 expression in WT MEF and (lack of) effect on C12-induced activation of caspase 3/7. WT MEF grown on coverglasses (A) or in plastic dishes (B) were infected with retroviruses expressing GFP or Bcl-2 + GFP. The retroviral vector DNA plasmids (MSCV-PIG) expressing GFP or Bcl2-IRES-GFP were obtained from Dr. A. Winoto (Univ. California, Berkeley). The Bcl2 vector has been described in Bassik et al, 2004. These plasmids were used to transfect Phoenix cells and produce retroviruses based on the system developed by Dr. G. Nolan (Stanford Univ., http://www.stanford.edu/group/nolan/retroviral_systems/phx.html). Briefly, Phoenix cells were plated and grown overnight, then transfected with retroviral vector and Nolan helper vectors. The culture media containing retroviruses was collected 24-48 hours post-transfection. The collected viral-containing media was filtered with 0.45 μm low protein-binding filter and kept frozen in small aliquots for future infection. WT MEF were plated and grown overnight to 50% confluence. Then the cells were infected by adding the retrovirus-containing media mixed with fresh media at 1:1 ratio. The expression of GFP and Bcl2-IRES-GFP was visualized by GFP fluorescence 24-48 hours after the infection. This Bcl-2 construct elicits efficient reduction of apoptosis in Jurkat cells (Bassik et al, 2004). A. Retroviruses expressing GFP and Bcl-2+GFP expressed in nearly 100% of cells on the coverglasses. B. Treatment of MEF with 50 μM C12 caused equivalent increases in caspase 3/7 activity in WT, GFP- and Bcl-2-infected MEF.
Fig. S2. C12-induced killing of WT and DKO NIH3T3 MEF. WT and DKO murine fibroblasts immortalized by the NIH3T3 spontaneous immortalization method (Zong et al, 2003) were used for measurements of PI uptake (cell death) or western blots to test for Bak/Bax expression. A. WT and DKO NIH3T3 cells were incubated with 10-50 μM C12 for 24 hrs then processed for PI uptake. There were no differences in cell killing between the two cell lines. B. Western blots for Bak and Bax of WT and DKO NIH3T3 cells confirmed Bak/Bax expression in WT but not DKO cells. Data are averages +/− SD, n = 3 expts in each case.
ACKNOWLEDGEMENTS
This research was funded by grants from the NIH (2PNE Y016241) (to TM) and R01 CA175003, K01 CA106599, and P20 RR018733 (to CL) and the Cystic Fibrosis Research, Inc - New Horizons (to TM). We are grateful to Russell Vance (UC Berkeley) for discussions, to Peter Haggie (UCSF) for discussions and to Horst Fischer and the Imaging Facility at Children’s Hospital Oakland Research Institute for help in using their Zeiss 710 LSM. We also think Drs Eileen White and Wei-Xing Zong for providing cell lines.
Footnotes
CONFLICT OF INTEREST The authors have no conflicts of interest.
REFERENCES
- Bender T, Martinou JC. Where killers meet--permeabilization of the outer mitochondrial membrane during apoptosis. Cold Spring Harb Perspect Biol. 2013;5:a011106. doi: 10.1101/cshperspect.a011106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 2004;23:1207–16. doi: 10.1038/sj.emboj.7600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen SB, Andersen A, Poulsen JC, Treiman M. Derivatives of thapsigargin as probes of its binding site on endoplasmic reticulum Ca2+ ATPase. Stereoselectivity and important functional groups. FEBS Lett. 1993;335:345–8. doi: 10.1016/0014-5793(93)80416-r. [DOI] [PubMed] [Google Scholar]
- DiMango E, Zar HJ, Bryan R, Prince A. Diverse Pseudomonas aeruginosa gene products stimulate respiratory epithelial cells to produce interleukin-8. J Clin Invest. 1995;96:2204–10. doi: 10.1172/JCI118275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Scorrano L. Source Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- Fu Z, Bettega K, Carroll S, Buchholz KR, Machen TE. Role of Ca2+ in responses of airway epithelia to Pseudomonas aeruginosa, flagellin, ATP, and thapsigargin. Am J Physiol Lung Cell Mol Physiol. 2007;292:L353–64. doi: 10.1152/ajplung.00042.2006. [DOI] [PubMed] [Google Scholar]
- Goldstein JC, Muñoz-Pinedo C, Ricci JE, Adams SR, Kelekar A, Schuler M, et al. Cytochrome c is released in a single step during apoptosis. Cell Death Differ. 2005;12:453–62. doi: 10.1038/sj.cdd.4401596. [DOI] [PubMed] [Google Scholar]
- Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nature Cell Biology. 2000;2:156–162. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- Huang H, Hu X, Eno CO, Zhao G, Li C, White C. An interaction between Bcl-xL and the voltage-dependent anion channel (VDAC) promotes mitochondrial Ca2+ uptake. J Biol Chem. 2013;288:19870–81. doi: 10.1074/jbc.M112.448290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hybiske K, Fu Z, Schwarzer C, Tseng J, Do J, Huang N, Machen TE. Effects of cystic fibrosis transmembrane conductance regulator and DeltaF508CFTR on inflammatory response, ER stress, and Ca2+ of airway epithelia. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1250–60. doi: 10.1152/ajplung.00231.2007. [DOI] [PubMed] [Google Scholar]
- Irie Y, Parsek MR. Quorum sensing and microbial biofilms. Curr Top Microbiol Immunol. 2008;322:67–84. doi: 10.1007/978-3-540-75418-3_4. [DOI] [PubMed] [Google Scholar]
- Jacobi CA, Schiffner F, Henkel M, Waibel M, Stork B, Daubrawa M, et al. Effects of bacterial N-acyl homoserine lactones on human Jurkat T lymphocytes-OdDHL induces apoptosis via the mitochondrial pathway. Int. J. Med. Microbiol. 2009;299:509–519. doi: 10.1016/j.ijmm.2009.03.005. [DOI] [PubMed] [Google Scholar]
- Jahoor A, Patel R, Bryan A, Do C, Krier J, Watters C, et al. Peroxisome proliferator-activated receptors mediate host cell proinflammatory responses to Pseudomonas aeruginosa autoinducer. J Bacteriol. 2008;190:4408–15. doi: 10.1128/JB.01444-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–62. doi: 10.1038/nature05111. [DOI] [PubMed] [Google Scholar]
- Kravchenko VV, Kaufmann GF, Mathison JC, Scott DA, Katz AZ, Grauer DC, et al. Modulation of gene expression via disruption of NF-kappaB signaling by a bacterial small molecule. Science. 2008;32:259–63. doi: 10.1126/science.1156499. [DOI] [PubMed] [Google Scholar]
- Kravchenko VV, Kaufmann GF, Mathison JC, Scott DA, Katz AZ, Wood MR, et al. N-(3-oxo-acyl)homoserine lactones signal cell activation through a mechanism distinct from the canonical pathogen-associated molecular pattern recognition receptor pathways. J Biol Chem. 2006;281:28822–30. doi: 10.1074/jbc.M606613200. [DOI] [PubMed] [Google Scholar]
- Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- Li H, Wang L, Ye L, Mao Y, Xie X, Xia C, et al. Influence of Pseudomonas aeruginosa quorum sensing signal molecule N-(3-oxododecanoyl) homoserine lactone on mast cells Med. Microbiol. Immunol. 2009;198:113–121. doi: 10.1007/s00430-009-0111-z. [DOI] [PubMed] [Google Scholar]
- Li L, Hooi D, Chhabra SR, Pritchard D, Shaw PE. Bacterial N-acylhomoserine lactone-induced apoptosis in breast carcinoma cells correlated with down-modulation of STAT3. Oncogene. 2004;23:4894–4902. doi: 10.1038/sj.onc.1207612. [DOI] [PubMed] [Google Scholar]
- Mikhailov V, Mikhailova M, Degenhardt K, Venkatachalam MA, White E, Saikumar P. Association of Bax and Bak homo-oligomers in mitochondria. Bax requirement for Bak reorganization and cytochrome c release. J Biol Chem. 2003;278:5367–76. doi: 10.1074/jbc.M203392200. [DOI] [PubMed] [Google Scholar]
- Mullauer FB, Kessler JH, Medema JP. Betulinic acid induces cytochrome c release and apoptosis in a Bax/Bak-independent permeability transition pore dependent fashion. Apoptosis. 2009;14:191–202. doi: 10.1007/s10495-008-0290-x. [DOI] [PubMed] [Google Scholar]
- Mullauer FB, Kessler JH, Medema JP. Betulinic acid, a natural compound with potent anticancer effects. Anticancer Drugs. 2010;21:215–27. doi: 10.1097/CAD.0b013e3283357c62. [DOI] [PubMed] [Google Scholar]
- Oakes SA, Opferman JT, Pozzan T, Korsmeyer SJ, Scorrano L. Regulation of endoplasmic reticulum Ca2+ dynamics by proapoptotic BCL-2 family members. Biochem Pharmacol. 2003;66:1335–40. doi: 10.1016/s0006-2952(03)00482-9. [DOI] [PubMed] [Google Scholar]
- Rumbaugh KP. Convergence of hormones and autoinducers at the host/pathogen interface. Anal Bioanal Chem. 2007;387:425–35. doi: 10.1007/s00216-006-0694-9. [DOI] [PubMed] [Google Scholar]
- Schuster M, Greenberg EP. A network of networks: quorum-sensing gene regulation in Pseudomonas aeruginosa. Int J Med Microbiol. 2006;296:73–81. doi: 10.1016/j.ijmm.2006.01.036. [DOI] [PubMed] [Google Scholar]
- Schwarzer C, Fu Z, Fischer H, Machen TE. Redox-independent activation of NF-kappaB by Pseudomonas aeruginosa pyocyanin in a cystic fibrosis airway epithelial cell line. J Biol Chem. 2008;283:27144–53. doi: 10.1074/jbc.M709693200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer C, Fu Z, Patanwala M, Hum L, Lopez-Guzman M, Illek B, et al. Pseudomonas aeruginosa biofilm-associated homoserine lactone C12 rapidly activates apoptosis in airway epithelia. Cell Microbiol. 2012;14:698–709. doi: 10.1111/j.1462-5822.2012.01753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer C, Wong S, Shi J, Matthes E, Illek B, Ianowski JP, et al. Pseudomonas aeruginosa Homoserine lactone activates store-operated cAMP and cystic fibrosis transmembrane regulator-dependent Cl- secretion by human airway epithelia. J Biol Chem. 2010;285:34850–63. doi: 10.1074/jbc.M110.167668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–9. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- Shiner EK, Terentyev D, Bryan A, Sennoune S, Martinez-Zaguilan R, Li G, et al. Pseudomonas aeruginosa autoinducer modulates host cell responses through calcium signaling. Cell Microbiol. 2006;8:1610. doi: 10.1111/j.1462-5822.2006.00734.x. [DOI] [PubMed] [Google Scholar]
- Smith RS, Fedyk ER, Springer TA, Mukaida N, Iglewski BH, Phipps RP. IL-8 production in human lung fibroblasts and epithelial cells activated by the Pseudomonas autoinducer N-3-oxododecanoyl homoserine lactone is transcriptionally regulated by NF-kappa B and activator protein-2. J Immunol. 2001;167:366–74. doi: 10.4049/jimmunol.167.1.366. [DOI] [PubMed] [Google Scholar]
- Smith RS, Kelly R, Iglewski BH, Phipps RP. The Pseudomonas autoinducer N-(3-oxododecanoyl) homoserine lactone induces cyclooxygenase-2 and prostaglandin E2 production in human lung fibroblasts: implications for inflammation. J Immunol. 2002;169:2636–42. doi: 10.4049/jimmunol.169.5.2636. [DOI] [PubMed] [Google Scholar]
- Speeckaert MM, Speeckaert R, Laute M, Vanholder R, Delanghe JR. Tumor necrosis factor receptors: biology and therapeutic potential in kidney diseases. Am J Nephrol. 2012;36:261–70. doi: 10.1159/000342333. [DOI] [PubMed] [Google Scholar]
- Tateda K, Ishii Y, Horikawa M, Matsumoto T, Miyairi S, Pechere JC, et al. The Pseudomonas aeruginosa autoinducer N-3-oxododecanoyl homoserine lactone accelerates apoptosis in macrophages and neutrophils. Infect Immun. 2003;71:5785–5793. doi: 10.1128/IAI.71.10.5785-5793.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine CD, Anderson MO, Feroz R, Papa FR, Haggie PM. X-Box binding protein 1 (XBP1s) is a critical determinant of Pseudomonas aeruginosa homoserine lactone-mediated apoptosis. PLoS Pathogens. 2013a;9:e1003576. doi: 10.1371/journal.ppat.1003576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine CD, Zhang H, Phuan PW, Nguyen J, Verkman AS, Haggie PM. Small molecule screen yields inhibitors of pseudomonas homoserine lactone-induced host responses. Cell Microbiol. 2013b 2013 Aug 2; doi: 10.1111/cmi.12176. doi: 10.1111/cmi.12176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikström E, Bui L, Konradsson P, Magnusson KE. The junctional integrity of epithelial cells is modulated by Pseudomonas aeruginosa quorum sensing molecule through phosphorylation-dependent mechanisms. Exp Cell Res. 2009;15:313–26. doi: 10.1016/j.yexcr.2008.10.044. [DOI] [PubMed] [Google Scholar]
- Vikström E, Bui L, Konradsson P, Magnusson KE. Role of calcium signalling and phosphorylations in disruption of the epithelial junctions by Pseudomonas aeruginosa quorum sensing molecule. Eur J Cell Biol. 2010;89:584–97. doi: 10.1016/j.ejcb.2010.03.002. [DOI] [PubMed] [Google Scholar]
- Wang X, Olberding KE, White C, Li C. Bcl-2 proteins regulate ER membrane permeability to luminal proteins during ER stress-induced apoptosis. Cell Death Differ. 2011;18:38–47. doi: 10.1038/cdd.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wilt LH, Kroon J, Jansen G, de Jong S, Peters GJ, Kruyt FA. Bortezomib and TRAIL: a perfect match for apoptotic elimination of tumour cells? Crit Rev Oncol Hematol. 2013;85:363–72. doi: 10.1016/j.critrevonc.2012.08.001. [DOI] [PubMed] [Google Scholar]
- Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J, Thompson CB. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis J. Cell Biol. 2003;162:59–69. doi: 10.1083/jcb.200302084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Bcl-2 expression in WT MEF and (lack of) effect on C12-induced activation of caspase 3/7. WT MEF grown on coverglasses (A) or in plastic dishes (B) were infected with retroviruses expressing GFP or Bcl-2 + GFP. The retroviral vector DNA plasmids (MSCV-PIG) expressing GFP or Bcl2-IRES-GFP were obtained from Dr. A. Winoto (Univ. California, Berkeley). The Bcl2 vector has been described in Bassik et al, 2004. These plasmids were used to transfect Phoenix cells and produce retroviruses based on the system developed by Dr. G. Nolan (Stanford Univ., http://www.stanford.edu/group/nolan/retroviral_systems/phx.html). Briefly, Phoenix cells were plated and grown overnight, then transfected with retroviral vector and Nolan helper vectors. The culture media containing retroviruses was collected 24-48 hours post-transfection. The collected viral-containing media was filtered with 0.45 μm low protein-binding filter and kept frozen in small aliquots for future infection. WT MEF were plated and grown overnight to 50% confluence. Then the cells were infected by adding the retrovirus-containing media mixed with fresh media at 1:1 ratio. The expression of GFP and Bcl2-IRES-GFP was visualized by GFP fluorescence 24-48 hours after the infection. This Bcl-2 construct elicits efficient reduction of apoptosis in Jurkat cells (Bassik et al, 2004). A. Retroviruses expressing GFP and Bcl-2+GFP expressed in nearly 100% of cells on the coverglasses. B. Treatment of MEF with 50 μM C12 caused equivalent increases in caspase 3/7 activity in WT, GFP- and Bcl-2-infected MEF.
Fig. S2. C12-induced killing of WT and DKO NIH3T3 MEF. WT and DKO murine fibroblasts immortalized by the NIH3T3 spontaneous immortalization method (Zong et al, 2003) were used for measurements of PI uptake (cell death) or western blots to test for Bak/Bax expression. A. WT and DKO NIH3T3 cells were incubated with 10-50 μM C12 for 24 hrs then processed for PI uptake. There were no differences in cell killing between the two cell lines. B. Western blots for Bak and Bax of WT and DKO NIH3T3 cells confirmed Bak/Bax expression in WT but not DKO cells. Data are averages +/− SD, n = 3 expts in each case.