

Abstract
Toll-like receptors (TLRs) mediate host responses to bacterial gene products. As the airway epithelium is potentially exposed to many diverse inhaled bacteria, TLRs involved in defense of the airways must be broadly responsive, available at the exposed apical surface of the cells, and highly regulated to prevent activation following trivial encounters with bacteria. We demonstrate that TLR2 is enriched in caveolin-1–associated lipid raft microdomains presented on the apical surface of airway epithelial cells after bacterial infection. These receptor complexes include myeloid differentiation protein (MyD88), interleukin-1 receptor–activated kinase-1, and TNF receptor–associated factor 6. The signaling capabilities of TLR2 are amplified through its association with the asialoganglioside gangliotetraosylceramide (Galβ1,2GalNAcβ1,4Galβ1,4Glcβ1,1Cer), which has receptor function itself for many pulmonary pathogens. Ligation of either TLR2 or asialoGM1 by ligands with specificity for either receptor, by Pseudomonas aeruginosa, or by Staphylococcus aureus stimulates IL-8 production through activation of NF-κB, as mediated by TLR2 and MyD88. Thus, TLR2 in association with asialo-glycolipids presented within the context of lipid rafts provides a broadly responsive signaling complex at the apical surfaces of airway cells to initiate the host response to potential bacterial infection.
Introduction
Epithelial cells lining the airway provide both barrier and signaling functions to protect against infection. The role of Toll-like receptors (TLRs) in signaling the presence of specific microbial components has been examined extensively in cells of hematopoietic origin, but their participation in the respiratory tract has not been well characterized (1). A fundamental property of airway epithelial cells is their polarization. In contrast to macrophages or dendritic cells, for airway cells the response to inhaled pathogens is initiated at the apical surface, and superficial stimulation, rather than invasion, is sufficient to induce cytokine and chemokine expression (2). Exactly which TLRs are available and function at the surface of airway cells is not established. The major pulmonary pathogens that cause airway inflammation and pneumonia through inhalation, organisms such as Staphylococcus aureus or Pseudomonas aeruginosa, would be expected to signal through TLR2, which responds to bacterial lipoproteins and peptidoglycan (3), and TLR4, which signals LPS (1). These TLRs and possibly others, along with co-receptors, adaptor proteins, and kinases, including myeloid differentiation protein (MyD88), interleukin-1 receptor–activated kinase-1 (IRAK-1), and TNF receptor–associated factor 6 (TRAF6), could provide a signaling cascade to mediate the epithelial response to superficial ligation of bacterial gene products (5).
A major property of TLR signaling, best illustrated for TLR4, is the participation of a receptor complex: the association of co-receptors, adaptor proteins, and signaling kinases. A simple interaction between LPS and TLR4 is not sufficient to activate NF-κB and downstream gene transcription, but requires MD2 (6) and preferably CD14 (7) and LPS-binding proteins as well as the more distal kinases (5). Less clear is what additional adaptor proteins or co-receptors are involved in amplifying TLR2-mediated responses. While there is evidence that S. aureus peptidoglycan interacts directly with TLR2 in vitro (8), the availability of TLR2 on the apical surface of airway cells has not been established, nor have co-receptors been identified.
A possible candidate for a TLR2 co-receptor is the asialoganglioside gangliotetraosylceramide (Galβ1,2GalNAcβ1,4Galβ1,4Glcβ1,1Cer) (asialoGM1). This glycolipid has an exposed GalNAcβ1-4Gal moiety that serves as a receptor for a large number of diverse pulmonary pathogens, including those commonly associated with airway infection: Streptococcus pneumoniae, S. aureus, and P. aeruginosa (9). Ligation of asialoGM1 by organisms or as mimicked by antibody initiates activation of an epithelial signaling cascade stimulating NF-κB and expression of IL-8 (2) and MUC-2 (10). Gram-positive and Gram-negative organisms, pili, and flagella have all been shown to activate epithelial signaling by ligation of asialoGM1 (11). We postulated that such an asialylated ganglioside (asialoGM1) could form a complex with a TLR, which would provide a transmembrane signaling component linking ligand recognition with downstream kinase activation.
Gangliosides, particularly GM1, are common components of lipid raft microdomains, including the specialized caveolin-1–containing lipid raft microdomains or caveolae found on airway epithelial cells (12). These lipid rafts provide a spatial compartmentalization of receptors and kinases that facilitates their interactions and the activation of downstream responses. The colocalization of gangliosides, caveolin-1, and signaling kinases in caveolae has been described in many cell types as mediating signal transduction (13). However, the presentation of TLRs within the context of caveolae has not been previously implicated in the host defense against respiratory tract infection. In the studies detailed herein, we demonstrate that TLR2 and asialoGM1 are presented on the apical surfaces of airway epithelial cells and are enriched in lipid raft microdomains after bacterial stimulation, providing a broadly responsive receptor complex to initiate host defense of the airways.
Results
TLR2 but not TLR4 is presented on the apical surfaces of airway epithelial cells.
The distribution of the major TLRs TLR2 and TLR4 was examined using confocal microscopy in 16HBE human airway cells grown in a polarized fashion at an air-liquid interface with intact tight junctions. TLR2 was found in an apical distribution quite distinct from that of TLR4, which was more diffusely distributed (Figure 1A). AsialoGM1 in these terminally differentiated cells was also apical, and asialoGM1 colocalized with TLR2 but not with TLR4. To establish that these cell lines accurately reflect the properties of cells in vivo, we also imaged the localization of TLR2 and TLR4 in epithelial cells in primary culture and found TLR2 colocalization with asialoGM1 (Figure 1B). Because TLR2 is glycosylated, we confirmed the specificity of the antibodies by using specific blocking peptides and demonstrating that anti-asialoGM1 did not recognize TLR2 when TLR2 was overexpressed in CHO cells (data not shown).
Figure 1.

Distribution of asialoGM1, TLR2, and TLR4 in infected airway cells. (A) Confocal microscopy was used to image monolayers of polarized 16HBE cells transfected with Flag epitope–tagged TLR2 or TLR4 and stimulated with P. aeruginosa PAO1. z-sections treated with anti-Flag labeled with TRITC (red) and with anti-asialoGM1 labeled with FITC (green) are shown. AsialoGM1 (aGM1) is apical and colocalizes with TLR2 (yellow) in discrete clusters along the apical surface of the monolayers. TLR4 is more diffuse, and colocalization with asialoGM1 is not appreciable. (B) Human airway cells in primary culture isolated from nasal polyps from a CF patient and stimulated with P. aeruginosa PAO1 were stained for asialoGM1 labeled with TRITC and TLR2 labeled with FITC and show abundant colocalization of these receptors.
More conclusive evidence that TLR2 is available on exposed, apical surfaces of airway epithelial cells was obtained in biotinylation experiments. Polarized 16HBE cells, both unstimulated and after exposure to S. aureus, were biotinylated and, after immunoprecipitation with streptavidin, Western hybridizations were performed to screen for components of the TLR signaling cascade (Figure 2A). TLR2 as well as the kinase TRAF6, which is involved in TLR-mediated responses, were present, as well as asialoGM1. Caveolin-1, the scaffolding protein that is often a component of signaling complexes associated with membrane lipids, was also present (14). Increased amounts of TLR2, caveolin-1, and especially IRAK-1 were surface biotinylated after bacterial stimulation.
Figure 2.
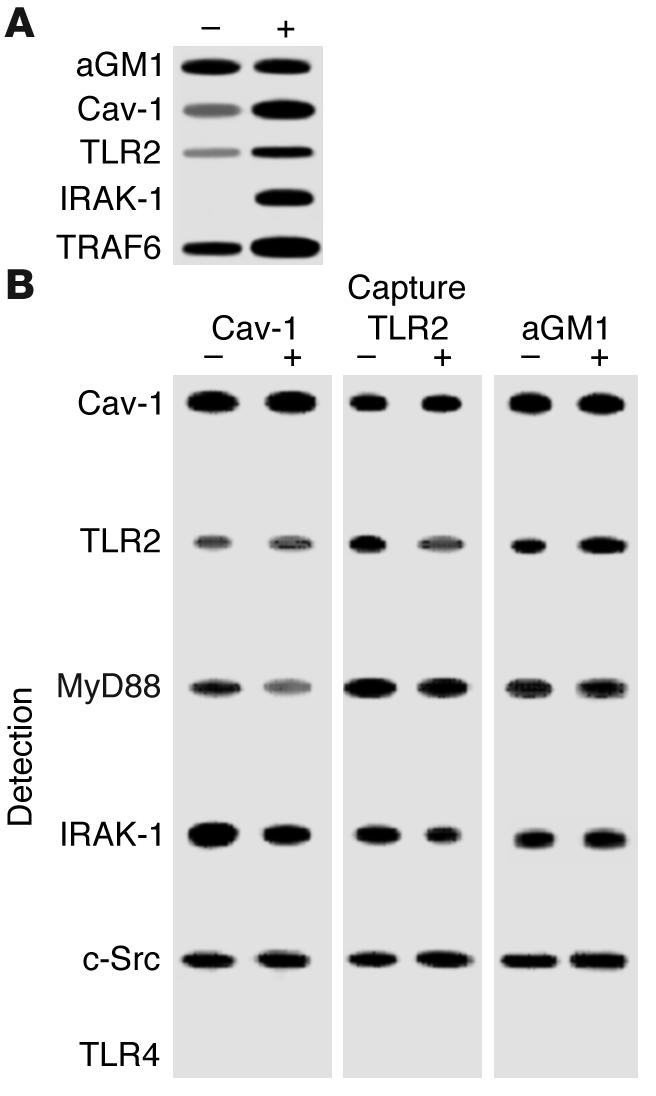
Identification of apically exposed components on polarized 16HBE cells exposed to bacteria. (A) Surface-exposed components of the airway cells were biotinylated under control conditions (–) and after a 1-hour exposure to S. aureus (+). After immunoprecipitation with streptavidin, Western hybridizations were done and asialoGM1, TLR2, and caveolin-1 (Cav-1), as well as the kinases IRAK-1 and TRAF6, were detected. (B) Coimmunoprecipitation studies demonstrate TLR2 but not TLR4 in a receptor complex along with asialoGM1. Coimmunoprecipitations of whole-cell lysates from control and S. aureus–stimulated cells were done using anti–caveolin-1, anti-TLR2, and anti-asialoGM1 as capture antibodies with screening for expected components of the TLR pathway, MyD88 and IRAK-1, as well as c-Src and TLR4.
A physical association between the receptors and kinases was further established by coimmunoprecipitation studies (Figure 2B). A lysate of 16HBE cells was immunoprecipitated with each of the capture antibodies indicated, including anti-asialoGM1, confirming the observed association of TLR2, caveolin-1, and IRAK-1. In addition, we identified MyD88 and c-Src in this complex. It is of note that caveolin-1 has a binding domain for c-Src, one of the kinases that is activated in asialoGM1-mediated signaling (4, 12), consistent with its close connection with the glycolipid receptor. TLR4 was not associated with asialoGM1. The colocalization of MyD88, IRAK-1, TRAF6, and TLR2 at the apical surface of the airway cells was demonstrated by confocal microscopy (Figure 3).
Figure 3.

Surface colocalization of MyD88, IRAK-1, and TRAF6 with TLR2 confirmed by confocal microscopy. After stimulation with S. aureus, permeabilized cells were stained with the kinases, each labeled with an Alexa Fluor 488–tagged secondary antibody (green). All were found at the cell surface, colocalized (yellow) with TLR2, labeled with an Alexa Fluor 594–tagged secondary antibody (red).
TLR2 is mobilized into lipid rafts after bacterial exposure.
The association of a ganglioside, caveolin-1, and signaling kinases has been well described in many signaling systems (13). The distribution of both caveolin-1 and GM1 on the apical surface of the polarized airway cells was visualized by confocal microscopy (Figure 4A). When associated with plasma membranes, these components often are compartmentalized into discrete structures called caveolae (14). Caveolae have been demonstrated in pulmonary cells (15, 16), although they have not previously been associated with bacterial infection. Airway epithelial monolayers incubated with P. aeruginosa were examined by electron microscopy for evidence of such structures. The flask-like structure typical of caveolae was readily apparent in the infected monolayers (Figure 4B). While the intact organisms are clearly too large to interact with the caveolae themselves, flagellin (55-kDa subunits) or pilin (15-kDa subunits), both of which bind to the asialoganglioside asialoGM1 (17, 18), could be readily accommodated by these structures. By flow cytometry, an increase in superficial caveolin-1 was readily detected in polarized cells following bacterial exposure (Figure 4C).
Figure 4.

Lipid rafts are involved in clustering of receptors and signaling. (A) Confocal z-section images demonstrate caveolin-1 labeled with Alexa Fluor 594 (red) and GM1, identified with cholera toxin β-subunit (CTB) conjugated to Alexa Fluor 488 (green), on the apical surfaces of polarized 16HBE cells permeabilized after stimulation with Pam3Cys-Ser-Lys4. (B) CF nasal polyp cells were infected with P. aeruginosa PAO1 (PA) and grown on semipermeable supports, and transmission electron micrograph were obtained after 3 hours of bacterial exposure. Arrow (Cav) indicates the flask-shaped electron-dense structures typical of caveolae (magnification, ∞30,000). (C) Flow cytometry was used to detect superficial caveolin-1 on polarized 16HBE cells after exposure to S. aureus. Unstim, unstimulated. (D) Aliquots of Triton-insoluble lysates of 16HBE cells obtained before (–) and after (+) stimulation with P. aeruginosa PAO1 were fractionated on discontinuous sucrose gradients (4–40%) and were immunoblotted with anti-flotillin, anti-caveolin, anti-TLR-2, anti-IRAK-1, or anti-asialoGM1. Downward arrow indicates raft fraction containing all the components after stimulation. (E) Aliquots of sucrose gradient fractions from cells treated with filipin prior to stimulation with P. aeruginosa PAO1 were immunoblotted with anti-flotillin, anti-TLR2, and anti-asialoGM1.
Biochemical evidence of lipid rafts in airway epithelial cells was obtained by the isolation of Triton-insoluble material from 16HBE cells before and after exposure to bacteria (19). This material was then fractionated on a discontinuous sucrose gradient and aliquots were screened for components of the receptor-signaling complex (Figure 4D). Flotillin was used as a marker for raft fractions (20). Caveolin-1 was distributed throughout the fractions, including those expected to represent lipid rafts. TLR2 was specifically mobilized into the raft fraction in cells stimulated with bacteria, as was IRAK-1 and asialoGM1. To confirm the compartmentalization of TLR2 into lipid rafts, we treated cells with filipin, which intercalates into lipid moieties disrupting lipid raft structures (21). Sucrose gradient fractions were similarly screened for TLR2, asialoGM1, and flotillin (Figure 4E). In filipin-treated cells, the partitioning of both flotillin and TLR2 in the sucrose fractions expected to include lipid rafts was lost. Instead, they were distributed throughout the Triton-insoluble material. AsialoGM1, as a lipid, was dispersed by the filipin treatment.
Given that superficial TLR2 has an active role in signaling from an apical lipid-raft complex, we postulated that both TLR2 and asialoGM1 would be co-mobilized to the cell surface in response to ligands with specificity for either receptor. There is a limited amount of surface asialoGM1 on normal human airway cells (18). Using flow cytometry, we quantified superficial asialoGM1 and TLR2 on human nasal polyp (HNP) airway cells before and after exposure to bacteria (Figure 5). In response to P. aeruginosa or S. aureus, there was a substantial increase in asialoGM1 as well as TLR2 on epithelial cells in primary culture (Figure 5A). Mobilization of these receptors in 16HBE cells was less substantial but was evident. No changes were detected in surface-accessible TLR4. We postulated that there may be reciprocal mobilization of both asialoGM1 and TLR2 induced by ligands with specificity for either receptor. Pretreatment of airway cells with monoclonal anti-asialoGM1 also resulted in increased surface expression of TLR2 and asialoGM1. Much greater mobilization of both asialoGM1 and TLR2 was observed after stimulation with the TLR2 agonist Pam3Cys-Ser-Lys4. There was no increase in superficial TLR4 in response to any of the ligands tested, including LPS (Figure 5B).
Figure 5.

Mobilization of TLR2 and asialoGM1 in response to bacteria or bacterial ligands. Flow cytometry was used to quantify exposed asialoGM1, TLR2, and TLR4 on primary (HNP) cells (A) or 16HBE cells (B) after stimulation with P. aeruginosa or S. aureus or with monoclonal anti-asialoGM1, Pam3Cys-Ser-Lys4 (Pam3Cys), a TLR2 ligand, or LPS, a TLR4 ligand. Peaks outlined with a thin black line indicate binding by secondary antibody alone; gray-shaded peaks represent the labeled population under control conditions; and peaks demarcated by the heavy black line represent the population after stimulation.
AsialoGM1-associated ligands signal through TLR2.
The availability of TLR2 on the apical surface of the airway epithelium suggests that TLR2-associated ligands released from lysed organisms, such as lipoproteins or peptidoglycan fragments, could readily activate the epithelial cells via interactions with caveolae. However, the presence of asialoGM1 in caveolae provides an additional receptor for bacterial components, including pili and flagella, that potently activate epithelial NF-κB and proinflammatory gene expression. The association of asialoGM1 and TLR2 receptors, along with the accessory proteins and kinases, suggested that ligands that recognize asialoGM1 and activate NF-κB or IL-8 expression do so through the TLR components MyD88 and TRAF6 (22). IL-8 expression induced by ligands with specificity for asialoGM1 should be inhibited by dominant negative (DN) mutations that interfere with TLR signaling. In human airway cells expressing a DN form of TLR2, activation of IL-8 by P. aeruginosa, S. aureus, or anti-asialoGM1 was inhibited by 48%, 49%, and 59%, respectively, compared with that of cells transfected with a vector expressing a wild-type TLR2 plasmid construct (P < 0.001 for each) (Figure 6A). Activation of an NF-κB–luciferase reporter in cells transfected with plasmids expressing TLR2, MyD88, or TLR4 DN mutations was similarly compared with that of cells transfected with an empty vector control plasmid (Figure 6B). Activation mediated by P. aeruginosa, S. aureus, anti-asialoGM1, or the TLR2 agonist Pam3Cys-Ser-Lys4 was significantly inhibited by the TLR2 DN and the MyD88 DN mutants (P < 0.001). A TLR4 DN mutation had no effect on the activation of NF-κB activity, and LPS (alone or in the presence of serum) failed to stimulate sufficiently to test the effects of the DN mutations (data not shown).
Figure 6.

Activation of NF-κB and IL-8 by bacteria or bacterial agonists is inhibited by DN mutations in TLR2 and MyD88. (A) IL-8 expression in 1HAEo- cells transfected with plasmids containing wild-type, DN TLR2, or a vector control was quantified by ELISA after exposure to P. aeruginosa PAO1, S. aureus RN6390, or anti-asialoGM1. Values represent the fold increase in IL-8 compared with that of unstimulated cells. IL-8 in cells stimulated by PAO1 was 1.9–2.5 ng/ml (*P < 0.001). (B) NF-κB luciferase activity in 1HAEo- cells transfected with plasmids expressing TLR2 DN or MyD88 DN constructs compared with that of cells transfected with the corresponding empty vector control was significantly inhibited after stimulation with P. aeruginosa PAO1, S. aureus RN6390, anti-asialoGM1, or Pam3Cys-Ser-Lys4 (*P < 0.001 for each). The TLR4 DN construct did not inhibit NF-κB luciferase activity. NF-κB luciferase activity for cells expressing the control vector was normalized for each stimulus and represents three- to fivefold increases over that of unstimulated cells. (C) Inhibition of IL-8 activation in the presence of filipin. IL-8 production induced by P. aeruginosa, S. aureus, anti-asialoGM1 (aGM1), or Pam3Cys-Ser-Lys4, but not TNF-α, was significantly reduced by filipin (P < 0.05, P < 0.001, P < 0.05, and P > 0.05, respectively).
We confirmed the involvement of the lipid raft structure in signaling by testing the effects of filipin, which intercalates into lipid moieties and interrupts ganglioside-dependent signaling (21). Bacterial activation of IL-8, as stimulated by P. aeruginosa, S. aureus, Pam3Cys-Ser-Lys4, or anti-asialoGM1, was significantly inhibited in the presence of filipin compared with that of untreated controls (P < 0.05, P < 0.001, P < 0.05, and P < 0.05, respectively). In contrast, expression of IL-8 induced by TNF-α was not significantly inhibited (Figure 6C). Nystatin and methyl-β-cyclodextrin had similar effects (data not shown).
Discussion
Airway epithelial cells are an important component of mucosal defenses, expressing chemokines and cytokines to direct the influx and activation of phagocytic cells in response to bacterial infection. Participation of the various TLRs in mediating these epithelial responses to inhaled bacteria is expected, although the details of their distribution and function in polarized cells with tight junctions has not been established previously. As the airways are often exposed to diverse organisms, the capability to respond to both Gram-negative and Gram-positive pathogens is critical. However, activation of epithelial signaling and recruitment of polymorphonuclear leukocytes is not innocuous to the host. Inflammation is clearly detrimental to the major physiological function of the airway epithelium in maintaining an open conduit for gas exchange. The airway surface is normally well protected by mucins, antimicrobial peptides, and other components of the innate defenses. Any bacteria or bacterial components that elude these defenses and reach the epithelial surface promptly activate epithelial proinflammatory signaling. Our data suggest that a receptor complex at the apical surface of airway cells initiates mobilization of signaling components as well as additional receptors in response to potential infection. Thus, the airway epithelium differs appreciably from the gastrointestinal mucosal, which tolerates chronic exposure to bacteria and is generally activated only in response to invasive pathogens and through different TLRs (23).
TLR2 seems to be especially important in airway signaling. TLR2 is a broadly responsive “pathogen-associated pattern-recognition receptor” activated by many different bacterial species (5). Our data indicate TLR2 signals both S. aureus and P. aeruginosa as well as isolated ligands such as Pam3Cys-Ser-Lys4 expected to interact directly with it. Although small amounts of TLR2 are present on the apical surface of polarized cells with tight junctions, TLR2 was promptly mobilized to the surface of the epithelial cells along with the asialoganglioside asialoGM1 after stimulation. The participation of TLR2 in epithelial defenses appears to be significantly enhanced by this association with asialoGM1, which has receptor function as well as providing a major structural component of the specialized lipid raft domains on the airway epithelium. By mobilizing TLR2 into specialized lipid raft microdomains, the recognition capabilities of the receptor complex are enhanced to include ligands with specificity for either the GalNAcβ1-4Gal moiety of the asialoganglioside or TLR2. In addition, caveolin-1 contributes its scaffolding function to bring the receptors and associated kinases in physical contiguity. The apical distribution of caveolin-1 and presence of caveolae in these 16HBE cells is similar to that of previous reports using this (16) and other airway epithelial cell lines (15, 24, 25). Biotinylation and coimmunoprecipitation experiments indicate that the major TLR-associated kinases and c-Src are made available within this apical receptor complex.
Signaling capabilities have also been associated with ligation of asialoGM1 (10, 26). Functioning as a receptor for many pulmonary pathogens (9), as well as for bacterial pili (2) and flagella (17), asialoGM1 activates MAPKs through Ras and Src to initiate NF-κB activation and IL-8 transcription or to stimulate MUC2 transcription (10). AsialoGM1 also serves as a receptor for the major Gram-positive pathogens S. pneumoniae (27) and S. aureus, which similarly evoke epithelial IL-8 expression (28). Particularly for these Gram-positive bacteria, not only do intact organisms bind asialoGM1, but they also shed multiple TLR2 agonists such as lipoteichoic acid and peptidoglycan to initiate proinflammatory responses (3). The recruitment of this broadly responsive receptor complex to the apical compartment of the airway cell provides a central transducer for diverse stimuli and multiple host responses. Activation of these downstream pathways is TLR2 dependent, as we demonstrated that asialoGM1-initiated activation of NF-κB is both TLR2 and MyD88 dependent. Although there is reciprocal mobilization of the asialoganglioside and TLR2 in response to agonists for either receptor, it is unclear if TLR2-mediated signaling is dependent upon the presence of the glycolipid. In vitro studies indicate that S. aureus peptidoglycan can interact directly with TLR2 (8). The mobilization of TLR2 into lipid rafts appears to be cholesterol dependent, as its localization to specific lipid raft fractions is lost in cells treated with filipin. A requirement for a lipid raft microdomain in TLR2 signaling is suggested by the effects of filipin in inhibiting activation of IL-8 expression by Pam3Cys-Ser-Lys4 but requires further experimental evidence. Instead, we propose that asialoGM1 provides, at a minimum, a co-receptor function that amplifies the signaling capabilities of TLR2.
Not only was TLR2 found to be associated with lipid raft microdomains, but it also appears to be presented within the more specialized lipid raft microdomains associated with caveolin-1. Caveolin-1 is present in membranes throughout the cell. Even in the absence of caveolae, the electron-dense structures observed by electron microscopy, caveolin-1 participates in lipid raft organization and signaling (13). Caveolae have been implicated previously in the pathogenesis of infection, mediating the internalization of viruses (29), bacterial pili (30), and toxins (21). A more general role for caveolae in providing the apical signaling complex that initiates host defenses in response to common respiratory pathogens has not been demonstrated previously. The importance of caveolin-1 in immune defenses was implied by the prominent pulmonary pathology exhibited by caveolin-1–null mice (31). The data presented herein suggest an explanation for these observations, as caveolae in the airways provide the compartmentalization necessary for TLR2 and associated kinases to efficiently signal bacterial infection.
In contrast to the primary role of TLR2 in airway epithelial signaling, TLR4 seems less involved, even in response to Gram-negative organisms such as P. aeruginosa or LPS by itself. TLR4 was abundant in the airway epithelial cells, but did not appear to function in signaling. Airway cells are poorly responsive to LPS, even when all of the required co-receptors and LPS-binding proteins are provided (11, 32). TLR4, in contrast to TLR2, was not found in association with caveolae at the apical surfaces of the cells; TLR4 was not present in the biotinylated complex, was not mobilized by either P. aeruginosa LPS or intact bacteria, and did not coimmunoprecipitate with caveolin-1 or asialoGM1. These observations were somewhat unexpected, as the presence of TLR4 in lipid rafts microdomains has been demonstrated previously in CHO cells, along with CD14 and heat shock proteins (33). Those studies clearly document the association of TLR4 and GM1 as well as a requirement for this association in LPS signaling. The pulmonary response to LPS provided systemically (rather than by inhalation) has been shown recently to be mediated by TLR4 expressed by pulmonary endothelial cells, not by cells of hematopoietic origin (34). Mice have profound immune responses to inhaled LPS, which presumably are initiated by alveolar macrophages. Thus, there is clearly a highly tissue-specific distribution and function of TLRs that is especially important in the lung.
Certain TLRs appear to have distinct functions in different tissues. In the airway epithelium, the signaling of infection is initiated at the apical surfaces of these polarized cells exposed to bacteria or their products. Activation of epithelial proinflammatory responses, whether stimulated by S. aureus or P. aeruginosa, appears to be handled predominantly by TLR2 presented at the surfaces of airway cells within the context of an asialoganglioside/lipid raft microdomain or caveolae. In diseases such as cystic fibrosis (CF), in which there is a CF transmembrane conductance regulator–associated increase in asialylated glycolipids such as asialoGM1 (35–37), there is likely to be enhanced TLR2 signaling, as suggested by the excessive epithelial IL-8 expression characteristic of CF epithelial cells (38–40). Strategies to ameliorate excessive proinflammatory responses to bacterial airway infection should include consideration of the role of TLR2 and asialoGM1-mediated signaling, even in the context of a Gram-negative infection.
Methods
Cell lines and reagents.
The 16HBE and 1HAEo- cells, human airway epithelial cell lines (D. Gruenert, University of Vermont, Burlington, Vermont, USA), were grown as detailed previously (26, 41). The 16HBE cells were grown on Transwell-Clear inserts (Corning Inc., Acton, New York, USA) in a polarized fashion with an air-liquid interface. Their epithelial properties, trans-epithelial resistance (800–1,200 ohms), and tight junctions have been established previously (41). They do not form cilia. For signaling studies, 1HAEo- cells were used. Although these cells do not form tight junctions, they have minimal endogenous proinflammatory activity. Primary airway epithelial cells isolated from HNPs were also grown on Transwell-Clear inserts as described previously (11). Rabbit polyclonal antibodies used were anti–caveolin-1, anti-TLR2, anti–IRAK-1, anti-MYD88, anti-TRAF6, and anti-TLR4 (Santa Cruz Biotechnology Inc., Santa Cruz, California, USA); anti-TLR2 antisera from Tularik (South San Francisco, California, USA); and anti-asialoGM1 (Wako, Richmond, Virginia, USA). Monoclonal antibodies used were anti-TLR2 (Imgenex, San Diego, California, USA), anti–c-Src (Santa Cruz Biotechnology Inc.), and anti-flotillin (BD Biosciences, San Diego, California, USA).
Confocal microscopy and immunofluorescence studies.
The 16HBE or human airway cells in primary culture harvested from HNPs were grown to confluence on Transwell-Clear filters (Corning-Costar) with an air-liquid interface to form polarized monolayers. Cells were fixed with 4% paraformaldehyde for 15 minutes at room temperature and were incubated with 5% normal serum blocking solution for 20 minutes at room temperature. Primary antibodies were added for 1 hour at room temperature followed by three 5-minute washes. Alexa Fluor 488– and Alexa Fluor 594–conjugated secondary antibodies (Molecular Probes, Eugene, Oregon, USA) were added for 1 hour and cells were washed three times. Membrane supports were removed from Transwell inserts using a scalpel and were mounted with Vectashield (Vector Laboratories Inc., Burlingame, California, USA) onto glass slides.
Biotinylation of cell surface proteins.
The 16HBE cells were grown in a polarized fashion with an air-liquid interface on Transwells (Corning-Costar) and were stimulated apically with 1 ∞ 108 S. aureus RN6390 for 1 hour, then were washed three times with cold PBS with Ca++/Mg++. A freshly prepared solution of 1 mg/ml EZ-Link Sulfo-NHS-LC-Biotin (Pierce, Rockford, Illinois, USA) in PBS with Ca++/Mg++ was added to the apical surface and cells were incubated on ice for 30 minutes. The following washes were performed to remove and “quench” the biotin: twice with PBS with Ca++/Mg++, once with 50 mM NH4Cl, and once with PBS with 1% BSA. Biotinylated cells were lysed on ice for 30 minutes using 60 mM n-octyl β-D-glucopyranoside in TBS (0.1 M Tris-HCl and 0.15 M NaCl, pH 7.8) with fresh protease inhibitors added. Lysates were harvested and incubated on ice another 30 minutes. After a 1-minute clearing spin at 10,000 g, the supernatant was collected and frozen. Protein concentrations were determined using the Micro BCA Protein Assay kit from Pierce, and a 2-mg aliquot was incubated with 60 ∝l of Immunopure Immobilized Streptavidin (Pierce) at 4°C overnight with gentle shaking. After three washes with lysis buffer, the agarose pellet was resuspended in 20 ∝l of sample dye buffer and the proteins in a 10-∝l sample were separated on a 4–12% Bis-Tris NUPAGE gel in MES buffer (Invitrogen, Carlsbad, California, USA) and were transferred to Immobilon-P (Millipore, Bedford, Massachusetts, USA) for Western hybridization.
Coimmunoprecipitation studies.
The 16HBE cells grown to confluence in 10-cm dishes were lysed for 30 minutes on ice with 60 mM n-octyl β-D-glucopyranoside in TBS (0.1 M Tris-HCl and 0.15 M NaCl, pH 7.8) with protease inhibitors. Lysed cells were harvested by scraping and were incubated on ice for an additional 30 minutes with occasional mixing. For pelleting of debris, tubes were centrifuged at 4°C for 10 minutes at 10,000 g. Supernatants were subjected to magnetic immunoprecipitation and separation using ∝MACS Protein A MicroBeads (Miltenyi Biotec, Auburn, California, USA) following the manufacturer’s directions. Briefly, 5 ∝l of anti-asialoGM1, anti-TLR2 (Santa Cruz Biotechnology Inc.), or anti–caveolin-1 was added to 300 ∝l of supernatant with 50 ∝l of Protein A Microbeads, and the sample was incubated on ice for 30 minutes. Immunolabeled lysate was applied to the ∝MACS separation column and a 20-mM Tris (low-salt) buffer was used. The eluted immunoprecipitate was analyzed by Western hybridization.
IL-8 assays.
Confluent monolayers of 1HAEo- cells “weaned” from serum for 24 hours were exposed to P. aeruginosa PAO1, S. aureus RN6390, Pam3Cys-Ser-Lys4, anti-asialoGM1, or TNF-α for 60 minutes with and without pretreatment for 30 minutes with filipin (0.5 ∝g/ml) (Sigma-Aldrich, St. Louis, Missouri, USA). Cells were washed and sterilized with gentamicin (100 ∝g/ml) and supernatants were harvested 18 hours later for IL-8 ELISA (R&D, Minneapolis, Minnesota, USA). For experiments utilizing expression constructs, cells were transfected with an empty vector control or with a TLR2 wild-type or mutant TLR2 DN expression vector (gifts from Jian Dong Li, House Ear Institute, Los Angeles, California, USA) using FuGENE 6.0 (Roche, Indianapolis, Indiana, USA) and were stimulated 18 hours later. Duplicate wells were treated with trypan blue for assessment of epithelial viability during the assay, which was more than 75%. Assays using S. aureus were done in the presence of 0.1% fetal calf serum. Each IL-8 data point was determined in quintuplicate and a mean and standard deviation were calculated. Statistical significance was determined using a one-way analysis of variance with Dunnett’s post test (GraphPad InStat version 3.0; GraphPad Software Inc., San Diego, California, USA) to test the null hypothesis that there was no difference in the amount of the outcome variable (IL-8 production) under each test condition compared with the untreated control.
Activation of NF-κB detected by luciferase reporter assay.
1HAEo- cells grown in 12-well plates to 70–80% confluence were transiently transfected using FuGENE 6.0 (Roche), pNF-κB-luc (Stratagene, La Jolla, California, USA), and TLR2 DN, MyD88 DN, or TLR4 DN expression vector or empty vector control (Jian Dong Li). After 24 hours of incubation, cells were stimulated with LPS for 20 hours or with S. aureus RN6390, P. aeruginosa PAO1, or Pam3Cys-Ser-Lys4 for 1 hour (in the presence of 0.1% serum). Luciferase assays were performed using the Dual-Luciferase Reporter Assay System (Promega, Madison, Wisconsin, USA). Data were plotted as the mean of sextuplicate samples and are representative of at least two independent experiments.
Electron microscopy.
HNP cells in primary culture from a CF patient were grown in a polarized fashion on Transwells and were stimulated with 1 ∞ 108 P. aeruginosa PAO1, fixed in 2.5% glutaraldehyde, and embedded for electron microscopy. Semithin sections 1 ∝m in thickness were cut and were stained with toludine blue for localization of cells. Thin sections 60 nm in thickness were cut on an MT-7000 ultramicrotome (Boeckeler Instruments Inc., Tucson, Arizona, USA) and were stained with uranyl acetate and lead citrate and analyzed on a JEOL EX-1200 electron microscope at 80 kV (JEOL USA Inc., Peabody, Massachusetts, USA).
Isolation of lipid rafts.
A modification of the method of Brown et al. was used to isolate lipid rafts (42). Confluent monolayers of 16HBE cells were grown in 150-mm dishes and were exposed to S. aureus RN6390 (1 ∞ 108 CFU/ml) for 1 hour and/or filipin 20 ∝g/ml for 30 minutes prior to bacterial stimulation. The cells were washed with cold PBS and lysed with 1% Triton X-100 in TNE buffer (25 mM TrisCl, pH 7.5, 150 mM NaCl, and 5 mM EDTA) plus protease inhibitors for 20 minutes on ice. Lysed cells were harvested and homogenized with a loose-fitting then a tight-fitting Dounce homogenizer. Whole cells and nuclei were removed by centrifugation at 3,000 g for 8 minutes. An equal volume of 80% sucrose in TNE was added to the supernatant. This 40% layer was overlaid with 30% and 4% sucrose in TNE to form a discontinuous gradient. Samples were centrifuged at 285,000 g for 24 hours at 4°C and 1-ml fractions were collected. Protein concentrations were determined using a Micro BCA Protein Assay Kit (Pierce) and 5 ∝g was run on a 4–12% bis-Tris NuPAGE gel with MES buffer (Invitrogen). After transfer to PVDF Immobilon-P (Millipore), blots were incubated in 5% skim milk blocking solution for 1 hour at room temperature for Western hybridization. Horseradish peroxidase–conjugated anti-rabbit, anti-goat, or anti-mouse IgG (Santa Cruz Biotechnology Inc.) and Western Lightning Chemiluminescence Reagent Plus (Perkin Elmer Life Sciences, Boston, Massachusetts, USA) or Femto SuperSignal (Pierce) were used for visualization.
Flow cytometry.
P. aeruginosa PAO1 or S. aureus RN6390 (1 ∞ 108 CFU/ml), anti-asialoGM1, Pam3Cys-Ser-Lys4, or LPS was added to 16HBE cells or primary cells harvested from HNPs and cells were incubated at 37°C for 1 hour, then the stimuli were removed with three to four PBS washes. After cells were incubated with 5% normal serum blocking solution for 15 minutes, primary antibodies were added for 1 hour at room temperature, and Alexa Fluor 488–conjugated secondary antibodies were added for 30 minutes, then samples were washed and incubated with HBSS plus 0.02% EGTA. Detached cells were removed and fixed in 1% paraformaldehyde. Cells were stored overnight at 4°C and then analyzed with a Becton Dickinson FACSCalibur using CellQuest software.
Acknowledgments
This work was funded by NIH grants RO1 DK39693 and HL56194 and the Cystic Fibrosis Foundation. We thank Kristy Brown for technical assistance with electron microscopy and the Herbert Irving Optical Microscopy facility at Columbia University.
Footnotes
Nonstandard abbreviations used: cystic fibrosis (CF); dominant negative (DN); gangliotetraosylceramide (Galβ1,2GalNAcβ1,4Galβ1,4Glcβ1,1Cer) (asialoGM1); human nasal polyp (HNP); interleukin-1 receptor–activated kinase-1 (IRAK-1); myeloid differentiation protein (MyD88); TNF receptor–associated factor 6 (TRAF6); Toll-like receptor (TLR).
Conflict of interest: The authors have declared that no conflict of interest exists.
References
- 1.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 2.DiMango E, Ratner AJ, Bryan R, Tabibi S, Prince A. Activation of NF-κB by adherent Pseudomonas aeruginosa in normal and cystic fibrosis respiratory epithelial cells. J. Clin. Invest. 1998;101:2598–2605. doi: 10.1172/JCI2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lien E, et al. Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J. Biol. Chem. 1999;274:33419–33425. doi: 10.1074/jbc.274.47.33419. [DOI] [PubMed] [Google Scholar]
- 4.Lien E, et al. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J. Clin. Invest. 2000;105:497–504. doi: 10.1172/JCI8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu. Rev. Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 6.da Silva Correia J, Soldau K, Christen U, Tobias PS, Ulevitch RJ. Lipopolysaccharide is in close proximity to each of the proteins in its membrane receptor complex: transfer from CD14 to TLR4 and MD-2. J Biol. Chem. 2001;276:21129–21135. doi: 10.1074/jbc.M009164200. [DOI] [PubMed] [Google Scholar]
- 7.Jiang Q, Akashi S, Miyake K, Petty HR. Lipopolysaccharide induces physical proximity between CD14 and toll-like receptor 4 (TLR4) prior to nuclear translocation of NF-κB. J. Immunol. 2000;165:3541–3544. doi: 10.4049/jimmunol.165.7.3541. [DOI] [PubMed] [Google Scholar]
- 8.Iwaki D, et al. The extracellular toll-like receptor 2 domain directly binds peptidoglycan derived from Staphylococcus aureus. J. Biol. Chem. 2002;277:24315–24320. doi: 10.1074/jbc.M107057200. [DOI] [PubMed] [Google Scholar]
- 9.Krivan HC, Roberts DD, Ginsburg V. Many pulmonary pathogenic bacteria bind specifically to the carbohydrate sequence GalNAcβ1-4Gal found in some glycolipids. Proc. Natl. Acad. Sci. U. S. A. 1988;85:6157–6161. doi: 10.1073/pnas.85.16.6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McNamara N, et al. ATP transduces signals from ASGM1, a glycolipid that functions as a bacterial receptor. Proc. Natl. Acad. Sci. U. S. A. 2001;98:9086–9091. doi: 10.1073/pnas.161290898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DiMango E, Zar HJ, Bryan R, Prince A. Diverse Pseudomonas aeruginosa gene products stimulate respiratory epithelial cells to produce interleukin-8. J. Clin. Invest. 1995;96:2204–2210. doi: 10.1172/JCI118275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smart EJ, et al. Caveolins, liquid-ordered domains, and signal transduction. Mol. Cell. Biol. 1999;19:7289–7304. doi: 10.1128/mcb.19.11.7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. J. Biol. Chem. 1998;273:5419–5422. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- 14.Harris J, Werling D, Hope JC, Taylor G, Howard CJ. Caveolae and caveolin in immune cells: distribution and functions. Trends Immunol. 2002;23:158–164. doi: 10.1016/s1471-4906(01)02161-5. [DOI] [PubMed] [Google Scholar]
- 15.Excoffon KJ, Moninger T, Zabner J. The coxsackie B virus and adenovirus receptor resides in a distinct membrane microdomain. J. Virol. 2003;77:2559–2567. doi: 10.1128/JVI.77.4.2559-2567.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newman GR, Campbell L, von Ruhland C, Jasani B, Gumbleton M. Caveolin and its cellular and subcellular immunolocalization in lung alveolar epithelium: implications for alveolar epithelial type 1 cell function. Cell. Tissue Res. 1999;295:111–120. doi: 10.1007/s004410051217. [DOI] [PubMed] [Google Scholar]
- 17.Feldman M, et al. Role of flagella in pathogenesis of Pseudomonas aeruginosa pulmonary infection. Infect. Immun. 1998;66:43–51. doi: 10.1128/iai.66.1.43-51.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saiman L, Prince A. Pseudomonas aeruginosa pili bind to asialoGM1 which is increased on the surface of cystic fibrosis epithelial cells. J. Clin. Invest. 1993;92:1875–1880. doi: 10.1172/JCI116779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwabuchi K, Handa K, Hakomori S. Separation of “glycosphingolipid signaling domain” from caveolin-containing membrane fraction in mouse melanoma B16 cells and its role in cell adhesion coupled with signaling. J. Biol. Chem. 1998;273:33766–33773. doi: 10.1074/jbc.273.50.33766. [DOI] [PubMed] [Google Scholar]
- 20.Dermine JF, et al. Flotillin-1-enriched lipid raft domains accumulate on maturing phagosomes. J. Biol. Chem. 2001;276:18507–18512. doi: 10.1074/jbc.M101113200. [DOI] [PubMed] [Google Scholar]
- 21.Orlandi PA, Fishman PH. Filipin-dependent inhibition of cholera toxin: evidence for toxin internalization and activation through caveolae-like domains. J. Cell Biol. 1998;141:905–915. doi: 10.1083/jcb.141.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qian Y, Commane M, Ninomiya-Tsuji J, Matsumoto K, Li X. IRAK-mediated translocation of TRAF6 and TAB2 in the interleukin–1-induced activation of NFκB. J. Biol. Chem. 2001;276:41661–41667. doi: 10.1074/jbc.M102262200. [DOI] [PubMed] [Google Scholar]
- 23.Melmed G, et al. Human intestinal epithelial cells are broadly unresponsive to Toll-like receptor 2-dependent bacterial ligands: implications for host-microbial interactions in the gut. J. Immunol. 2003;170:1406–1415. doi: 10.4049/jimmunol.170.3.1406. [DOI] [PubMed] [Google Scholar]
- 24.Ehrhardt C, et al. 16HBE14o- human bronchial epithelial cell layers express P-glycoprotein, lung resistance-related protein, and caveolin-1. Pharm. Res. 2003;20:545–551. doi: 10.1023/a:1023230328687. [DOI] [PubMed] [Google Scholar]
- 25.Bradbury NA, et al. Characterization of the internalization pathways for the cystic fibrosis transmembrane conductance regulator. Am. J. Physiol. 1999;276:L659–L668. doi: 10.1152/ajplung.1999.276.4.L659. [DOI] [PubMed] [Google Scholar]
- 26.Ratner AJ, et al. Cystic fibrosis pathogens activate Ca2+-dependent mitogen-activated protein kinase signaling pathways in airway epithelial cells. J. Biol. Chem. 2001;276:19267–19275. doi: 10.1074/jbc.M007703200. [DOI] [PubMed] [Google Scholar]
- 27.Sundberg-Kovamees M, Holme T, Sjogren A. Interaction of the C-polysaccharide of Streptococcus pneumoniae with the receptor asialo-GM1. Microb. Pathogen. 1996;21:223–234. doi: 10.1006/mpat.1996.0057. [DOI] [PubMed] [Google Scholar]
- 28.Heyer G, et al. Staphylococcus aureus agr and sarA functions are required for invasive infection but not inflammatory responses in the lung. Infect. Immun. 2002;70:127–133. doi: 10.1128/IAI.70.1.127-133.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderson HA, Chen Y, Norkin LC. Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol. Biol. Cell. 1996;7:1825–1834. doi: 10.1091/mbc.7.11.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shin JS, Gao Z, Abraham SN. Involvement of cellular caveolae in bacterial entry into mast cells. Science. 2000;289:785–788. doi: 10.1126/science.289.5480.785. [DOI] [PubMed] [Google Scholar]
- 31.Razani B, Lisanti MP. Caveolin-deficient mice: insights into caveolar function and human disease. J. Clin. Invest. 2001;108:1553–1561. doi:10.1172/JCI200114611. doi: 10.1172/JCI14611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cario E, et al. Lipopolysaccharide activates distinct signaling pathways in intestinal epithelial cell lines expressing Toll-like receptors. J. Immunol. 2000;164:966–972. doi: 10.4049/jimmunol.164.2.966. [DOI] [PubMed] [Google Scholar]
- 33.Triantafilou M, Miyake K, Golenbock DT, Triantafilou K. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J. Cell Sci. 2002;115:2603–2611. doi: 10.1242/jcs.115.12.2603. [DOI] [PubMed] [Google Scholar]
- 34.Andonegui G, et al. Endothelium-derived Toll-like receptor-4 is the key molecule in LPS-induced neutrophil sequestration into lungs. J. Clin. Invest. 2003;111:1011–1020. doi:10.1172/JCI200316510. doi: 10.1172/JCI16510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kube D, Adams L, Perez A, Davis PB. Terminal sialylation is altered in airway cells with impaired CFTR-mediated chloride transport. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001;280:L482–L492. doi: 10.1152/ajplung.2001.280.3.L482. [DOI] [PubMed] [Google Scholar]
- 36.Barasch J, et al. Defective acidification of intracellular organelles in cystic fibrosis. Nature. 1991;352:70–73. doi: 10.1038/352070a0. [DOI] [PubMed] [Google Scholar]
- 37.Poschet JF, et al. Molecular basis for defective glycosylation and Pseudomonas pathogenesis in cystic fibrosis lung. Proc. Natl. Acad. Sci. U. S. A. 2001;98:13972–13977. doi: 10.1073/pnas.241182598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Venkatakrishnan A, et al. Exaggerated activation of nuclear factor-κB and altered IκB-β processing in cystic fibrosis bronchial epithelial cells. Am. J. Resp. Cell Mol. Biol. 2000;23:396–403. doi: 10.1165/ajrcmb.23.3.3949. [DOI] [PubMed] [Google Scholar]
- 39.Tirouvanziam R, et al. Inflammation and infection in naive human cystic fibrosis airway grafts. Am. J. Resp. Cell Mol. Biol. 2000;23:121–127. doi: 10.1165/ajrcmb.23.2.4214. [DOI] [PubMed] [Google Scholar]
- 40.Heeckeren A, et al. Excessive inflammatory response of cystic fibrosis mice to bronchopulmonary infection with Pseudomonas aeruginosa. J. Clin. Invest. 1997;100:2810–2815. doi: 10.1172/JCI119828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rajan S, et al. Pseudomonas aeruginosa induction of apoptosis in respiratory epithelial cells: analysis of the effects of cystic fibrosis transmembrane conductance regulator dysfunction and bacterial virulence factors. Am. J. Resp. Cell Mol. Biol. 2000;23:304–312. doi: 10.1165/ajrcmb.23.3.4098. [DOI] [PubMed] [Google Scholar]
- 42.Brown DA, Rose JK. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 1992;68:533–544. doi: 10.1016/0092-8674(92)90189-j. [DOI] [PubMed] [Google Scholar]