

Abstract
For a long time, the most important inflammatory demyelinating diseases of the central nervous system (CNS), for example, multiple sclerosis (MS) and neuromyelitis optica (NMO), were extremely hard to differentiate, often with severe consequences for affected patients. This changed with the discovery of NMO‐immunoglobulin G (IgG), a specific autoantibody which was detected in the vast majority of NMO patients, and with the demonstration that this autoantibody targets aquaporin 4 (AQP4), a water channel found on astrocytes in the CNS. These findings paved the way for the generation of experimental models of NMO. This chapter will discuss the contribution of experimental models to NMO research and what key questions remain to be addressed.
Keywords: animal models, aquaporin 4, astrocytes, neuromyelitis optica
Relationship between human disease and experimental animal models
Although first reports about neuromyelitis optica (NMO) appeared over 100 years ago 11, 19, a clear distinction between NMO and multiple sclerosis (MS) only became possible in the past decade, when Lennon et al discovered a serum autoantibody marker of NMO, the so‐called NMO‐immunoglobulin G (IgG) 34 antibody, and subsequently identified aquaporin 4 (AQP4) as the target structure of these antibodies on astrocytes 33. Since then, our knowledge about NMO has grown exponentially.
NMO is a severe autoimmune disease of the central nervous system (CNS), which is characterized by the formation of large, often necrotic lesions preferentially affecting the optic nerves and spinal cord (Figure 1). Astrocytes are the prime target of the immune‐system in NMO patients 33. These cells express large amounts of the water channel AQP4, which are recognized by AQP4‐specific autoantibodies in the vast majority of NMO patients. Some patients show only a limited form of NMO typically characterized by recurrent attacks of either optic neuritis or longitudinally extensive transverse myelitis in the setting of AQP4 antibody seropositivity. These patients are referred to as having NMO spectrum disorder (NMOSD) 27. It is likely, however, that NMOSD and NMO are the same disease.
Figure 1.
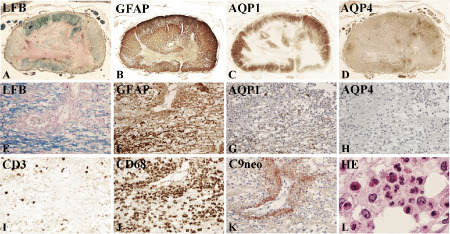
Pathology of neuromyelitis optica NMO. A–D. Cervical spinal cord from an NMO patient, showing large destructive lesions, which mainly affect the central portions of the cord and shows complete loss of myelin (A), of glial fibrillary acidic protein (GFAP) (B), of aquaporin (AQP) 1 (C) and of AQP 4 (D); more peripheral portions of the lesions reveal in part preserved astrocytes with variable loss of AQP 1 and complete loss of AQP 4; x: 6.6; E–L. Active lesion of NMO with partial loss of myelin (E), more pronounced loss of GFAP (F), severe loss of AQP 1 (G) and complete loss of AQP 4 (H); such lesions contain T cells (I), abundant numbers of macrophages (J), activated complement (C9neo antigen) present in a rosette‐like manner around vessels (k), and abundant granulocytes and eosinophils (L). E–K: × 200; l: × 1000. LFB = luxol fast blue.
Based on the availability of a known circulating autoantibody and a defined target antigen, one would have expected that the development of appropriate animal models would be a straightforward task. However, the simple transfer of AQP4 antibodies into naïve animals was not sufficient to induce experimental NMO 8. This is somewhat akin to the human situation wherein patients may harbor AQP4‐specific serum antibodies for many years without showing clinical signs of NMO 41. As the factors needed for NMO‐IgG to induce clinically overt disease are unknown, it remains challenging to induce experimental NMO. Modeling NMO is further complicated by the fact that NMO‐IgG recognizes conformational epitopes of AQP4 3 that are not available when recombinant, unfolded protein is used for immunization. The epitopes require immunogen consisting of properly folded AQP4, which is not soluble or requires toxic concentrations of triton for solubilization. Further, when membrane lysates of AQP4‐transfected cells are used for immunization, they can trigger unwanted antibody responses against other membrane components of the transfectants as well (own observations). Given all these obstacles, there currently is no reproducible experimental NMO model with long‐term, robust AQP4‐specific antibody expression available, let alone with a spontaneous onset of AQP4‐specific, antibody‐mediated CNS pathology. Despite these limitations, it is possible to create experimental models that reproduce important aspects of human NMO and to use them for addressing key questions raised by clinicians and pathologists about the pathogenic significance of AQP4‐specific autoantibodies in NMO, the contribution of different cells and cytokines to lesion formation, and the role of AQP4‐specific T cells in the disease process.
“Pathogenic or epiphenomenon?”—The significance of AQP4‐specific autoantibodies revealed by animal models
The clinical observation that most NMO/NMOSD patients harbor serum autoantibodies against AQP4 33, 34 immediately raised two important questions: (i) Are the antibodies just an epiphenomenon of the disease, unrelated to the initiation of NMO lesions, or are they pathogenic? (ii) And if pathogenic, under which conditions do the antibodies enter the CNS to initiate the formation of astrocyte‐destructive lesions? The first question was relatively easy to address in experimental models. As NMO patients who experience steroid‐unresponsive severe exacerbation are often treated with plasma exchange, this provides large amounts of NMO–IgG‐containing plasma, which can be further purified and used for experimental purposes. Because these NMO‐IgGs recognize AQP4 on the surface of living astrocytes 33 and react with AQP4 in humans 33, rats 8, and mice 33, their pathogenic potential can be easily tested in experimental animals. First attempts to induce experimental NMO by injecting NMO‐IgG into the circulation/peritoneum of healthy rodents led to NMO‐IgG seropositive animals but failed to produce CNS lesions 8. These early experiments clearly demonstrated that an intact blood–brain barrier (BBB) efficiently shields the CNS from the entry of antibodies. Even the leaky BBB of immature rodents 8, 47 or a pertussis toxin‐ or lipopolysaccharide‐permeabilized BBB of adult animals 47 did not permit lesion formation with AQP4 loss in NMO‐IgG seropositive animals. Therefore, NMO animal models were designed in which NMO‐IgG could reach the CNS parenchyma either in the context of CNS inflammation, when the BBB was opened, or via direct intracerebral injection, thereby circumventing the BBB.
NMO/experimental autoimmune encephalomyelitis (EAE) models
The first set of experimental models was based on two observations: (i) the presence of inflammatory cells in NMO lesions 36; and (ii) on previous experiences with studying the pathogenicity of other CNS antigen‐specific IgGs, using antibodies directed against the myelin oligodendrocyte glycoprotein (MOG) recognized on the surface of myelin sheaths and oligodendrocytes 31, 35. Here we knew from older studies that these antibodies were pathogenic when introduced into the CNS of Lewis rats in the course of EAE, a paradigm of T cell‐mediated inflammatory disease of the CNS. EAE can be induced either by activating CNS antigen‐specific T cells by adjuvant‐assisted immunization with the respective CNS antigen (active EAE) or by injecting in vitro‐activated, CNS antigen‐specific T cells into naïve syngeneic rats (passive EAE). When the animals show the first clinical signs of CNS inflammation (ie, weight loss or mild neurological symptoms like the loss of tail tonicity), the BBB is open and facilitates the entry of serum proteins into the CNS parenchyma. When the immune system is supplemented at this time point with MOG‐specific antibodies, these IgGs cross through the opened BBB and enter the CNS parenchyma, bind to the surface of myelin sheaths, fix complement and initiate demyelination 31, 35. Using a similar approach, we induced EAE and supplemented the immune system with NMO–IgG‐containing AQP4‐specific antibodies at the time of first clinical symptoms, thereby permitting these antibodies to enter into the CNS. These antibodies were shown to bind to the surface of astrocytes in a rosette‐like pattern very similar to the pattern seen in human NMO lesions, fix complement at these sites and trigger the formation of astrocyte‐destructive lesions 3, 8, 23. Hence, these NMO/EAE models clearly showed that the AQP4‐specific antibodies found in NMO patients were pathogenic and not merely epiphenomena of the disease process. Moreover, CNS pathology in both active and passive NMO/EAE was similar to the initial tissue alterations observed in some NMO lesions. NMO/EAE lesions were characterized by massive infiltration of neutrophils at lesion sites, along with complement deposition on subpial and perivascular astrocyte processes and profound loss of astrocytes 3, 8. At later stages characterized by the presence of macrophages/activated microglia containing myelin degradation products and/or by overt demyelination 3, secondary oligodendrocyte destruction was also observed (Figure 2). The NMO/EAE model clearly demonstrates that T cells are required to open the BBB for the entry of antibodies and complement, which themselves are not sufficient to initiate lesions, as learned from NMO‐IgG transfer into animals with a developmentally leaky 8, 47 or a pertussis toxin‐ or lipopolysaccharide‐permeabilized BBB 47, but also require additional effector mechanisms for the initiation of astrocyte‐destructive lesions. Hence, NMO/EAE is a suitable model to study the initial stages of lesion formation in NMO. However, this model also has disadvantages. First, it requires large amounts of patient‐derived NMO‐IgG (10 mg) for a single NMO/EAE animal. Second, NMO/EAE in Lewis rats is a TH1 cell‐driven CNS disease, whereas NMO in humans may have a TH17 bias 16. Although mice show this TH17 bias 40, they cannot currently be used for the induction of NMO/EAE, as human NMO‐IgG can only fix and activate rat 3, 8, but not murine complement 49.
Figure 2.
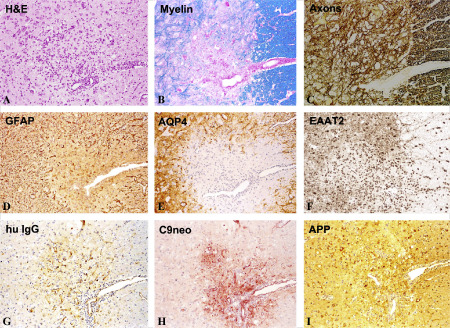
Pathology of experimental neuromyelitis optica (NMO) in the rat. Activation of myelin basic protein (MBP) reactive T‐cells by immunization with MBP/complete Freund's adjuvant (CFA) and intraperitoneal injection of human aquaporin 4 antibody containing NMO‐immunoglobulin G (IgG), beginning at the first signs of clinical symptoms and repeated 24 h later. The animals were sacrificed 48 h after the first NMO‐IgG application. As in NMO lesions in humans, the lesions are infiltrated by lymphocytes and macrophages (A), there is some loss of myelin (B), but no loss of axons in the early stage (C); astrocytes, reactive for glial fibrillary acidic proteins (GFAP) are partially lost in the lesions (D) and there is even more widespread loss of aquaporin 4 (E); the excitatory amino acid transporter 2 (EAAT2) is also lost in areas of aquaporin 4 loss (F); within active lesions there is deposition of human immunoglobulin (G) and activated complement (C9neo antigen; H); immunocytochemistry for amyloid precursor protein shows axonal spheroids in the lesions, indicating ongoing axonal injury (I); × 60. APP = amyolid precursor protein; H&E = hematoxylin and eosin.
NMO‐IgG/complement intracerebral injection models
In this model system, the BBB was not opened by the effects of CNS antigen‐specific T cells. Rather, this was bypassed by direct, repeated infusions of NMO‐IgG and human complement into the cerebral hemisphere or ventricular system of experimental mice 49. The pathogenicity of human NMO‐IgG was apparent in these models. The first signs of astrocyte injury appeared 12 h after the injections and included loss of AQP4 expression, glial cell edema, some myelin breakdown and early axonal injury. Seven days later, perivascular deposition of activated complement components, loss of AQP4 expression, loss of astrocytes and extensive demyelination as well as neuronal cell death were observed. By this time point, extensive inflammatory cell infiltration could also be observed, dominated by mononuclear and polymorphonuclear cells 49. Because the BBB is circumvented by intracerebral injection in this experimental model, T cells are not needed to open it. Consequently, macrophage‐ and granulocyte‐infiltrated lesions with a comparable loss of AQP4 reactivity and demyelination were also seen after injection of NMO‐IgG and human complement into the brains of T cell‐deficient nude mice 51. This experimental model also revealed the importance of neutrophils for lesion formation, as neuroinflammation, myelin loss and AQP4 loss were reduced in neutropenic mice and in mice treated with neutrophil protease inhibitors 50.
Like the NMO/EAE model described earlier, the NMO‐IgG/complement‐infusion model has both advantages and disadvantages. Because this model requires much less NMO‐IgG than the NMO/EAE models, it is an ideal tool for therapeutic studies such as the initial testing of small molecules to inhibit binding of NMO‐IgG to its cellular targets in vivo 57, 58 or of drugs inhibiting neutrophil effector molecules 50. Because this model works in mice, studies can be extended to the large zoo of transgenic or knockout mice currently available, which will certainly give further insight into the role of individual molecules in lesion formation (or its prevention). The disadvantages of this model are:
-
(i)
NMO‐IgG cannot fix and activate murine complement, which necessitates co‐injections of human complement (with the possible risk of obscuring the action of naturally occurring rodent complement inhibitors, which might not work on human molecules); and
-
(ii)
the target tissue has to be manipulated by repeated injections of molecules into the mouse brain, often at multiple sites and in relatively large volumes, which might alter the susceptibility of the CNS to additional inflammatory stimuli.
Cytokine‐injection NMO models
The animal models described earlier clearly reveal that NMO‐IgG is pathogenic in sufficiently high concentration in the CNS parenchyma and when it is able to fix complement upon binding to its target structures in the CNS. Both models were also clearly associated with inflammation 3, 8, 49. This raises the question of whether pro‐inflammatory cytokines and chemokines also contribute to lesion formation with AQP4 loss in the presence of NMO‐IgG and complement. To address this point, the cytokines interleukin‐1 beta, tumor necrosis factor alpha, interleukin‐6, interferon gamma or the chemokine CXCL2 were stereotactically injected into the striatum of NMO–IgG‐seropositive rats. One day later, all injected cytokines and chemokines led to profound leakage of immunoglobulins into the injected hemisphere, but only interleukin‐1 beta triggered the formation of perivascular, neutrophil‐infiltrated lesions with AQP4 loss and complement‐mediated astrocyte destruction distant from the needle tract 26. These effects of interelukin‐1 beta were at least in part mediated by its action on the BBB, as treatment of rat brain endothelial cells with interleukin‐1 beta, but not with any other cytokine or chemokine applied at the same concentration and over the same period of time, caused profound up‐regulation of granulocyte‐recruiting and supporting molecules in these cells 26. Moreover, injections of interleukin‐1 beta caused higher numbers of blood vessels with perivascular, cellular C1q reactivity than any other cytokine tested. These data clearly confirmed that interleukin‐1 beta facilitates the entry of neutrophils and the breakdown of the BBB. Because activated microglia/macrophages in active NMO lesions contain more interleukin‐1 beta than those found in active MS lesions 26, this cytokine could be an important secondary factor in lesion formation, possibly by paving the way for rapid lesion growth and amplified immune cell recruitment to this site 26. This model also affords an opportunity to study the contribution of defined single cytokines (instead of the cytokine cocktails secreted by immune cells) to lesion initiation and/or evolution in NMO–IgG‐seropositive rats. Unfortunately, this model also relies on surgical manipulation of the CNS.
Cumulatively, all experimental models described earlier clearly revealed that NMO‐IgG is pathogenic when it can overcome the constraints placed by an impermeable BBB and when additional cellular/molecular effector mechanisms are in place to amplify immune cascade.
The quest for pathogenic AQP4‐specific T cells in animal models
Having established that AQP4‐specific antibodies of NMO patients were pathogenic, investigators moved on to study AQP4‐specific T cell responses. Several questions were raised. Do these T cells only facilitate antibody production, or can they also induce inflammatory lesions in the CNS? Lewis rats were used to help address these questions. Because all autoimmune T cell responses in these animals were restricted by major histocompatibility complex (MHC) class II (RT1.BL) products, we used an epitope‐prediction model 62 to identify AQP4 peptides binding to these MHC class II molecules. We found two perfect epitope matches. The first epitope, AQP4220–228, contributed to the extracellular loop E of AQP4, while the second epitope, AQP4296–304, was contained in the intracellular C‐terminal domain of AQP4 45. When Lewis rats were immunized with peptides containing these epitope sequences, there was no evidence for clinical or histological CNS disease, probably because of the low numbers of specific T cells in the immune repertoire of the immunized animals. However, when Lewis rats were challenged with large numbers of activated T cells recognizing any one of the selected AQP4 peptides, the situation was completely different. Then, inflammatory lesions along the entire neuraxis were seen, especially at sites of high AQP4 expression like the glial lamellae of the basal/lateral hypothalamus or spinal cord dorsal horn at the thoracic or lumbar/sacral level, although the optic system was spared. In all inflammatory lesions, astrocytes remained intact and AQP4 expression was preserved 45. The picture changed when AQP4‐specific T cells were acting in NMO–IgG‐seropositive animals. Then, the lesions were more numerous and larger, and perivascular lesions with neutrophil infiltration and AQP4 loss ensued 45. Overall, however, the encephalitogenic potential of the different AQP4 peptide‐specific T cell lines was very low and did not culminate in clinical deficits 45. Instead, another peculiarity of the AQP4‐specific T cells became obvious, especially after co‐transfer of these cells with NMO‐IgG. Inflammation was also observed in two other AQP4‐expressing organ systems: in kidneys, which contain AQP4‐positive collecting ducts 56 and developed interstitial nephritis, and in muscles containing AQP4‐positive fast‐twitch skeletal muscle fibers 22, which revealed foci of inflammation limited to their connective tissue compartment 45. To date, renal disease has not been described in NMO patients, but there are a few reports of NMO patients presenting with elevated levels of creatine kinase as a marker for muscle injury 10, 12, 55, 69.
Many questions concerning AQP4‐specific T cell reactivities remain and have to be addressed further in T cell transfer‐induced animal models of CNS inflammation. For example, we do not know whether the AQP4 epitopes used so far are dominantly generated in the course of natural processing of AQP4 by antigen‐presenting cells in Lewis rats 45. AQP4‐specific T cells have also been characterized in mice 20, 40, and it will probably become clear soon which strains of rat or mice are better suited to mimic the lesion tropism observed in NMO. However, we cannot yet exclude an additional possibility: that lesions in NMO patients are initiated by T cells with other, more CNS‐selective antigen specificities, especially as the AQP4‐specific T cells tested so far also induced inflammation in peripheral organs. Future experiments will hopefully answer some of these questions.
Modeling AQP4‐antibody‐negative NMO
All the animal models described above dealt with antibody or T cell questions arising from the most frequent subset of NMO patients: those who are seropositive for AQP4‐specific, pathogenic antibodies. However, there are also patients who fulfill all criteria for diagnosis of NMO on the basis of clinical and radiological findings, but are AQP4‐antibody‐seronegative. Some of these patients were shown to harbor serum autoantibodies against MOG 21, 28, 37, 48. Although it is currently not clear whether these antibodies affect disease progression or severity 63, it is tempting to speculate that they contribute to lesions with myelin loss and therefore may be pathogenic. This speculation is supported by the fact that these MOG‐specific antibodies were identified in cell‐based assays and recognized “their” specific antigen on the surface of MOG‐transfected cells 21, 28, 37, 48, which is a prerequisite for binding anti‐MOG antibodies to their target structure in the intact organism. This binding profile resembles that seen in the murine monoclonal antibody 8‐18C5, an anti‐MOG antibody with a known pathogenic role in the induction of demyelinated lesions 31, 35.
Because it is unresolved whether anti‐MOG antibody‐positive NMO patients respond in the same way to currently available treatment options as anti‐AQP4 antibody‐positive NMO patients 63, therapeutic studies for this subtype of NMO may become important. Such studies would greatly benefit from the use of double‐transgenic mice overrepresenting MOG‐specific T cells and demyelinating MOG‐specific antibodies in their immune repertoire. These have been produced in two different laboratories 6, 30. These double‐transgenics are present on the genetic background of C57BL/6 mice and develop an EAE‐like neurological syndrome described as “spontaneous opticospinal encephalomyelitis (OSE)” 30 or “Devic‐like disease” 6. In this experimental model, large numbers of animals spontaneously develop paralytic disease with inflammatory, demyelinating lesions in optic nerves and spinal cords 6, 30.
Current animal models and patient reality
The key finding of all NMO experimental models has been the pathogenicity of NMO‐IgG in combination with complement, the contribution of T cells, other mononuclear cells, granulocytes and interleukin‐1 beta to the formation of astrocyte‐destructive lesions, and the development of lesions at NMO‐typical sites in double‐transgenic mice harboring MOG‐specific T cells and demyelinating MOG‐specific antibodies. However, notable differences exist between humans and rodent models which may impact findings.
First and foremost, the target structures of pathogenic antibody responses, for example, the astrocytes of the central gray matter, differ between rodents and humans. Both rodent and human astrocytes express AQP4, which is an important prerequisite for all experiments done so far. While AQP4 seems to be the only aquaporin in rat 39 or mouse 13 astrocytes, human astrocytes additionally express AQP1 39, a water channel that may partially compensate for the loss of AQP4 reactivity and function (Figure 3). In addition, rodent and human astrocytes differ in size. Human protoplasmic gray matter astrocytes have a 27‐fold greater volume and a 10‐fold increase in numbers of processes compared with their rodent counterparts 42. Moreover, while a single human protoplasmic astrocyte supports and modulates the function of ∼2 million synapses, its rodent counterpart only supports ∼90 000 42. Because of this increased complexity, damage to human astrocytes might affect larger areas of tissue and more adjacent cells than damage caused to rodent astrocytes. This may be one of several factors accounting for the proportionally larger lesions in NMO patients compared with lesions generated in experimental rodent models.
Figure 3.

Expression of aquaporin (AQP) 1 in rat and human brain tissue. In the brain of rats (and mice), AQP1 is only expressed on epithelial cells of the chroroid plexus, while astrocytes are AQP1‐negative (A); this is different in humans, where AQP1 is abundantly expressed on white matter astrocytes and on single cells or small clusters of astrocytes in the grey matter (B,C); A, B: × 30; C: × 150.
Second, humans and rodents also differ with regard to their polymorphonuclear cells. For example, although total white blood cell count is similar in humans, rats and mice, the percentage of neutrophils is dramatically different. In humans, neutrophils represent ~45%–75% of all leukocytes in peripheral blood 29, whereas in Lewis rats they constitute approximately 35% 70 and in mice, 5%–27% 7, 71 of leukocytes. Interestingly, the percentage of neutrophils in the blood is a determinant of the size of lesions associated with loss of AQP4 reactivity. Mice, when rendered neutrophilic by injections with granulocyte‐colony stimulating factor (G‐CSF) prior to the intrahemispheric injection of NMO‐IgG and human complement, had significantly larger areas of AQP4 loss and myelin loss than animals receiving only saline 50. Moreover, NMO patients have more neutrophils among their peripheral leukocytes during acute exacerbation than during remission 50. Notably, the inadvertent administration of G‐CSF in an NMO patient caused a marked rise in peripheral neutrophil count followed by a severe attack of NMO 17.
The other type of polymorphonuclear cells that differ between rodents and humans are the eosinophils. These cells were described in NMO lesions 36 and are also occasionally seen in lesions of murine models for anti‐MOG antibody‐positive NMO 30, but are absent in the rat models of anti‐AQP4 antibody‐positive NMO 3, 8, 24. However, this may reflect an anomaly of the animal strains used so far, because Lewis rats are also unable to recruit eosinophils to CNS lesions provoked by the combination of MOG‐specific T cells and MOG‐specific antibodies, while Brown Norway and Dark Agouti rats are able to do so 1, 54.
Third, there are still pronounced differences in the location of lesions between AQP4–antibody‐positive NMO patients with predominantly opticospinal lesions and their experimental rodent counterparts, which display lesions throughout the CNS (with or without sparing of the optic system), or at sites of intrahemispheric injections 3, 8, 24. These differences between patients and models can easily be explained by technical issues. It is well established that the location of inflammatory CNS lesions in EAE results from a combination of antigen availability and T cell specificity 5. However, because the target antigen of T cells‐initiating lesions in anti‐AQP4 antibody‐positive NMO patients is currently unknown, surrogate CNS antigen‐specific T cells must be used for the induction of NMO/EAE, and these T cells target slightly different CNS areas. Moreover, the infusion of NMO‐IgG and human complement and the injection of cytokines are best done in easily accessible, robust areas, which makes striatum and ventricular systems better experimental choices than optic nerves or spinal cord.
Finally, rats and mice are not able to show one particular symptom of NMO that may herald the formation of optic neuritis and transverse myelitis in a number of affected patients: intractable vomiting 2, 46. This symptom is caused by lesions in the brain stem adjoining or within the area postrema and the medullary floor of the fourth ventricle 46. However, although NMO/EAE models frequently reveal lesions at these sites, mice and rats lack the anatomical capability to vomit 67.
In summary, there are differences in size and location of lesions between human NMO patients and the relevant experimental models. Some of these differences may be overcome by modifications and refinements of current models, or they will have to be accepted as limitations when using rodent models. In spite of these limitations in the existing models, we have already learned a great deal about factors and mechanisms which may contribute to the initiation of lesions in NMO patients. These models have confirmed in vitro data suggesting that AQP4‐specific autoantibodies are pathogenic. Furthermore, current models underscore the critical prerequisite for AQP4‐IgG access to the CNS parenchyma across the BBB in order to bind to the extracellular domain of AQP4 on astrocytes and activate complement.
These models also reinforce the important role of T cells, granulocytes, microglia/macrophages and pro‐inflammatory cytokines in lesion formation and lesion expansion. Lastly, the encephalitogenic potential of AQP4‐specific T cells has been demonstrated.
Future animal models
The next generation of NMO animal models needs to address several key points. There is an urgent need for chronic models. So far, all experimental models for AQP4 antibody‐positive NMO described earlier require the transfer of human NMO‐IgG into rats or mice. While this approach works well to replicate early events of lesion formation, it will inevitably fail to mimic more chronic stages of NMO. The reason for this is quite simple: repeated transfer of immunoglobulins from different species will result in serum sickness, an immune complex‐mediated systemic illness 52. This dilemma may be resolved with the use of genetically modified animals capable of producing pathogenic, AQP4‐specific autoantibodies.
The manipulation of experimental animals should also be minimized. Currently available experimental NMO models depend not only on the presence of human NMO‐IgG, but also (in mice) on the application of human complement or (in rats) on the injection of large numbers of CNS antigen‐specific T cells or adjuvant‐assisted immunization with CNS antigens. The latter approach might be especially problematic as complete Freund's adjuvants (CFA), which is typically used for immunizations, contains components of the avirulent H37Ra strain of mycobacterium tuberculosis. According to a recent systematic study regarding para‐infectious NMO syndromes, associations with varicella zoster virus or mycobacterium tuberculosis/mycobacterium pneumonia have been reported 53. Because mycobacteria express proteins with sequence homology to AQP4 14, 43, 53, immunization procedures employing CFA might induce T cell responses against such proteins, which may explain an observation by one laboratory that “pre‐treatment with CFA alone was sufficient for AQP4‐Ab to induce astrocytic damage in vivo” 24.
To date, in all NMO animal models created, the focus of interest was just on one antigen specificity, for example, AQP4. However, many NMO patients have coexisting autoimmune diseases, most frequently (in more than 40% of all cases) systemic lupus erythematosus or Sjögren syndrome 44, 65, but also to lesser extent, myasthenia gravis 18, 32, autoimmune thyroid disease, type I diabetes, rheumatoid arthritis 32, and others. Consequently, NMO patients often harbor multiple autoantibodies specific for the relevant coexisting diseases. Hence, the underlying mechanisms for this autoimmune phenotype and even possible consequences of additional antibodies for the disease process should be explored.
In addition, the pathogenicity of human, NMO‐patient‐derived, AQP4‐specific T cell clones should be investigated. Are these T cells pathogenic, and are they able to direct CNS inflammation to typical NMO sites in the CNS? To address these points, humanized mouse models similar to the ones produced by Lars Fugger and colleagues might become the research tool of choice 38, that is, transgenic mice expressing three different human components involved in antigen presentation and T cell recognition: the HLA‐DPB1*0501 molecule, which is an MHC class II gene found in individuals of Japanese 61, 68 or Southern Han Chinese 60 descent; or the HLA‐DRB1*0301/HLA‐DRB3*0202 molecule, which has been described in American patients with NMO 59; the variable domains of T cell receptor alpha and beta chains derived from an NMO–patient‐derived T‐cell clone recognizing a single immunodominant AQP4 peptide in the context of such HLA molecules 59, and the human CD4 coreceptor. Because human and mouse AQP4 have a 93% identity at the amino acid level (using the respective GenBank sequences GI:18490380 and GI:1857925 for comparison), and because mice and man process antigens in a similar way 38, such an experiment might be feasible.
Future experimental NMO animal models should also consider the contribution of genetic factors to NMO pathogenesis. AQP4–antibody‐seropositive NMO patients are predominantly women; most frequently show relapsing/remitting disease courses, display longitudinally extensive lesions in the spinal cord and/or the optic system, and have astrocyte‐destructive lesions as the pathological substrate. However, differences between patient cohorts exist 27. For example, Afro‐Caribbean patients are typically younger at disease onset and are more likely to develop visual disability than Caucasians or Japanese Asians 27, while motor disabilities, wheelchair dependence and death from NMO prevail in Caucasians 27. Differences in clinical course and disability are also seen in EAE experiments using different strains of rat or mouse. Therefore, it is very likely that similar differences are also observed when different experimental NMO models are established.
Finally, the contribution of environmental factors should be more fully addressed. It is striking that among the most prevalent inflammatory demyelinating diseases of the CNS, MS predominates in Europe and Northern America, while NMO predominates in Asia and the Caribbean 64. Environmental factors could in part account for this observation. According to a recent study, clonally expanded AQP4‐specific T cells of NMO patients display cross‐reactivity with an ABC transporter molecule of Clostridium perfringens 59, a common pathogen responsible for food poisoning 9. This finding immediately leads to two questions: (i) Are we facing a situation similar to one previously observed in MS patients, in which T cell clones specific for myelin basic protein were shown to cross‐react with peptides derived from the Herpes simplex virus, Epstein–Barr virus, influenza type A virus, adenovirus type 12 or Pseudomonas aeruginosa 66 and none of the pathogens identified could be correlated with the onset of MS? Then, the cross‐reactivity of human AQP4‐specific T cells with a C. perfringens‐derived epitope would just be another example of the fact that a single T cell receptor recognizes several distinct, structurally related peptides 66. (ii) Or does it indicate a real involvement of C. perfringens in the disease process, as has been noted before for mycobacteria tuberculosis/pneumonia 53? If so, immunization of appropriate (perhaps even humanized) experimental animals with C. perfringens should lead not only to AQP4‐specific T cell responses, but also to the production of AQP4‐specific, pathogenic autoantibodies.
Environmental factors contributing to the development of NMO, however, do not necessarily have to be pathogens. They can also be commensal microbes, as the composition and molecular makeup of gut microbiota may change with diet. A very impressive example of the contribution of nutrition to gut microbiota is the evolutionary recent transfer of genes for carbohydrate‐active enzymes (porphyranases) from marine bacteria thriving on red algae of the genus Porphyra to commensal gut bacteria of the Japanese population. This transfer most likely resulted from the consumption of nori (Porphyra spp.), traditionally used in Japan to prepare sushi 15. Interestingly, changes in lifestyle from a Japanese traditional style to a modern Western style, including changes in diet, may underlie the currently observed shift in frequency from Japanese‐type MS (opticospinal MS, possibly including NMO/NMOSD, as the study was performed before AQP4‐antibody testing became available in 2004/2005) to Western‐type MS (conventional MS with lesions at multiple sites of the CNS including cerebrum, cerebellum and brain stem) from 1:0.5 in patients born in the 1920s to 1:4 in patients born after the 1970s 25.
Commensal microbes can cooperate with CNS autoantigens to trigger autoimmune CNS disease. This cooperation has recently been shown in MOG‐specific T cell receptor transgenic mice which spontaneously developed EAE under specific pathogen‐free conditions, but not under completely germ‐free conditions 4. However, previously germ‐free mice came down with EAE again, when their gut was recolonized with conventional commensal microbiota. Hence, gut microbes contribute to the recruitment and activation of autoantibody‐producing B cells from the immune repertoire 4, and it will be interesting to determine the role of gut microbes in anti‐AQP4 responses. The tools for such studies, AQP4‐specific T cell receptor transgenic animals, may soon be available.
Outlook
Ten years ago, the prognosis for NMO patients was very poor. NMO could not be properly distinguished from MS, and the diagnostic value and pathogenicity of AQP4‐specific antibodies was yet to be discovered. Today, we know that the vast majority of NMO/NMOSD patients are seropositive for AQP4‐specific antibodies, that these antibodies are pathogenic and cooperate with complement, encephalitogenic T cells, neutrophils and cytokines in the formation of large granulocyte‐infiltrated, astrocyte‐destructive lesions, and that NMO patients harbor expanded populations of potentially pathogenic AQP4‐specific T cells. Ongoing research focused on developing relevant NMO experimental models is expected to provide new insights regarding the critical points involved in the transition from an AQP4 antibody‐seropositive state to overt clinical disease, as well as lead to the identification of novel therapeutic targets with improved efficacy and limited toxicity. A multidisciplinary effort between clinicians, pathologists and basic scientists will hopefully soon lead to this reality.
Acknowledgments
The authors were supported by the Austrian Science Fund [grant numbers P21581‐B09 and P25240‐B24 to MB and I916‐B13 (International Programme, Eugène Devic European Network) to HL].
References
- 1. Adelmann M, Wood J, Benzel I, Fiori P, Lassmann H, Matthieu JM et al (1995) The N‐terminal domain of the myelin oligodendrocyte glycoprotein (MOG) induces acute demyelinating experimental autoimmune encephalomyelitis in the Lewis rat. J Neuroimmunol 63:17–27. [DOI] [PubMed] [Google Scholar]
- 2. Apiwattanakul M, Popescu BF, Matiello M, Weinshenker BG, Lucchinetti CF, Lennon VA et al (2010) Intractable vomiting as the initial presentation of neuromyelitis optica. Ann Neurol 68:757–761. [DOI] [PubMed] [Google Scholar]
- 3. Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C et al (2009) Intrathecal pathogenic anti‐aquaporin‐4 antibodies in early neuromyelitis optica. Ann Neurol 66:617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C et al (2011) Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 479:538–541. [DOI] [PubMed] [Google Scholar]
- 5. Berger T, Weerth S, Kojima K, Linington C, Wekerle H, Lassmann H (1997) Experimental autoimmune encephalomyelitis: the antigen specificity of T lymphocytes determines the topography of lesions in the central and peripheral nervous system. Lab Invest 76:355–364. [PubMed] [Google Scholar]
- 6. Bettelli E, Baeten D, Jager A, Sobel RA, Kuchroo VK (2006) Myelin oligodendrocyte glycoprotein‐specific T and B cells cooperate to induce a Devic‐like disease in mice. J Clin Invest 116:2393–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boxio R, Bossenmeyer‐Pourie C, Steinckwich N, Dournon C, Nusse O (2004) Mouse bone marrow contains large numbers of functionally competent neutrophils. J Leukoc Biol 75:604–611. [DOI] [PubMed] [Google Scholar]
- 8. Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M et al (2009) Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo . Ann Neurol 66:630–643. [DOI] [PubMed] [Google Scholar]
- 9. Canard B, Cole ST (1989) Genome organization of the anaerobic pathogen Clostridium perfringens . Proc Natl Acad Sci U S A 86:6676–6680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Deguchi S, Deguchi K, Sato K, Yunoki T, Omote Y, Morimoto N et al (2012) HyperCKemia related to the initial and recurrent attacks of neuromyelitis optica. Intern Med 51:2617–2620. [DOI] [PubMed] [Google Scholar]
- 11. Devic E (1894) Myélite subaigue compliquée de névrite optique. Bull Méd 8:1033–1034. [Google Scholar]
- 12. Di Filippo M, Franciotta D, Massa R, Di Gregorio M, Zardini E, Gastaldi M et al (2012) Recurrent hyperCKemia with normal muscle biopsy in a pediatric patient with neuromyelitis optica. Neurology 79:1182–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gao J, Tan M, Gu M, Marshall C, Ding J, Hu G, Xiao M (2012) Cellular localization of aquaporin‐1 in the human and mouse trigeminal systems. PLoS ONE 7:e46379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Graber DJ, Levy M, Kerr D, Wade WF (2008) Neuromyelitis optica pathogenesis and aquaporin 4. J Neuroinflammation 5:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hehemann JH, Correc G, Barbeyron T, Helbert W, Czjzek M, Michel G (2010) Transfer of carbohydrate‐active enzymes from marine bacteria to Japanese gut microbiota. Nature 464:908–912. [DOI] [PubMed] [Google Scholar]
- 16. Ishizu T, Osoegawa M, Mei FJ, Kikuchi H, Tanaka M, Takakura Y et al (2005) Intrathecal activation of the IL‐17/IL‐8 axis in opticospinal multiple sclerosis. Brain 128 (Pt 5):988–1002. [DOI] [PubMed] [Google Scholar]
- 17. Jacob A, Saadoun S, Kitley J, Leite M, Palace J, Schon F, Papadopoulos MC (2012) Detrimental role of granulocyte‐colony stimulating factor in neuromyelitis optica: clinical case and histological evidence. Mult Scler 18:1801–1803. [DOI] [PubMed] [Google Scholar]
- 18. Jarius S, Paul F, Franciotta D, de Seze J, Munch C, Salvetti M et al (2012) Neuromyelitis optica spectrum disorders in patients with myasthenia gravis: ten new aquaporin‐4 antibody positive cases and a review of the literature. Mult Scler 18:1135–1143. [DOI] [PubMed] [Google Scholar]
- 19. Jarius S, Wildemann B (2013) The history of neuromyelitis optica. J Neuroinflammation 10:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kalluri SR, Rothhammer V, Staszewski O, Srivastava R, Petermann F, Prinz M et al (2011) Functional characterization of aquaporin‐4 specific T cells: towards a model for neuromyelitis optica. PLoS ONE 6:e16083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kezuka T, Usui Y, Yamakawa N, Matsunaga Y, Matsuda R, Masuda M et al (2012) Relationship between NMO‐antibody and anti‐MOG antibody in optic neuritis. J Neuroophthalmol 32:107–110. [DOI] [PubMed] [Google Scholar]
- 22. King LS, Kozono D, Agre P (2004) From structure to disease: the evolving tale of aquaporin biology. Nat Rev Mol Cell Biol 5:687–698. [DOI] [PubMed] [Google Scholar]
- 23. Kinoshita M, Nakatsuji Y, Kimura T, Moriya M, Takata K, Okuno T et al (2009) Neuromyelitis optica: passive transfer to rats by human immunoglobulin. Biochem Biophys Res Commun 386:623–627. [DOI] [PubMed] [Google Scholar]
- 24. Kinoshita M, Nakatsuji Y, Kimura T, Moriya M, Takata K, Okuno T et al (2010) Anti‐aquaporin‐4 antibody induces astrocytic cytotoxicity in the absence of CNS antigen‐specific T cells. Biochem Biophys Res Commun 394:205–210. [DOI] [PubMed] [Google Scholar]
- 25. Kira J, Yamasaki K, Horiuchi I, Ohyagi Y, Taniwaki T, Kawano Y (1999) Changes in the clinical phenotypes of multiple sclerosis during the past 50 years in Japan. J Neurol Sci 166:53–57. [DOI] [PubMed] [Google Scholar]
- 26. Kitic M, Hochmeister S, Wimmer I, Bauer J, Misu T, Mader S et al (2013) Intrastriatal injection of interleukin 1 beta triggers the formation of neuromyelitis optica‐like lesions in NMO‐IgG seropositive rats. Acta Neuropathol Commun 1:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kitley J, Leite MI, Nakashima I, Waters P, McNeillis B, Brown R et al (2012) Prognostic factors and disease course in aquaporin‐4 antibody‐positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain 135 (Pt 6):1834–1849. [DOI] [PubMed] [Google Scholar]
- 28. Kitley J, Woodhall M, Waters P, Leite MI, Devenney E, Craig J et al (2012) Myelin‐oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology 79:1273–1277. [DOI] [PubMed] [Google Scholar]
- 29. Kratz APM, Basner RC, Einstein AJ (2012) Appendix: laboratory values of clinical importance. In: Harrison's Online Featuring the Complete Contents of Harrison's Principles of Internal Medicine. Longo DLFA, Kasper DL, Hauser SL, Jameson JL, Loscalzo J (eds), Chapter 60. The McGraw‐Hill Companies: New York. Available at: http://accessmedicine.com/content.aspx?aid=9113657 (accessed 19 November 2013). [Google Scholar]
- 30. Krishnamoorthy G, Lassmann H, Wekerle H, Holz A (2006) Spontaneous opticospinal encephalomyelitis in a double‐transgenic mouse model of autoimmune T cell/B cell cooperation. J Clin Invest 116:2385–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lassmann H, Brunner C, Bradl M, Linington C (1988) Experimental allergic encephalomyelitis: the balance between encephalitogenic T lymphocytes and demyelinating antibodies determines size and structure of demyelinated lesions. Acta Neuropathol (Berl) 75:566–576. [DOI] [PubMed] [Google Scholar]
- 32. Leite MI, Coutinho E, Lana‐Peixoto M, Apostolos S, Waters P, Sato D et al (2012) Myasthenia gravis and neuromyelitis optica spectrum disorder: a multicenter study of 16 patients. Neurology 78:1601–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR (2005) IgG marker of optic‐spinal multiple sclerosis binds to the aquaporin‐4 water channel. J Exp Med 202:473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K et al (2004) A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 364:2106–2112. [DOI] [PubMed] [Google Scholar]
- 35. Linington C, Bradl M, Lassmann H, Brunner C, Vass K (1988) Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am J Pathol 130:443–454. [PMC free article] [PubMed] [Google Scholar]
- 36. Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM et al (2002) A role for humoral mechanisms in the pathogenesis of Devic's neuromyelitis optica. Brain 125 (Pt 7):1450–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mader S, Gredler V, Schanda K, Rostasy K, Dujmovic I, Pfaller K et al (2011) Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflammation 8:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Madsen LS, Andersson EC, Jansson L, krogsgaard M, Andersen CB, Engberg J et al (1999) A humanized model for multiple sclerosis using HLA‐DR2 and a human T‐cell receptor. Nat Genet 23:343–347. [DOI] [PubMed] [Google Scholar]
- 39. Misu T, Hoeftberger R, Fujihara K, Wimmer I, Takai Y, Nishiyama S et al (2013) Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol 125:815–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nelson PA, Khodadoust M, Prodhomme T, Spencer C, Patarroyo JC, Varrin‐Doyer M et al (2010) Immunodominant T cell determinants of aquaporin‐4, the autoantigen associated with neuromyelitis optica. PLoS ONE 5:e15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nishiyama S, Ito T, Misu T, Takahashi T, Kikuchi A, Suzuki N et al (2009) A case of NMO seropositive for aquaporin‐4 antibody more than 10 years before onset. Neurology 72:1960–1961. [DOI] [PubMed] [Google Scholar]
- 42. Oberheim NA, Wang X, Goldman S, Nedergaard M (2006) Astrocytic complexity distinguishes the human brain. Trends Neurosci 29:547–553. [DOI] [PubMed] [Google Scholar]
- 43. Papazisi L, Gorton TS, Kutish G, Markham PF, Browning GF, Nguyen DK et al (2003) The complete genome sequence of the avian pathogen Mycoplasma gallisepticum strain R(low). Microbiology 149 (Pt 9):2307–2316. [DOI] [PubMed] [Google Scholar]
- 44. Pittock SJ, Lennon VA, de Seze J, Vermersch P, Homburger HA, Wingerchuk DM et al (2008) Neuromyelitis optica and non organ‐specific autoimmunity. Arch Neurol 65:78–83. [DOI] [PubMed] [Google Scholar]
- 45. Pohl M, Fischer MT, Mader S, Schanda K, Kitic M, Sharma R et al (2011) Pathogenic T cell responses against aquaporin 4. Acta Neuropathol (Berl) 122:21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Popescu BF, Lennon VA, Parisi JE, Howe CL, Weigand SD, Cabrera‐Gomez JA et al (2011) Neuromyelitis optica unique area postrema lesions: nausea, vomiting, and pathogenic implications. Neurology 76:1229–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ratelade J, Bennett JL, Verkman AS (2011) Intravenous neuromyelitis optica autoantibody in mice targets aquaporin‐4 in peripheral organs and area postrema. PLoS ONE 6:e27412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rostasy K, Mader S, Hennes E, Schanda K, Gredler V, Guenther A et al (2012) Persisting myelin oligodendrocyte glycoprotein antibodies in aquaporin‐4 antibody negative pediatric neuromyelitis optica. Mult Scler 19:1052–1059. [DOI] [PubMed] [Google Scholar]
- 49. Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC (2010) Intra‐cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 133 (Pt 2):349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Saadoun S, Waters P, Macdonald C, Bridges LR, Bell BA, Vincent A et al (2011) T cell deficiency does not reduce lesions in mice produced by intracerebral injection of NMO‐IgG and complement. J Neuroimmunol 235:27–32. [DOI] [PubMed] [Google Scholar]
- 51. Saadoun S, Waters P, MacDonald C, Bell BA, Vincent A, Verkman AS, Papadopoulos MC (2012) Neutrophil protease inhibition reduces neuromyelitis optica‐immunoglobulin G‐induced damage in mouse brain. Ann Neurol 71:323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sandhu A, Harford A, Singh P, Alas E (2012) Is thymoglobulin or rituximab the cause of this serum sickness? A case report of serum sickness dilemma and literature review. Case Rep Med 2012:234515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sellner J, Hemmer B, Muhlau M (2010) The clinical spectrum and immunobiology of parainfectious neuromyelitis optica (Devic) syndromes. J Autoimmun 34:371–379. [DOI] [PubMed] [Google Scholar]
- 54. Storch MK, Stefferl A, Brehm U, Weissert R, Wallstrom E, Kerschensteiner M et al (1998) Autoimmunity to myelin oligodendrocyte glycoprotein in rats mimics the spectrum of multiple sclerosis pathology. Brain Pathol 8:681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Suzuki N, Takahashi T, Aoki M, Misu T, Konohana S, Okumura T et al (2010) Neuromyelitis optica preceded by hyperCKemia episode. Neurology 74:1543–1545. [DOI] [PubMed] [Google Scholar]
- 56. Terris J, Ecelbarger CA, Marples D, Knepper MA, Nielsen S (1995) Distribution of aquaporin‐4 water channel expression within rat kidney. Am J Physiol 269 (Pt 2):F775–F785. [DOI] [PubMed] [Google Scholar]
- 57. Tradtrantip L, Zhang H, Anderson MO, Saadoun S, Phuan PW, Papadopoulos MC et al (2012) Small‐molecule inhibitors of NMO‐IgG binding to aquaporin‐4 reduce astrocyte cytotoxicity in neuromyelitis optica. FASEB J 26:2197–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tradtrantip L, Zhang H, Saadoun S, Phuan PW, Lam C, Papadopoulos MC et al (2011) Anti‐aquaporin‐4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann Neurol 71:314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Varrin‐Doyer M, Spencer CM, Schulze‐Topphoff U, Nelson PA, Stroud RM, Cree BA, Zamvil SS (2012) Aquaporin 4‐specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter. Ann Neurol 72:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang H, Dai Y, Qiu W, Zhong X, Wu A, Wang Y et al (2011) HLA‐DPB1 0501 is associated with susceptibility to anti‐aquaporin‐4 antibodies positive neuromyelitis optica in southern Han Chinese. J Neuroimmunol 233:181–184. [DOI] [PubMed] [Google Scholar]
- 61. Warabi Y, Matsumoto Y, Hayashi H (2007) Interferon beta‐1b exacerbates multiple sclerosis with severe optic nerve and spinal cord demyelination. J Neurol Sci 252:57–61. [DOI] [PubMed] [Google Scholar]
- 62. Wauben MH, van der Kraan M, Grosfeld‐Stulemeyer MC, Joosten I (1997) Definition of an extended MHC class II‐peptide binding motif for the autoimmune disease‐associated Lewis rat RT1.BL molecule. Int Immunol 9:281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Willison HJ, Linington C (2012) Antibodies to MOG in NMO: a seasoned veteran finds a new role. Neurology 79:1198–1199. [DOI] [PubMed] [Google Scholar]
- 64. Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG (2007) The spectrum of neuromyelitis optica. Lancet Neurol 6:805–815. [DOI] [PubMed] [Google Scholar]
- 65. Wingerchuk DM, Weinshenker BG (2012) The emerging relationship between neuromyelitis optica and systemic rheumatologic autoimmune disease. Mult Scler 18:5–10. [DOI] [PubMed] [Google Scholar]
- 66. Wucherpfennig KW, Strominger JL (1995) Molecular mimicry in T cell‐mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell 80:695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yamamoto K, Matsunaga S, Matsui M, Takeda N, Yamatodani A (2002) Pica in mice as a new model for the study of emesis. Methods Find Exp Clin Pharmacol 24:135–138. [DOI] [PubMed] [Google Scholar]
- 68. Yamasaki K, Horiuchi I, Minohara M, Kawano Y, Ohyagi Y, Yamada T et al (1999) HLA‐DPB1*0501‐associated opticospinal multiple sclerosis: clinical, neuroimaging and immunogenetic studies. Brain 122 (Pt 9):1689–1696. [DOI] [PubMed] [Google Scholar]
- 69. Yokoyama N, Niino M, Takahashi T, Matsushima M, Maruo Y (2012) Seroconversion of neuromyelitis optica spectrum disorder with hyperCKemia: a case report. Eur J Neurol 19:e143. [DOI] [PubMed] [Google Scholar]
- 70. Rat phenome database of the National BioResource Project for the Rat in Japan. Avalable at: http://www.anim.med.kyoto‐u.ac.jp/nbr/strainsx/hematology_list.aspx (accessed 19 November 2013).
- 71. Mouse phenome database. The Jackson Laboratory, Bar Harbor, ME. Available at: http://phenome.jax.org/db/qp?rtn=views/measplot&brieflook=6211&projhint=Peters1 (accessed 18 November 2013).