

Abstract
For allogeneic cell therapies to reach their therapeutic potential, challenges related to achieving scalable and robust manufacturing processes will need to be addressed. A particular challenge is producing lot-sizes capable of meeting commercial demands of up to 109 cells/dose for large patient numbers due to the current limitations of expansion technologies. This article describes the application of a decisional tool to identify the most cost-effective expansion technologies for different scales of production as well as current gaps in the technology capabilities for allogeneic cell therapy manufacture. The tool integrates bioprocess economics with optimization to assess the economic competitiveness of planar and microcarrier-based cell expansion technologies. Visualization methods were used to identify the production scales where planar technologies will cease to be cost-effective and where microcarrier-based bioreactors become the only option. The tool outputs also predict that for the industry to be sustainable for high demand scenarios, significant increases will likely be needed in the performance capabilities of microcarrier-based systems. These data are presented using a technology S-curve as well as windows of operation to identify the combination of cell productivities and scale of single-use bioreactors required to meet future lot sizes. The modeling insights can be used to identify where future R&D investment should be focused to improve the performance of the most promising technologies so that they become a robust and scalable option that enables the cell therapy industry reach commercially relevant lot sizes. The tool outputs can facilitate decision-making very early on in development and be used to predict, and better manage, the risk of process changes needed as products proceed through the development pathway. Biotechnol. Bioeng. 2014;111: 69–83. © 2013 Wiley Periodicals, Inc.
Keywords: allogeneic cell therapy manufacture, stem cells, single-use cell expansion, microcarriers, cell factories, bioprocess economics
Introduction
Allogeneic stem cell therapies will potentially be able to treat a broad range of unmet medical needs ranging from dry eye related macular degeneration to acute myocardial infarction. They are particularly promising for treating large patient numbers as they are obtained from a universal donor and are thus more suited to manufacturing at large scale. To date, commercialized allogeneic stem cell therapies include Prochymal (Osiris, Columbia, MD) for graft-versus-host disease, approved in Canada and New Zealand, and Cartistem (Medipost, Seoul, Korea) for osteoarthritis, approved in South Korea. However, several products have faced challenges achieving scalable, robust, and cost-effective manufacturing processes (Brandenberger et al., 2011; Griffith and Naughton, 2002; Kirouac and Zandstra, 2008; Ratcliffe et al., 2011; Rowley et al., 2012). This has contributed to several notable failures due to manufacturing concerns such as high cost of goods (COG), high process variability, and loss of efficacy upon scale-up (Brandenberger et al., 2011). Hence the commercial feasibility of cell therapies is underpinned by the need to solve the manufacturing challenges posed by large-scale production. This article investigates the technical innovation required in cell expansion technologies for cell therapy products to realize their commercial potential and achieve the manufacturing success of biopharmaceuticals.
Biopharmaceuticals, such as monoclonal antibodies (mAbs), have benefited from the availability of large-scale bioprocessing technologies and the associated economies of scale (e.g., Birch and Racher, 2006; Farid, 2006; Kelley, 2007). However, this is not currently the case for allogeneic cell therapy manufacture due to its relative infancy as well as the inherent complexities of manufacturing living cells as the final product. In contrast to mAbs, only a limited number of cases of xeno-free cell culture media for cell therapy products have been reported (Lindroos et al., 2009; Rajala et al., 2010). Additionally, the culture can also include feeder cells to promote growth. Although cell lines used to generate mAbs are adapted to suspension culture, most cell therapies require adherent culture. This introduces challenges for scale-up to commercial demands. Following expansion and potential differentiation of cell therapy products, the cells are typically washed and centrifuged for cell concentration and recovery. Large-scale centrifuges such as disc stacks (Kempken et al., 1995) used in mAb manufacture are not suitable for the processing of shear-sensitive cells, and hence the use of alternative single-use recovery equipment such as closed continuous fluidized bed centrifuges (e.g., kSep® Systems, Durham, NC) is being explored. The transportation of cell therapy products is also more complex since the product cannot be lyophilized (freeze-dried) as is the case with mAbs; cell therapy products are typically delivered either fresh or cryopreserved (frozen). Both delivery options necessitate costly cold chains with the fresh state requiring careful and timely inventory management between manufacturing sites and the clinic and the cryopreserved state requiring clinics to maintain the frozen state in expensive freezers until the time of patient administration. Cryopreservation is more typical for allogeneic cell therapy treatments given the benefits of “off-the-shelf” inventory when creating many doses per lot to treat large numbers of patients. Considering facility design distinctions, although single-use technologies are increasingly being adopted in mAb manufacture for vessels typically below 2000L, their adoption is essential in cell therapy bioprocessing due to sterility concerns (Lapinskas, 2010). In contrast to the well-established mAb sector, the poor automation, labor-intensive, and more open nature of cell therapy manufacture makes it more prone to operator-mediated variability (Lopez et al., 2010), and contamination risks. Closed and automated technologies are now becoming available to address the need for greater process robustness and reproducibility.
Table I highlights several allogeneic cell therapy treatments and their current phase of development. Most are in the clinical trials stage. To date, allogeneic cell therapy products in development have mainly consisted of mesenchymal stem cells or mesenchymal progenitor cells derived from the bone-marrow (Kebriaei et al., 2009; Goldschlager et al., 2011; Vaes et al., 2012), umbilical-cord blood (Jung et al., 2011), liver (Christ and Stock, 2012), or adipose (DelaRosa et al., 2012). Promising results have also been observed for cells differentiated from embryonic stem cells such as retinal pigment epithelial cells (Schwartz et al., 2012), or from fetuses for neuronal stem cell production (Miljan and Sinden, 2009; Tamaki et al., 2002). Table I highlights that the doses (cells/patient) typically used for cell therapy products currently range from 105 cells for indications such as dry eye related macular degeneration to 109 cells for liver disease, GvHD or cardiac disease (e.g., infarction; Reinecke et al., 2008). Accordingly, a maximum dose of 109 cells is investigated in this study. Most companies in clinical trials are using 2-D multi-layer vessels (e.g., 10-layer Cell Factories (Nunc, ThermoFischer Scientific, Waltham, MA) CellSTACKs (Corning Incorporated Life Sciences, Tewksbury, MA) as their main expansion technology (Rowley et al., 2012). It is estimated that up to 1012–1013 cells will need to be produced per lot to meet commercial demands of high dose cell therapies. This would represent the use of 10,000–100,000 10-layer vessels per lot (values calculated by the model described in this article). However, only 50–100 vessels can be handled per lot due to the need to perform labor-intensive handling tasks, rendering this type of system unsuitable for large-scale production.
Table I.
Characteristics of allogeneic cell therapies currently under development
| Indication | Cell types under investigation | Dose for clinical trials (cells/dose)a |
|---|---|---|
| Acute kidney injury | Bone-marrow derived hMSCs | 2 × 108 [1] |
| Acute myocardial infarction | Bone marrow or other nonembryonic tissue source-derived Multistem | 0.2, 0.5, 1 × 108 [2] |
| Chronic Discogenic Lumbar Back Pain | Bone-marrow derived adult mesenchymal precursor cells | 0.6–1.8 × 107 [1] |
| Congestive heart failure | Bone-marrow derived adult mesenchymal precursor cells | 1.5 × 108 [3] |
| Critical limb ischemia | Placenta-derived PLX-PAD stromal cells (hMSC-like) | 1.5–3 × 108 [4] |
| Crohn's disease | Adipose-derived expanded stem cells (eASCs); Bone-marrow-derived hMSC | 2, 4 × 107 [5]; 6–12 × 108 [6] |
| Dry eye related macular degeneration | Embryonic Stem Cell-Derived Retinal Pigment Epithelial (RPE) Cells | 0.5–2 × 105 [1] |
| Graft vs. host disease | Umbilical cord blood-derived hMSC; Bone-marrow-derived hMSC; Bone marrow or other nonembryonic tissue source-derived Multistem | 1–5 × 108 [1]; 1.6 × 109 [1]; 0.5–1 × 109 [7] |
| Intracerebral hemorrhage (ICH) | Bone-marrow derived hMSCs | 7.8 × 106 [1] |
| Ischemic stroke | Human foetal brain stem cell-derived hNSC; Bone-marrow-derived hMSC; Bone-marrow derived hMSC | 2 × 107 [8]; 0.5 −1.5 × 108 [1]; 2 × 108 [1] |
| Liver disease | Adipose-derived stromal cells | 0.1–1 × 109 [1] |
| Osteoarthritis | Bone-marrow- derived hMSC; Umbilical cord blood-derived hMSCs (hUCB-MSCs) | 5–15 × 107 [1]; 3.5 × 107 [11] |
| Peripheral vascular diseases | Menstrual blood-derived Endometrial regenerative cells (hMSC-like) | 0.25–1 × 108 [1] |
| Prostate cancer | Prostate tumour-derived cancer cell line | 2–4 × 107 [12] |
| Rheumatoid arthiritis aggravated | Adipose-derived expanded stem cells (eASCs) | 1–4 × 108 [1] |
| Spinal cord injury | hESC-derived oligodendrocyte progenitor cells; Foetal-derived hNSCs; Brain-derived hNSCs | 2 × 106 [13]; 2 × 107 [10]; 1 × 108 [13] |
| Type I diabetes | Bone-marrow-derived hMSC | 6 × 108 [6] |
| Type II diabetes | Bone-marrow derived adult mesenchymal precursor cells | 0.1, 0.3, 1, 2 × 108 [14] |
| Ulcerative colitis | Bone-marrow-derived multipotent adult progenitor cell (MAPC) | 1.8 × 108 [11,15] |
All doses are relevant to the phase of the trial reported in the literature source indicated in brackets. 100kg body weight was assumed where relevant.
2. Penn et al. (2012).
9. WIPO: WO/2008/002523.
11. http://advbiols.com/documents/Bravery-AreBiosimilarCellTherapiesPossible.pdf and http://clinicaltrials.gov/ct2/show/record/NCT01041001 (500 μl/cm2 of cartilage defect at 5 × 106 cells/ml, and assuming the area for treatment is similar to that for knee sports injury (Mason and Dunnill, d) i.e. 2 × 7 cm2 or 14 cm2.
14. http://www.mesoblast.com/newsroom/asx-announcements/archives/ (10 November 2011).
The need for closed systems to limit the potential points of contamination, to produce more cells per unit footprint, and for greater upstream production control has driven the production of compact multi-layered systems (e.g., HYPERStack (Corning, Incorporated Life Sciences, Tewksbury, MA)), multi-layer bioreactors (e.g., Integrity Xpansion unit (ATMI, Danbury, CT)), and scalable microcarrier-based bioreactor systems. To successfully meet higher future demands, it is necessary to determine the practical and economic feasibility of each technology.
To date, there has been a limited number of studies addressing impact on costs and expansion technology limitations in the cell therapy industry. Hambor (2012) identified the probable need for increasing automation and controlled bioreactor systems for the production of clinical grade cell therapy products. Automation has been advanced by the introduction of robotically controlled equipment such as TAP Biosystems' SelectT and CompactT systems, which increase the potential of T flasks. Prior studies estimating the number and type of expansion technologies required to meet a demand (Rowley et al., 2012; Want et al., 2012) were solely based on technical inputs such as surface area, size, and density. In another study, based on interviews and various model assumptions, Malik (2012) estimated the cost to produce allogeneic cell therapy products for a fixed demand of 2,500 doses/year, where a single dose represented 108 mesenchymal stem cells using T-flasks with automation.
This article presents an integrated decisional tool combining bioprocess economics and optimization for addressing cell therapy manufacturing challenges. The detailed cost model accounts for both technical inputs such as media requirements as well as financial inputs such as resource costs. The bioprocess economics model presented in this article focuses on the upstream processing cost of goods (COGUSP) components that are expected to be more affected by the choice of different expansion technologies for allogeneic cell therapy manufacture, that is, raw materials (particularly cell culture media and single-use technologies), labor, and depreciation of equipment directly related to the cell expansion step. It also incorporates QC costs associated with lot release testing such that different manufacturing options in terms of lot size and number of lots per year can be compared.
The novelty of this work lies in the integration of bioprocess economics and optimization approaches to systematically assess the economic competitiveness of planar and microcarrier-based cell expansion technologies, predict the optimal and most cost-effective technology for different scales, identify gaps in the available technologies and predict future performance targets necessary to meet commercial demands for cell therapies.
Tool Description
Overview
The allogeneic cell therapy manufacturing challenge addressed in this article is to meet a demand determined by estimated dose (number of cells) with a process where performance can be determined by seeding and harvest cell densities, number of expansion stages, and yield. The goal is to identify the optimal type and size of cell expansion technology to be used at each expansion stage, to help ensure that the COGUSP is minimized and demand targets are met.
A decisional tool was developed to address this challenge, which integrates a bioprocess economics model, an optimization algorithm and a database, and is implemented in C# (C-sharp) using the .NET framework (Microsoft® Visual Studio 2010, Microsoft Corporation, Redmond, WA) linked to Microsoft® Access (Microsoft Corporation). The bioprocess economics model predicts multiple technical and financial performance measures of a particular process configuration. The optimization algorithm generates alternative process configurations and uses the bioprocess economics model to evaluate each alternative. The database stores input data to be used by the bioprocess economics model and optimization algorithm in addition to the output data that results from running those procedures. The tool also comprises a graphical user interface. The focus of this article is on the development and application of the core components of the optimization framework for the cell expansion stage in allogeneic cell therapy manufacture, as illustrated in Figure 1.
Figure 1.
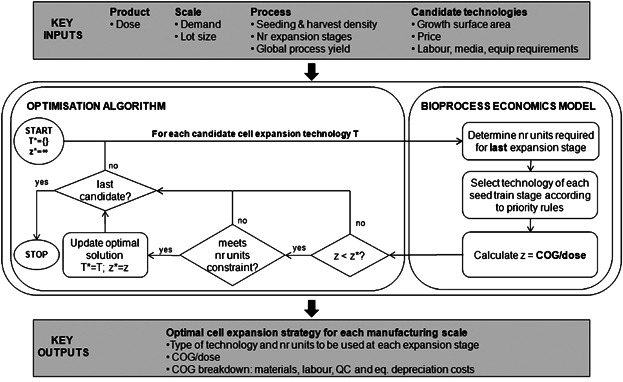
Cell expansion optimization framework.
Bioprocess Economics Model
The bioprocess economics model was configured to perform equipment sizing and resource consumption calculations and consequently to determine the value of the upstream processing COG per dose (COGUSP/dose) of a particular process configuration. In this article, COGUSP/dose comprised the annual direct (materials, labor and QC) and indirect (equipment depreciation) operating costs divided by the annual product output (number of doses/year). Key to the evaluation of the cost of a cell expansion process is the type of technology used and the number of units necessary to obtain the required number of cells. For a given product with dose M (cells/dose) and harvest density 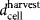 (cells/cm2) and a manufacturing lot size
(cells/cm2) and a manufacturing lot size 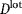 (doses/lot), the number of units of a particular technology i required for the last cell expansion stage N,
(doses/lot), the number of units of a particular technology i required for the last cell expansion stage N, 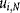 was calculated by the bioprocess economics model as follows:
was calculated by the bioprocess economics model as follows:
| (1) |
where y is the overall yield of the downstream operations (e.g., volume reduction, filling) and ai is the growth surface area (cm2) per technology i unit. For microcarrier-based systems using single-use bioreactors (SUB), the value of ai is calculated by:
| (2) |
where amicrocarrier is the growth surface area per gram of microcarrier (cm2/g), cmicrocarrier is the concentration at which the microcarriers are seeded into the bioreactor (g/L), Vbior,i is the total volume of the bioreactor and λ is the bioreactor working volume ratio.
The type of technology to be used in the expansion seed train was determined by a set of rules that take into account the compatibility between different technology types. The number of technology units i to be used in the expansion seed train (stage j = 1, …, N − 1) was calculated by:
| (3) |
where T is the technology used in stage j + 1 and 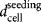 is the cell seeding density (cells/cm2).
is the cell seeding density (cells/cm2).
Once the type of technology and number of units to be used at each expansion stage were defined, the bioprocess economics model calculated the value of the objective function COGUSP/dose as follows:
| (4) |
where 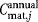 ,
, 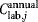 ,
, 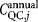 , and
, and 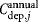 are the total annual material, labor, QC, and equipment depreciation costs, respectively, for each expansion stage and Dannual is the annual demand (doses/year). The demand can be obtained by different combinations of lot size (doses/lot) and number of lots per year, as explored in the case study.
are the total annual material, labor, QC, and equipment depreciation costs, respectively, for each expansion stage and Dannual is the annual demand (doses/year). The demand can be obtained by different combinations of lot size (doses/lot) and number of lots per year, as explored in the case study.
The material costs were designed to account for the consumables and media directly used in each expansion stage:
| (5) |
where pconsum,i, Vmedia,i, and ai are the unit consumables price, the media requirements (mL/cm2) and the surface area of technology i, respectively, pmedia is the media price and Lannual is the number of lots per year. For microcarrier-based systems, the material costs included media costs, the cost of the microcarriers and the costs with SUB bags:
| (6) |
where pmicrocarier is the unit price of microcarriers ($/g) and pSUB,i is the price of a SUB bag of size i.
Labor costs were based on the total operator time required to perform manual cell expansion tasks including seeding, feeding, and harvesting:
| (7) |
where tseed,i, tfeed,i, and tharvest,i represent the time required for an operator to perform the manual operations associated with seeding, feeding, and harvesting of cell expansion vessels, 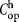 is the labor hourly wage, and β is a multiplier to account for other labor costs (e.g., supervisors and management).
is the labor hourly wage, and β is a multiplier to account for other labor costs (e.g., supervisors and management).
QC costs comprised the range of studies required for testing a lot prior to release and a fixed value (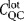 ) was incurred per batch:
) was incurred per batch:
| (8) |
The indirect costs considered here were the equipment depreciation costs for equipment directly related to the handling of the cell expansion technologies. This value is proportional to the total facility-dependent overhead costs. The cost of ancillary equipment (e.g., controllers, automation units), incubators and biosafety cabinets was calculated taking into account their capacity and unit price and the total was divided by the depreciation period to obtain the annual equipment depreciation costs:
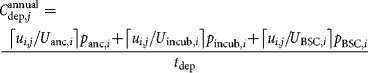 |
(9) |
where Uanc,i, Uincub,i, and UBSC,i are the capacities of the different types of equipment in terms of number of units of technology i each can handle per lot, panc,i, pincub,i, and pBSC,i are the corresponding prices and tdep is the depreciation period. The capacity of a biosafety cabinet (UBSC,i) was derived assuming that it could only be used by one operator at a time.
The value of COGUSP/dose was then used by the optimization algorithm to select the most cost-effective technology, as described in the following section.
Optimization Algorithm
The optimization algorithm implemented in the tool consisted of an enumeration procedure that screened through all the cell expansion technologies and selected the most cost-effective alternative for meeting pre-defined manufacturing constraints. The cost-effectiveness was evaluated by the bioprocess economics model, as described in the previous section. The manufacturing constraints defined the maximum number of cell expansion technology units that could be handled per lot in the last expansion stage such that:
| (10) |
where umax is a user-defined parameter that can have different values for different technology types. As shown in Figure 1, the optimal solution determined by the tool was the expansion technology that gave rise to the lowest COGUSP/dose within the manufacturing constraints defined by Equation 2012. In some cases, there may be scenarios where none of the candidate technologies are able to meet the maximum number of units constraint, and this is discussed in the Results and Discussion section.
Case Study Setup
An industrially relevant case study was set up to illustrate and examine the ability of the proposed tool to discover optimal cell expansion strategies for the design of cell therapy manufacturing processes. The case study focuses on therapies using mesenchymal stem cells derived from bone marrow. Different allogeneic cell therapy products are considered, with doses within the range identified from Table I and with potential for high commercial demands of up to 500,000 doses/year (e.g., assuming a 10% market share of a 5 million patient population, as indicated for heart disease by Mason and Dunnill, 2009). The goal of the study was to investigate which commercially available technologies would be the most cost-effective for meeting production demands. This analysis would allow for resources to be allocated appropriately for relevant experimental validation and optimization of the most promising technologies at earlier stages of development, thus potentially reducing risk.
Table II presents the different planar technologies evaluated for cell expansion and specific characteristics of each candidate, generated using information from literature, vendor communications, as well as advice sought from industrial experts so as to capture trade-offs in surface area, cost, equipment, and labor requirements. Six types of planar technologies were considered and generic names were given: T-flasks (T), multi-layers (L), compact flasks (cT), compact multi-layers (cL), multi-layer bioreactors (bL), and hollow-fiber bioreactors (HF). Examples of associated commercial names are shown in the footnote of Table II. Each type of technology is sized by surface area or the number of layers and this is represented by numerical values (e.g., T175 is a T-flask with 175 cm2 of surface area and L-10 has 10 layers). The use of additional automation equipment is indicated by the suffix “(aut),” as in the case of L-40 and cL-120. It is assumed that these two technologies have a similar footprint and 4 units can be handled simultaneously by a robot (automated cell factory manipulator, ACFM) performing seeding (filling) and harvesting (emptying) operations. The use of microcarriers in SUBs was also considered as a candidate technology for cell expansion but only for those demand scenarios where the use of planar systems would exceed the maximum number of units imposed by Equation 2012. This was implemented to reflect the current industrial preference for planar cell expansion technologies. The parameters used for different SUB sizes are presented in Table III. The values for the surface area (cm2/g) and density (g/L) of the microcarrier-based technology assumed in the case study were based on literature data. Ranges of 360–5,500 cm2/g and 3.3–9.3 g/L have been reported in the literature for the expansion of adult stem cells using non-porous microcarriers, as shown in Table IV. The mid-point values of 2,930 cm2/g and 6.3 g/L were used for the microcarrier surface area and density, respectively. Although these values were initially used to estimate the required size and number of SUBs to meet large demands, a sensitivity analysis was subsequently carried out in order to determine the impact of the variation of the microcarrier surface area on the optimal expansion strategy across different demand scenarios.
Table II.
Key parameters for candidate planar cell expansion technologies
| Type | Name | Surface area, ai (cm2) | Consumables unit price, pconsum ($) | Media req., Vmedia (mL/cm2) | Labor requirements (time per operator to handle max # units) | Requires biosafety cabinet | Incubator capacity, Uincub (# units) | Ancillary control and automation equipment | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Seed time, tseed (h) | Feed time, tfeed (h) | Harvest time, tharvest (h) | Max # unitsa | Capacity, Uanc (# units) | Price, panc ($) | |||||||
| T-flasks | T175 | 175 | 9 | 0.25 | 0.38 | 0.38 | 0.75 | 10 | Y | 100 | — | — |
| T225 | 225 | 10 | 0.25 | 0.38 | 0.38 | 0.75 | 10 | Y | 100 | — | — | |
| T500 | 500 | 15 | 0.40 | 0.38 | 0.38 | 0.75 | 10 | Y | 100 | — | — | |
| Multi-layersb | L-1 | 636 | 60 | 0.25 | 0.15 | 0.15 | 0.30 | 1 | Y | 60 | — | — |
| L-2 | 1,272 | 73 | 0.25 | 0.15 | 0.15 | 0.30 | 1 | Y | 60 | — | — | |
| L-5 | 3,180 | 241 | 0.25 | 0.20 | 0.20 | 0.40 | 1 | Y | 24 | — | — | |
| L-10 | 6,360 | 507 | 0.25 | 0.25 | 0.25 | 0.50 | 1 | N | 12 | — | — | |
| L-40 (aut) | 25,440 | 1,265 | 0.25 | 0.08 | 0.08 | 0.17 | 4 | N | 16b | 16 | 425,000 | |
| Compact flasksd | cT | 1,720 | 19 | 0.33 | 0.38 | 0.38 | 0.75 | 10 | Y | 100 | — | — |
| Compact multi-layerse | cL-12 | 6,000 | 575 | 0.22 | 0.20 | 0.20 | 0.40 | 1 | N | 24 | — | — |
| cL-36 | 18,000 | 1,050 | 0.22 | 0.25 | 0.25 | 0.50 | 1 | N | 12 | — | — | |
| cL-120 (aut) | 60,000 | 3,000f | 0.20 | 0.08 | 0.08 | 0.17 | 4 | N | 16c | 16 | 425,000 | |
| Multi-layer bioreactorsg | bL-10 | 6,360 | 2,506 | 0.27 | 0.75 | 0.25 | 0.50 | 1 | N | 6 | 1 | 56,000 |
| bL-50 | 31,800 | 5,586 | 0.19 | 0.75 | 0.25 | 0.50 | 1 | N | 4 | 1 | 56,000 | |
| bL-180 | 114,480 | 13,986 | 0.17 | 0.75 | 0.25 | 0.75 | 1 | N | 2 | 1 | 56,000 | |
| Hollow fiber bioreactorsh | HF | 21,000 | 12,000 | 0.37 | 0.20 | 0 | 0.20 | 1 | N | — | 1 | 150,000 |
Max # units = Maximum number of units that can be handled by one operator simultaneously.
For example, Cell Factory systems (Nunc), CellSTACK (Corning).
It is assumed that L-40 and cL-120 use a specific incubator, while the other systems use a typical double-stack incubator.
For example, HYPERFlask (Corning).
For example, HYPERStack (Corning).
Price of cL-120 not available, calculated based on cL-36 price ($25/layer).
For example, Integrity Xpansion (ATMI).
For example, Quantum (TerumoBCT).
Table III.
Key process and cost assumptions used in the case study
| Process parameter | Value |
|---|---|
| Process data | |
| Number of expansion stages (N) | 4 |
Seeding density 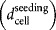
|
3,000 cells/cm2 |
Harvest density 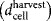
|
25,000 cells/cm2 |
| Overall process yield (y) | 85% |
| Maximum # units/lot (umax for planar technologies) | 80 |
| Maximum #SUBs/lot (umax for microcarriers) | 8 |
| Microcarrier surface area (amicrocarrier) | 2930 cm2/g |
| Microcarrier seed concentration (cmicrocarrier) | 6.3 g/L |
| Single-use bioreactor working volume ratio (λ) | 75% |
| Cost parameter | Value |
|---|---|
| Cost data | |
| Cell culture media (pmedia) | $150/L |
| Microcarriers (pmicrocarriers) | $5/g |
| Single-use bioreactor bag (pSUB(Vbior)) | $2,000 (20L); $4,500 (200L); $5,850 (500L); $8,850 (1000L); $10,500 (2000L) |
| Single-use bioreactor support equipment (panc for microcarriers (Vbior)) | $185,000 (20L); $215,000 (200L); $320,000 (500L); $425,000 (1000L); $575,000 (2000L) |
| L-40/cL-120 incubator (pincub for L-40 and cL120 systems) | $30,000 |
| Double stack incubator (pincub for other systems) | $17,835 |
| Biosafety cabinet (pBSC) | $17,000 |
Operating labor 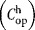
|
$200/h |
QC testing 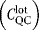
|
$10,000/lot |
| Other labor cost multiplier (β) | 2.2 |
| Depreciation period (tdep) | 10 years |
Table IV.
Reported ranges for microcarrier surface area and density values for mesenchymal stem cells
| Type of microcarrier | Surface area (cm2/g) | Density (g/L) |
|---|---|---|
| Non-porous | 360–5,500 | 3.3–9.3 |
| Porous | 11,000–15,000 | 1–5 |
Non-porous = Cytodex I, II, III (GE Healthcare), MicroHex (Thermo Fisher Scientific), and Plastic (SoloHill Engineering).
Porous = CultiSpher S and G (Percell Biolytica), Cytopore II (GE Healthcare).
The tool was run for different scenarios in terms of annual demand (1,000–500,000 doses/year) and manufacturing lot size (50–10,000 doses/lot) in order to determine the most cost-effective cell expansion technologies and identify the limits of existing technologies. The key process and cost parameters used in the model for the case study are shown in Table III.
Results and Discussion
This section presents insights from the cost modeling and optimization studies for adherent cell expansion technologies for allogeneic cell therapies. The economic competitiveness of commercially available planar and microcarrier-based cell expansion technologies is initially discussed across different production scales. A sensitivity analysis study is then presented to identify the most critical model parameters and hence the key cost drivers. A technology S-curve is proposed to visualize the performance trajectories of each technology. The analysis is extended to identify targets for microcarrier-based technologies to meet potential commercial lot sizes.
Economic Competitiveness of Commercially Available Cell Expansion Technologies
The tool was used initially to determine the cost-effectiveness of commercially available planar expansion technologies for different scales. Figure 2 illustrates how the optimal cell expansion technology changes across a matrix of demands and lot sizes for four different doses (106–109 cells/dose); the doses are representative of the range of doses reported for allogeneic cell therapy treatments in Table I. Figure 2e shows the required number of lots necessary to meet the demand, for each combination of demand and lot size. As the focus of the article is on commercial manufacturing, only manufacturing options with a minimum of 10 and a maximum of 200 lots/year were considered. Each individual matrix cell shows the optimal technology for a particular combination of demand and lot size, and the number of units required per lot (within brackets) for the final stage of the expansion process train. For L-40 and cL-120 the values shown within brackets represent the number of automated units used (i.e., number of robots (ACFM) handling up to 4 units each).
Figure 2.
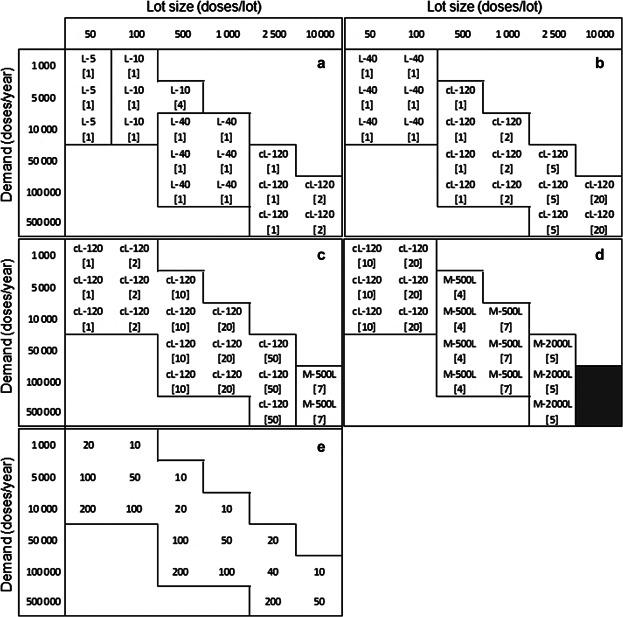
Optimal cell expansion technologies across a matrix of demands and lot sizes for a dose of (a) 106 cells, (b) 107 cells, (c) 108 cells, and (d) 109 cells. Each matrix cell shows the name of the optimal technology for a particular combination of demand and lot size and the number of units required per lot (inside brackets). For L-40 and cL-120 the value inside brackets represents the number of automated units required (i.e., number of sets of 4 units). The use of microcarriers was allowed only when the maximum number of units was exceeded for all planar technologies. The gray areas represent production scenarios that cannot be met by any candidate technology. Matrix (e) shows the number of lots run per year for each combination of demand and lot size.
A closer examination of Figure 2a reveals trends in the characteristics of the optimal technologies. Along the matrix diagonal, the cells per lot increase and the optimal technologies have increasing surface areas per unit. Moving vertically down the matrix in Figure 2a, the number of lots per year increases and the optimal technologies switch to those that can be automated. For example, for a lot size of 500 doses (Fig. 2a) the tool predicts a shift in competitiveness from four L-10 vessels to one ACFM with four L-40 units. This is because labor costs increase with the number of lots and thus, having automation reduces its contribution to the final COGUSP/dose value. In addition, as the dose size increases (Fig. 2a–d), a greater number of cells is produced per lot and the matrices show the increasing need for technologies with larger surface areas at smaller lot sizes. This is observed, for example, by the solution space of the matrix shifting to the left when moving from Figure 2a (106 cells/dose) to Figure 2b (107 cells/dose).
The data used to generate the optimal matrices were further examined so as to understand the inflection points where the ranking of competing technologies switches (Fig. 3a) and the associated cost drivers influencing the selection of the optimal technologies (Fig. 3b). Figure 3a presents a cost comparison between L-40 and cL-120 in Figure 2b (107 cells/dose) across the matrix row with a demand of 10,000 doses/year. The figure also includes a comparison with L-10 vessels, as this technology represents the typical manufacturing option currently being used in industrial settings. Figure 3b illustrates that for small lot sizes of 50 doses/lot, QC costs dominate the COGs (>50%) followed by labor (21–33%) and material costs (14–25%), due to the high number of lots (200) necessary to meet the demand. The fixed equipment depreciation costs are not significant since they are spread over several lots. The optimal solution L-40 (represented by point A) achieves cost savings through minimizing material costs given its lower consumable unit price relative to cL-120. L-10 has the largest proportion of labor costs due to the manual handling of the vessels, while L-40 and cL-120 are automated. For the opposite scenario of fewer but larger batches (1,000 doses/lot), the technology ranking switches from L-40 to cL-120. QC costs no longer dominate since they are proportional to the number of lots. Larger lots require a higher number of vessels and hence the optimal technology, cL-120 (point B), achieves cost savings by minimizing the total number of units required, given its larger surface area per unit. The use of two automated units (cL-120), instead of five (L-40), results in lower equipment depreciation and labor costs. (Although material costs dominate at these larger scales, they are similar for both technologies at this lot size and demand combination.)
Figure 3.
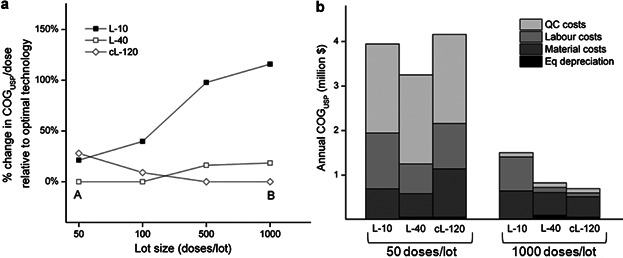
Comparison between L-10, L-40, and cL-120 for a fixed demand of 10,000 doses/year and across different lot sizes for a dose of 107 cells in terms of (a) % change in COGUSP/dose relative to optimal technology and (b) COGUSP structure. (a) and (b) are the optimal solutions for lot sizes of 50 and 1,000 doses.
The impact on COGUSP/dose of using L-10 vessels instead of the optimal technologies determined by the tool is significant, with a 17% increase in the COGUSP/dose value relative to the optimal solution for 50 doses/lot and 138% for 1,000 doses/lot. Labor costs dominate due to the manual handling of large numbers of units. For 1,000 doses/lot, 74 units/lot are required and therefore the maximum number of units constraint is still met (80). However for a dose of 108 cells, 740 units would be required per lot leading not only to the violation of the maximum number of units constraint but also to a 232% increase in COGUSP/dose relative to the optimal strategy. These insights are valuable for a company currently using L-10 vessels for an early phase product since they provide greater understanding of the financial and logistical impact of continuing to use such vessels for commercial production. Hence such analysis can be used to anticipate early in development, the commercial feasibility of processes and thus prioritize investment and development efforts in alternative technologies.
Point B in Figure 3a represents the most cost-effective manufacturing strategy for meeting the demand of 10,000 doses/year for a product with 107 cells/dose and this highlights the importance of an efficient equipment and facility utilization to lower depreciation costs per dose. The optimal manufacturing configuration for a lot size of 1,000 doses/lot is to run 10 lots/year using two automated cL-120 units/lot for the final (4th) stage of cell expansion. The expansion seed train generated by the tool for this configuration, using the rules defined in the Bioprocess Economics Model section, was: three T-500 units for stage 1, six cT units for stage 2, and four cL-36 units for stage 3. The model assumed that enough cells are available from a master cell bank to seed the T-flasks at stage 1.
Microcarrier-based systems were only used in production scenarios that could not be met by any planar technologies because the number of units required per lot exceeded the maximum limit constraint (i.e., 80 units), as shown in the last column of Figure 2c and in most columns of Figure 2d. However, the tool outputs also identified the demands where the base case non-porous microcarrier-based systems would also exceed the number of units constraint (i.e., 8 units) as illustrated by the gray area in Figure 2d. This emphasizes the need for technologies with larger growth surface area for expansion and indicates that available technologies are not feasible for large lot sizes with high dose products. Microcarrier SUBs are more capable of matching very high demands and lot sizes, but the gray region indicates that a gap still exists for theoretical maximum lot size scenarios (Fig. 2d). The use of seven 500L SUBs per lot allows the manufacture of up to 5,000 doses/lot for a dose of 108 cells (Fig. 2c) but this value drops to 1,000 doses/lot for higher dose values of 109 cells (Fig. 2d). To be able to meet the maximum demand of 10,000 doses it would be necessary to run 17 × 2000L SUBs per lot, which violates the constraint imposed on the maximum number of SUBs that can run in parallel 2011.
The previous analysis assumed that planar cell expansion technologies were preferred over microcarrier-based systems. If no such preference existed, then the cost savings of using microcarriers in SUBs could be substantial. For example, the use of 1 × 20L SUB instead of 1 × cL-120 for the production of 500 × 107 cells/lot (Fig. 2b, lot size = 500, demand = 100,000) would represent a decrease in COGUSP of 40%, while the use of 1 × 1000L SUB instead of 50 × cL-120 to produce 2.5 × 1011 cells/lot (Fig. 2c, lot size = 2,500, demand = 50,000) would allow savings of 73%.
Sensitivity Analysis to Identify Key Cost Drivers
A sensitivity analysis on the key yield parameters (harvest density and DSP yield) and resource costs was carried out to identify the most critical parameters in the bioprocess economics model that influence the cost outcome. The tornado diagrams in Figure 4 represent the impact of changing each of the parameters by ±30% on COGUSP/dose. Figure 4a–c represents the use of different planar cell expansion technologies (L-10, Fig. 4a; L-40, Fig. 4b; cL-120, Fig. 4c) for the manufacturing scenario highlighted in Figure 3 with dose = 107 cells, annual demand = 10,000 doses and lot size = 1,000 doses. Figure 4d shows the tornado diagram for a production scenario using 2000L SUB with microcarriers, where microcarrier-related model parameters were also included.
Figure 4.
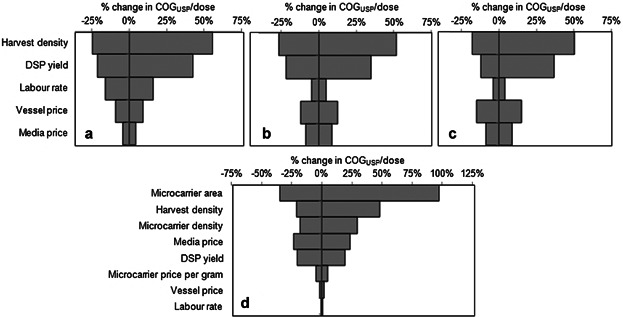
Tornado diagrams showing the sensitivity of COGUSP/dose to the key bioprocess economics model parameters. Results are shown for manufacturing scenarios where the following cell expansion technologies are used per lot in the base case scenario: (a) 74 × L-10 vessels, (b) 19 × L-40 handled by five ACFMs, (c) 8 × cL-120 handled by two ACFMs, (d) 5 × M-2000L bioreactors with microcarriers. The corresponding values of dose, demand, and lot size are: (a), (b), (c) dose = 107 cells, demand = 10,000 doses/year, lot size = 1,000 doses/lot, (d) dose = 109 cells, demand = 50,000 doses/year, lot size = 2,500 doses/lot. The base case values of each parameter are shown in Table III. For each parameter the base case values were changed by ±30% to generate the plots. The vertical axis intersects the horizontal axis at the base case value in each diagram.
For planar technologies, the harvest density (amount of cells harvested per cm2) was the parameter with the highest impact on the value of COGUSP/dose. This is to be expected since the harvest density dictates the yield of the cell expansion technologies and hence the number of expansion units required. Of the resource costs, the labor rate was found to be significant for L-10 (Fig. 4a) due to the labor-intensive nature of the handling tasks but not so important for L-40 and cL-120 (Fig. 4b and c) due to the automation of these tasks. Instead, the vessel price was found to be the most significant resource cost driving the COGUSP/dose value for the larger automated vessels (L-40 and cL-120). For manufacturing scenarios where microcarriers are used in 2000L SUB, the most critical model parameter affecting COGUSP/dose was found to be the microcarrier surface area (Fig. 4d), followed by harvest density and microcarrier density. All these parameters influenced the amount of cells that could be obtained from a particular setting leading to an increase or decrease in the number of units required to meet the demand. The impact of changes in these critical parameters on the performance of the microcarrier option was then explored further (see Figs. 5 and 7). Media price was also found to be significant due to the larger volumes of media handled in SUB relative to planar technologies.
Figure 5.
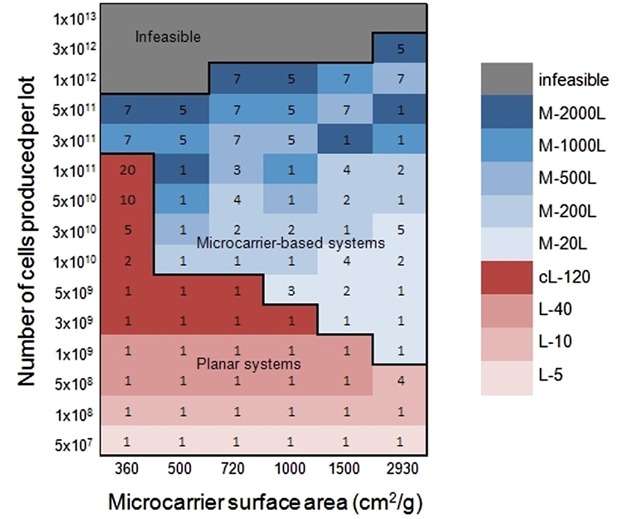
Impact of microcarrier surface area on the optimal cell expansion strategy across different lot sizes (number of cells produced per lot). The numbers inside the plot represent the number of units of the optimal technology required for the last expansion stage, for each combination of microcarrier surface area and number of cells produced per lot. For L-40 and cL-120, the value represents the number of automated units required (i.e., number of sets of 4 units). The gray areas represent production scenarios that cannot be met by any candidate technology.
Figure 7.
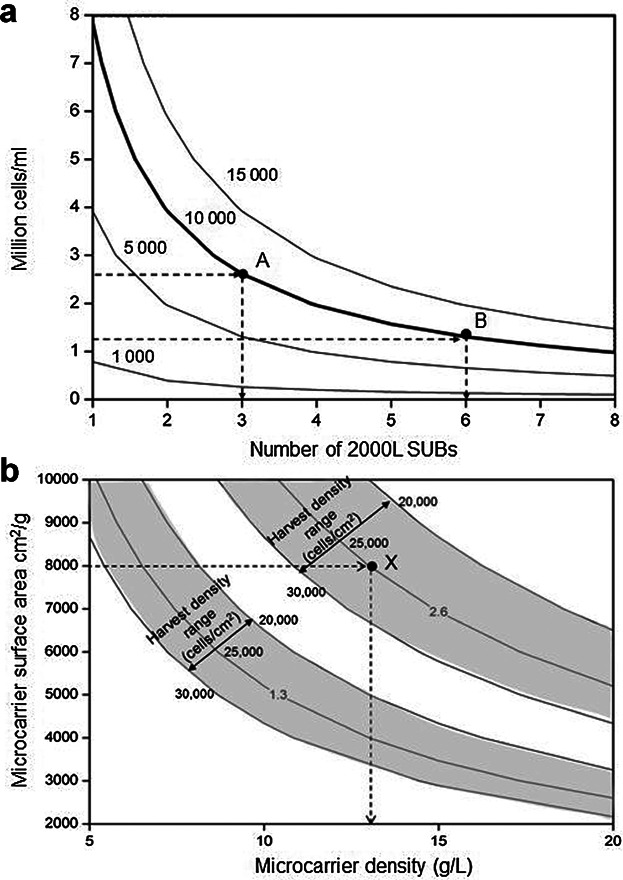
Contour plots showing characteristics of required future microcarrier performance. (a) Billion cells per lot achieved as a function of the number of 2000L SUBs used and the million cells/mL present in the microcarrier culture. The bold line represents the target of 10,000 billion cells/lot that can be achieved using different configurations including points A and B. (b) Million cells/mL achieved in a microcarrier culture as a function of the microcarrier density and surface area. The shaded areas highlight zones with the same value of million cells/mL (1.3 or 2.6) that can be achieved with harvest densities ranging from 20,000 cells/cm2 (upper limit of shaded area) to 30,000 cells/cm2 (lower limit of shaded area). X represents a possible setup to achieve 2.6 × 106 cells/mL.
Although the base case process analyzed in the article is representative of current mesenchymal stem cells manufacturing processes, experimental validation of the key model parameters would be necessary to apply the proposed modeling framework to a specific cell therapy process.
The microcarrier-SUB results were obtained assuming an average non-porous microcarrier surface area of 2,930 cm2/g. Although this is within the range of values found in the literature for different microcarrier types, it might represent an overestimation of the performance of some non-porous systems. Hence, the sensitivity analysis of this parameter was extended by assigning to it different values within the range 360–2,930 cm2/g and running the optimization algorithm to find the optimal expansion technology, across different lot sizes. In this study no preference of planar technologies over microcarriers was included and the results are presented in Figure 5. The last column of the heat map corresponds to a microcarrier surface area of 2,930 cm2/g, that is, the value assumed for the previous analysis, and shows that planar technologies are only optimal for smaller lot sizes of up to 5 × 108 cells. For production scenarios of 1 × 109 to 3 × 1010 cells/lot, small 20L SUBs with microcarriers become the most cost-effective option and for higher production scales the use of larger SUBs is required. As the microcarrier surface area decreases (moving to the left in the plot), a larger number of SUBs or SUBs with larger sizes are required and planar technologies gain competitiveness. The left extreme of the heat map represents the worst performance of microcarriers in terms of surface area. In this scenario, microcarrier-based systems are only selected when the maximum number of units is exceeded by planar technologies. Finally, the gray area has a similar interpretation as in Figure 2d, that is, it represents large production scales for doses of 109 cells that cannot be met by any of the candidate technologies.
As indicated in Table I, some cell therapy products currently under development have doses in the order of 109 cells and thus a step improvement in the performance of existing cell expansion technologies is necessary so as to avoid future failures related to manufacturing and scale-up issues.
Technology S-Curve for Cell Therapy Manufacture
Technology S-curves illustrate the introduction, growth and maturation of innovations and have been used to analyze the evolution of technologies in several industry sectors varying from semiconductors to renewable energy sources (Bowden, 2004; Schilling and Esmundo, 2009). For cell expansion technologies, a conceptual illustration of a technology S-curve was created by plotting the performance of each technology in terms of billion cells achieved per lot (when using the maximum number of units per lot) against R&D effort/investment. The x-axis represents qualitatively the R&D effort required for a company currently using T-flasks to change to other cell expansion technologies.
Figure 6 shows a technology S-curve built using the data and assumptions of the tool for candidate cell expansion technologies considered in the case study. The details of the upper and lower limits for each technology in the S-curve and the corresponding COGUSP estimates are presented in Table V. For smaller lot sizes using T-flasks the COGUSP range is $49–240/million cells while for larger lots using multi-layers the range decreases to $15–62/million cells. Significant economies of scale exist for very large lot sizes using microcarriers with COGUSP values as low as $0.7–3.2/million cells being achieved. (It should be noted that these are the operating costs for the cell expansion step and associated QC lot release costs and that only the equipment depreciation component of the facility-dependent indirect costs are included). For a dose of 109 cells the best case COGUSP values translate into $700/dose for microcarriers and $15,000/dose for multi-layer vessels. Given reimbursement values could be in the order of $25,000/patient this would result in values of COGUSP as % sales of 3% for microcarrier-based processes to ∼60% for multi-layer processes (assuming single dose products). In the biologics industry typical values for COG as % sales have been reported to range from 15% to 40% (Smith, 2012) in order to recover R&D, sales and marketing costs whilst achieving attractive profit margins. Assuming allogeneic cell therapies will have gross margins in line with biologics (Smith, 2012) and that COGUSP represents at least 50% of the total COG, this would translate into COGUSP targets for the allogeneic cell therapy industry of $1,875–5,000/dose. Hence for the high cell doses of 109 cells/dose, only the microcarrier-based processes would meet this cost target and allow for a successful business model, as predicted by the optimization tool.
Figure 6.
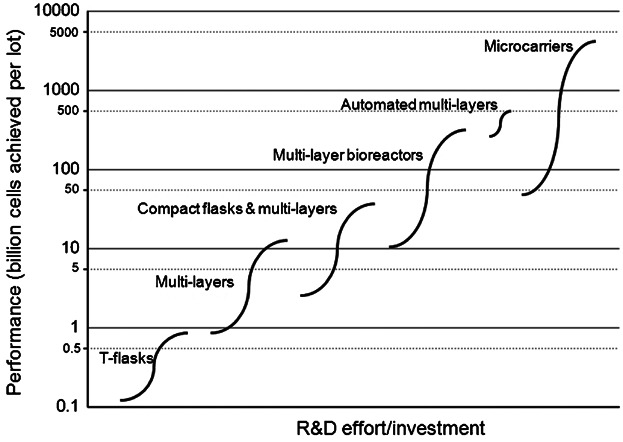
Conceptual illustration of a technology S-curve showing the evolution of expansion technologies used in cell therapy manufacture. The limits of each S-curve correspond to the amount of cells achieved by the smallest and largest size of each technology type when using the maximum number of units (80 for planar and 8 for microcarriers). Automated multi-layers refer to L-40 and cL-120. The x-axis represents qualitatively the R&D effort required for a company currently using T-flasks to change to other cell expansion technologies.
Table V.
Limits of S-curve and associated COGUSP values
| Technology type | Lower limit | Upper limit | ||||
|---|---|---|---|---|---|---|
| Size | Performance (109 cells/lot) | COGUSPa ($/106 cells) | Size | Performance (109 cells/lot) | COGUSPa ($/106 cells) | |
| T-flasks | 80 × T75 | 0.1 | 240 | 80 × T500 | 0.9 | 49 |
| Multi-layers | 80 × L-1 | 1 | 62 | 80 × L-10 | 11 | 15 |
| Compact flasks and multi-layers | 80 × cT | 3 | 19 | 80 × cL-36 | 31 | 8.5 |
| Multi-layer bioreactors | 80 × bL-10 | 13 | 39 | 80 × bL-180 | 229 | 9.2 |
| Automated multi-layers | 80 × 4 × L-40 | 173 | 6.5 | 80 × 4 × cL-120 | 408 | 5.0 |
| Microcarriers | 8 × M-20L | 47 | 3.2 | 8 × M-2000L | 4,708 | 0.7 |
COGUSP values shown here are based on the direct costs (material, labor, QC testing) and indirect costs (equipment depreciation only) of the cell expansion process and assuming overheads are spread over 10 lots/year for all scenarios.
The conceptual S-curve exhibits a similar trend to a typical technology adoption curve where a limit in the performance forces the introduction of new technologies. It shows that the adoption of new technologies for cell therapy manufacture is driven by the need to produce a larger amount of cells. Analyzing cell expansion technologies from a technology S-curve demonstrated that each technology covers approximately one log of performance in terms of lot size (billion cells per lot) before being replaced by a newer technology. In the case of microcarrier-based systems, two logs are covered due to the wide range of SUB-sizes considered in this analysis (from 20L to 2000L). The S-curve also shows that a technology gap exists for meeting the largest anticipated commercial lot sizes (1013 cells). The top performance target value of 10,000 billion cells corresponds to the production scenario of the last column of the matrix shown in Figure 2d and it is evident from the S-curve that none of the technologies considered in this study are able to achieve it. Existing planar technologies are unable to achieve the next log demand (1,000 billion cells) nor the largest value (10,000 billion). Given the nature of microcarrier technologies and their potential for improvement, an analysis was carried out to explore how the performance target could be achieved in terms of operating parameters. This study is described in the following section.
Future Performance Targets for Microcarrier Applications
The current performance level of the microcarrier-based technology analyzed in this article is 0.5 million cells/mL. This is the result of using non-porous microcarriers with a surface area of 2,930 cm2/g, a density of 6.3 g/L and assuming a harvest cell density of 25,000 cells/cm2. The production target of 10,000 billion cells/lot can be achieved through different combinations of total bioreactor capacity and cell concentration in the microcarrier culture. The contours in Figure 7a represent the number of billion cells achieved per lot (in the body of the graph) as a function of the cell concentration (million cells/mL) present in the microcarrier culture and the number of SUBs used per lot. The bold contour represents the target of 10,000 billion cells/lot. This graph can be used to drive facility design given the level of performance of a particular technology. For example, if the microcarrier technology is able to achieve a 5.2-fold increase in cell concentration to 2.6 million cells/mL, then a total of three SUBs per lot would be required (represented by point A on the graph). However, if only a 2.6-fold increase to 1.3 million cells/mL was possible, then six SUBs would need to be used per lot. Note that in order to meet the maximum production target of 10,000 billion cells/lot using the current performance level of 0.5 million cells/mL it would be necessary to run 17 × 2,000L SUBs per lot, thus significant cost savings could be achieved by improving the microcarrier performance. This approach allows companies to explore the trade-off between the cell concentration achieved in a microcarrier culture and the number of SUBs to run per lot and to identify the operating conditions most suitable for a particular process.
As the cell concentration depends on several process parameters including harvest cell density per area, microcarrier surface area and microcarrier density, the impact of each of them on the performance target was investigated and a visual tool to aid this decision-making process was generated. Figure 7b illustrates the million cells/mL achieved in a microcarrier culture as a function of the microcarrier density and surface area, and highlights the impact of the harvest cell density per area on those parameters. Two shaded areas are shown and these correspond to the windows of operation to achieve 2.6 and 1.3 million cells/mL, that is, to implement the strategies represented by points A and B of Figure 7a, respectively. A possible setup to achieve 2.6 million cells/mL is shown by point X; assuming a microcarrier with a surface area of 8,000 cm2/g and a typical harvest density of 25,000 cells/cm2, then the required microcarrier density would be around 13 g/L. This represents a substantial increase relative to current performance levels, where values up to 5,500 cm2/g and 9.3 g/L have been reported for the expansion of adult stem cells in non-porous microcarriers (Table IV). Investment in improving the microcarrier surface area could be an option, such as the use of porous microcarriers with surface areas of 11,000–15,000 cm2/g (Table IV). Although this is a valuable guide, normally at small-scale, several microcarriers would have to be screened for attachment, expansion, and optimal harvest from the microcarriers. Important properties of microcarriers include size, degree of porosity, and charge density. Sart et al. (2010) indicated that porous microcarriers are more suitable for the propagation of mesenchymal stem cells than solid supports, and this was thought to be due to the protective effect of porous microcarriers shielding cells from shear. On the other hand, it may be harder to harvest the cells from porous microcarriers such as Cultispher S (Hyclone), than non-porous equivalents such as Cytodex III. Thus, further experimental optimization would have to be performed to validate the use of porous microcarriers for the expansion of stem cells and to resolve difficulties associated with cell productivities and harvesting.
The challenges of scaling-up mammalian cell culture have been addressed in the past by the mAb/recombinant protein industry. Taking those lessons onboard and investing in the development of alternative and more scalable technologies, such as suspension culture, will be critical if cell therapy products are to achieve the commercial manufacturing success of biopharmaceuticals.
Conclusion
This article presents an integrated decisional tool combining a bioprocess economics simulation engine with an optimization procedure to identify optimal expansion technologies for cell therapies. The application of the tool to an industrially relevant case study on the production of allogeneic cell therapies highlighted how the competitiveness of alternative systems varies with production scale and identified limits of available expansion technologies and technology gaps. Further analysis also indicated the fold increase in performance that would be required to reach maximum target demands. Emphasis was placed on the use of visualization methods to present optimal solutions across a range of scales and windows of operation for future performance targets. A technology S-curve for cell expansion was derived with data from the tool and facilitated the characterization of industry trends and identification of technology gaps. The modeling insights can be used to prioritize the focus of future R&D investment so as to improve the performance of the most promising technologies so that they become a robust and scalable option that enables the cell therapy industry reach commercially relevant lot sizes. The tool outputs can facilitate decision-making very early on in development and may be used to predict, and better manage, the risk of process changes needed as products proceed through the development pathway. Future research will include the extension of the tool to include downstream operations and the development of case studies to address different types of allogeneic and autologous cell therapies.
Acknowledgments
Financial support from the Technology Strategy Board (UK) and Lonza is gratefully acknowledged. Constructive feedback and technical advice from industrial experts at Lonza, ATMI, ThermoFisher, and Corning is gratefully acknowledged. UCL hosts the EPSRC Centre for Innovative Manufacturing in Emergent Macromolecular Therapies with Imperial College and a consortium of industrial and government users.
References
- Birch JR, Racher AJ. Antibody production. Adv Drug Deliver Rev. 2006;58:671–685. doi: 10.1016/j.addr.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Bowden MJ. Moore's Law and the technology S-curve. Curr Issues Technol Manage. 2004;8(1):1–4. [Google Scholar]
- Brandenberger R, Burger S, Campbell A, Fong T, Lapinskas E, Rowley JA. Cell therapy bioprocessing. BioProcess Int. 2011;9(Suppl. 1):30–37. [Google Scholar]
- Christ B, Stock P. Mesenchymal stem cell derived hepatocytes for functional liver replacement. Front Immunol. 2012;3(168):1–10. doi: 10.3389/fimmu.2012.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DelaRosa O, Dalemans W, Lombardo E. Toll-like receptors as modulators of mesenchymal stem cells. Front Immunol. 2012;3(182):1–8. doi: 10.3389/fimmu.2012.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farid SS. Established bioprocesses for producing antibodies as a basis for future planning. Adv Biochem Eng Biotechnol. 2006;101:1–42. doi: 10.1007/10_014. [DOI] [PubMed] [Google Scholar]
- Frauenschuh S, Reichmann E, Ibold Y, Goetz PM, Sittinger M, Ringe J. A microcarrier-based cultivation system for expansion of primary mesenchymal stem cells. Biotechnol Prog. 2007;23:187–193. doi: 10.1021/bp060155w. [DOI] [PubMed] [Google Scholar]
- Goldschlager T, Rosenfeld JV, Ghosh P, Itescu S, Blecher C, McLean C, Jenkin G. Cervical interbody fusion is enhanced by allogeneic mesenchymal precursor cells in an ovine model. SPINE. 2011;36:615–623. doi: 10.1097/BRS.0b013e3181dfcec9. [DOI] [PubMed] [Google Scholar]
- Griffith LG, Naughton G. Tissue engineering: Current challenges and expanding opportunities. Science. 2002;295:1009–1014. doi: 10.1126/science.1069210. [DOI] [PubMed] [Google Scholar]
- Hambor JE. Bioreactor design and bioprocess controls for industrialized cell processing: Bioengineering strategies and platform technologies. BioProcess Int. 2012;10:22–33. [Google Scholar]
- Jung KH, Uhm Y-K, Lim YJ, Yim SV. Human umbilical cord blood-derived mesenchymal stem cells improve glucose homeostasis in rats with liver cirrhosis. Int J Oncol. 2011;39:137–143. doi: 10.3892/ijo.2011.1016. [DOI] [PubMed] [Google Scholar]
- Kelley B. Very large scale monoclonal antibody purification: The case for conventional unit operations. Biotechnol Prog. 2007;23:995–1008. doi: 10.1021/bp070117s. [DOI] [PubMed] [Google Scholar]
- Kempken R, Preissmann A, Berthold W. Assessment of a disc stack centrifuge for use in mammalian cell separation. Biotechnol Bioeng. 1995;46:132–138. doi: 10.1002/bit.260460206. [DOI] [PubMed] [Google Scholar]
- Kirouac DC, Zandstra PW. The systematic production of cells for cell therapies. Cell Stem Cell. 2008;3:369–381. doi: 10.1016/j.stem.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Kebriaei P, Isola L, Bahceci E, Holland K, Rowley S, McGuirk J, Devetten M, Jansen J, Herzig R, Schuster M, Monroy R, Uberti J. Adult human mesenchymal stem cells added to corticosteroid therapy for the treatment of acute Graft-versus-Host Disease. Biol Blood Marrow Transplant. 2009;15:804–811. doi: 10.1016/j.bbmt.2008.03.012. [DOI] [PubMed] [Google Scholar]
- Lapinskas E. Scaling up research to commercial manufacturing. Chem Eng Prog. 2010;106:S44–S55. [Google Scholar]
- Lindroos B, Boucher S, Chase L, Kuokkanen H, Huhtala H, Haataja R, Vemuri M, Suuronen R. Serum-free, xeno-free culture media maintain the proliferation rate and multipotentiality of adipose stem cells in vitro. Cytotherapy. 2009;11:958–972. doi: 10.3109/14653240903233081. [DOI] [PubMed] [Google Scholar]
- Lopez F, Di Bartolo C, Piazza T, Passannanti A, Gerlach JC, Gridelli B, Triolo F. A quality risk management model approach for cell therapy manufacturing. Risk Anal. 2010;30:1857–1871. doi: 10.1111/j.1539-6924.2010.01465.x. [DOI] [PubMed] [Google Scholar]
- Malik N. Allogeneic versus autologous stem-cell therapy. BioPharm Int. 2012;25:36–40. [Google Scholar]
- Mason C, Dunnill P. Quantities of cells used for regenerative medicine and some implications for clinicians and bioprocessors. Regen Med. 2009;4:153–157. doi: 10.2217/17460751.4.2.153. [DOI] [PubMed] [Google Scholar]
- Meyers VM, Zayzafoon M, Douglas JT, McDonald JM. RhoA and cytoskeletal disruption mediate reduced osteoblastogenesis and enhanced adipogenesis of human mesenchymal stem cells in modeled microgravity. J Bone Miner Res. 2005;20:1858–1866. doi: 10.1359/JBMR.050611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miljan EA, Sinden JD. Stem cell treatment of ischemic brain injury. Curr Opin Mol Ther. 2009;11:394–403. [PubMed] [Google Scholar]
- Penn MS, Ellis S, Gandhi S, Greenbaum A, Hodes Z, Mendelsohn FO, Strasser D, Ting AE, Sherman W. Adventitial delivery of an allogeneic bone marrow-derived adherent stem cell in acute myocardial infarction: Phase I clinical study. Circ Res. 2012;110:304–311. doi: 10.1161/CIRCRESAHA.111.253427. [DOI] [PubMed] [Google Scholar]
- Rajala K, Lindroos B, Hussein SM, Lappalainen RS, Pekkanen-Mattila M, Inzunza J, Rozell B, Miettinen S, Narkilahti S, Kerkelä E, Aalto-Setälä K, Otonkoski T, Suuronen R, Hovatta O, Skottman H. A defined and xeno-free culture method enabling the establishment of clinical-grade human embryonic, induced pluripotent and adipose stem cells. PLoS ONE. 2010;5:1–14. doi: 10.1371/journal.pone.0010246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliffe E, Thomas RJ, Williams DJ. Current understanding and challenges in bioprocessing of stem cell-based therapies for regenerative medicine. Br Med Bull. 2011;100:135–155. doi: 10.1093/bmb/ldr037. [DOI] [PubMed] [Google Scholar]
- Reinecke H, Minami E, Zhu W-Z, Laflamme MA. Cardiogenic differentiation and transdifferentiation of progenitor cells. Circ Res. 2008;103:1058–1071. doi: 10.1161/CIRCRESAHA.108.180588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley J, Abraham E, Campbell A, Brandwein H, Oh S. Meeting lot-size challenges of manufacturing adherent cells for therapy. BioProcess Int. 2012;10:16–22. [Google Scholar]
- Rubin PJ, Bennett JM, Doctor JS, Tebbets BM, Marra KG. Collagenous microbeads as a scaffold for tissue engineering with adipose-derived stem cells. Plast Reconstr Surg. 2007;120:414–424. doi: 10.1097/01.prs.0000267699.99369.a8. [DOI] [PubMed] [Google Scholar]
- Sart S, Schneider Y-J, Agathos SN. Influence of culture parameters on ear mesenchymal stem cells expanded on microcarriers. J Biotechnol. 2010;150:149–160. doi: 10.1016/j.jbiotec.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Schilling MA, Esmundo M. Technology S-curves in renewable energy alternatives: Analysis and implications for industry and government. Energ Policy. 2009;37:1767–1781. [Google Scholar]
- Schwartz SD, Hubschman JP, Heilwell G, Franco-Cardenas V, Pan CK, Ostrick RM, Mickunas E, Gay R, Klimanskaya I, Lanza R. Embryonic stem cell trials for macular degeneration: A preliminary report. Lancet. 2012;379:713–720. doi: 10.1016/S0140-6736(12)60028-2. [DOI] [PubMed] [Google Scholar]
- Smith DM. Assessing commercial opportunities for autologous and allogeneic cell-based products. Regen Med. 2012;7:721–732. doi: 10.2217/rme.12.40. [DOI] [PubMed] [Google Scholar]
- Tamaki S, Eckert K, He D, Sutton R, Doshe M, Jain G, Tushinski R, Reitsma M, Harris B, Tsukamoto A, Gage F, Weissman I, Uchida N. Engraftment of sorted/expanded human central nervous system stem cells from foetal brain. J Neurosci Res. 2002;69:976–986. doi: 10.1002/jnr.10412. [DOI] [PubMed] [Google Scholar]
- Vaes B, Craeye D, Pinxteren J. Quality control during manufacture of a stem cell therapeutic. BioProcess Int Suppl Cell Ther Anal. 2012;10:50–55. [Google Scholar]
- Want AJ, Nienow AW, Hewitt CJ, Coopman K. Large-scale expansion and exploitation of pluripotent stem cells for regenerative medicine purposes: Beyond the T flask. Regen Med. 2012;7:71–84. doi: 10.2217/rme.11.101. [DOI] [PubMed] [Google Scholar]
- Whitford WG, Fairbank A. Considerations in scale-up of viral vaccine production. BioProcess Int. 2011;9:16–28. [Google Scholar]
- Wu Q-F, Wu C-T, Dong B, Wang L-S. Cultivation of human mesenchymal stem cells on macroporous CultiSpher G microcarriers. J Exp Hematol/Chin Assoc Pathophysiol. 2003;11:15–21. [PubMed] [Google Scholar]
- Yang Y, Rossi FMV, Putnins EE. Ex vivo expansion of rat bone marrow mesenchymal stromal cells on microcarrier beads in spin culture. Biomaterials. 2007;28:3110–3120. doi: 10.1016/j.biomaterials.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Zayzafoon M, Gathings WE, McDonald JM. Modeled microgravity inhibits osteogenic differentiation of human mesenchymal stem cells and increases adipogenesis. Endocrinology. 2004;145:2421–2432. doi: 10.1210/en.2003-1156. [DOI] [PubMed] [Google Scholar]