

Abstract
Previous mass spectrometry analysis of cerebrospinal fluid (CSF) has allowed the identification of a panel of molecular markers that are associated with Alzheimer’s disease (AD). The panel comprises Amyloid beta, Apolipoprotein E, Fibrinogen alpha chain precursor, Keratin type I cytoskeletal 9, Serum albumin precursor, SPARC-like 1 protein and Tetranectin. Here we report the development and implementation of immunoassays to measure the abundance and diagnostic capacity of these putative biomarkers in matched lumbar CSF and blood plasma samples taken in life from individuals confirmed at post-mortem as suffering from AD (n = 10) and from screened ‘cognitively healthy’ subjects (n = 18). The inflammatory components of Alzheimer’s disease were also investigated. Employment of supervised learning techniques permitted examination of the interrelated expression patterns of the putative biomarkers and identified inflammatory components, resulting in biomarker panels with a diagnostic accuracy of 87.5% and 86.7% for the plasma and CSF datasets respectively. This is extremely important as it offers an ideal high-throughput and relatively inexpensive population screening approach. It appears possible to determine the presence or absence of AD based on our biomarker panel and it seems likely that a cheap and rapid blood test for AD is feasible.
Keywords: Alzheimer’s disease, biomarker, blood plasma, cerebrospinal fluid
Introduction
Neurodegenerative disorders, and in particular Alzheimer’s disease (AD), are having an increasing impact on our society. By 2040 dementia, of which AD is the most common cause, is predicted to affect 81.1 million individuals worldwide [1]. Although AD therapeutics including Donepezil, Rivastigmine, Galantamine and Memantine are available, AD remains incurable as these drugs only provide some symptomatic relief and do not modify disease progression [2,3]. As AD is associated with a progressive decline in health, earlier detection may result in enhanced therapeutic success [3]. Current diagnostic procedures are, however, inadequate for early disease detection or the accurate differentiation of AD from other forms of dementia. Provision of enhanced cost-effective diagnostic strategies is vital and a biochemical test that is diagnostic and predictive of AD would help achieve this aim.
Investigations into biomarkers for AD have, to date, failed to identify a single molecule capable of fully depicting the disease state. Genetically AD is one of the best characterised diseases [4-9], however only approximately 24% of heritability has been resolved [10] and possession of genetic indicators (e.g. the E4 allele of the APOE gene) does not guarantee disease onset [11]. Diagnosis based solely on a genetic marker would, therefore, result in many inaccurate diagnoses. Studies into individual AD protein biomarkers have tended to focus on the constituents of amyloid beta deposits and neurofibrillary tangles as these are the characteristic hallmarks of the disease [12] but such studies have demonstrated varying levels of success [13-16]. A recent study examining amyloid beta peptide 1-42 (Aβ42) has, however, lent support to the belief that the underlying causative factors of AD are initiated many years before the symptoms of the late-onset form of AD (LOAD) manifest [17]. In this study, cerebrospinal fluid (CSF) concentrations of Aβ42 were shown to have reached pathological levels 5-10 years prior to conversion from mild cognitive impairment (MCI) to AD, highlighting the huge benefits that biomarker identification could have in the early diagnosis of AD. However, as is the case with single biomarkers in most complex disorders, Aβ42 does not appear to have the necessary power to act as a stand-alone AD marker. The findings of this study indicate that 10% of individuals with MCI who had pathological levels of Aβ42 did not go on to develop AD. As such these measurements would need to be utilised in conjunction with additional diagnostic procedures and it may be that a panel of biomarkers incorporating Aβ42 may be more appropriate.
Vafadar-Isfahani and colleagues recently identified a panel of CSF biomarkers capable of differentiating between healthy individuals and those with AD [18]. Comprising Amyloid beta, Apolipoprotein E, Fibrinogen alpha chain precursor, Keratin type I cytoskeletal 9, Serum albumin precursor, SPARC-like 1 protein and Tetranectin, the diagnostic performance of this biomarker panel was found to improve as more markers were sequentially added to the model for diagnosis i.e. the effect was additive suggesting that all the markers are necessary for accurate diagnosis. The panel of markers also demonstrated its potential utility in early diagnosis of AD by mapping individuals with Mild Cognitive Impairment (MCI) at an intermediate point between samples from healthy and AD individuals. To enable routine screening of a population, however, it would be preferable for any biochemical test developed to be analysed in blood plasma as this is a far less invasive clinical sample to collect from a patient than CSF. In this study we first aim to identify the components of the recently identified CSF AD biomarker panel [18] in blood plasma. We then proceed to determine their plasma and CSF concentrations and assess their potential utility as diagnostics tools. As Tau is considered one of the foremost AD biomarkers, we have included it in this study along with Clusterin which was recently identified as a potential blood plasma biomarker for AD [19].
Materials and methods
Patient samples
Sample cohorts used in this study were obtained from the Oxford Project to Investigate Memory and Ageing (OPTIMA; University of Oxford, UK). The OPTIMA study received approval from the Central Oxford Ethics Committee and all individuals gave informed written consent to participate in the study. For 10 patients with a clinical diagnosis of probable AD, ‘definite’ AD was diagnosed pathologically by the established CERAD criteria. The 18 control subjects were cognitively screened annually for at least 3 years and 6 came to autopsy and were classified as CERAD ‘negative’. See a recent report for a brief description of the OPTIMA cohort, CSF sampling procedure and post-mortem analysis [20]. The average interval between CSF sampling and death was 2090 days in the controls and 1806 days in the AD patients.
Protein detection by immunoassay: ELISA
SPARCL1 was analysed by sandwich ELISA using capture and biotinylated detection antibodies from Creative Biomart (CAB-701MH) and R&D Systems (BAF2728) respectively. The wells of ELISA plates were coated overnight with 5 μg/ml capture antibody (CAB-701MH in PBS). Following this and every subsequent incubation wells were washed three times with PBS-0.05% Tween20 (PBST). Antibody-coated wells were blocked with PBST-1% BSA for 1 hr, 50 μl sample (either clinical sample or protein standard) was added for 2 hr and then biotinylated detection antibody (BAF2728) was added at 500 ng/ml for 2 hr. The reaction was developed using streptavidin-HRP and TMB substrate, stopped with 1 M HCl and read at 450 nm. Concentrations of SPARCL1 in the samples were determined from a standard curve generated with recombinant SPARCL1 (R&D Systems; 2728-SL). Keratin 9 was detected according to the manufacturer’s protocol using an ELISA kit purchased from antibodies-online GmbH (ABIN417500). ELISA kits against β-Amyloid peptide 1-40 (Aβ40) and Aβ42 were purchased from Wako Chemicals GmbH (298-64601 and 296-64401) and used according to manufacturer’s instructions.
Antibodies and recombinant proteins
Appropriate pairs of monoclonal capture antibody and biotinylated polyclonal detection antibody and a corresponding recombinant protein standard were sourced as displayed in Table 1 and verified by western blot and ELISA.
Table 1.
Immunoassay Reagents
| Target | Capture Antibody | Recombinant Protein | Detection Antibody | |||
|---|---|---|---|---|---|---|
|
| ||||||
| Supplier | Product | Supplier | Product | Supplier | Product | |
| ApoE | Abcam | ab1907 | Merck | 178468 | Abcam | ab24274 |
| Clusterin | AbD Serotec | MCA2612 | R&D Systems | 2937-HS | R&D Systems | BAF2937 |
| Fibrinogen α-chain | Abcam | ab19079 | Thermo Scientific | RP-43142 | Abcam | ab34546 |
| Tau | Abcam | ab80579 | Abcam | ab72489 | Thermo Scientific | MN1000B |
| Tetranectin | Abcam | ab51883 | R&D Systems | 5170-CL | R&D Systems | BAF5170 |
Details of the antibody and protein reagents used in the development of Luminex assays for this study.
Antibody coupling to microspheres
COOH-coated fluorescently dyed microspheres (Bio-Plex) were purchased from Bio-Rad (Hercules, CA). Bead regions 011, 020, 027, 033 and 042 were assigned to ApoE, Tetranectin, Fibrinogen alpha chain, Clusterin and Tau respectively. Monoclonal capture antibodies were coupled to the microspheres using the Bio-Plex Amine Coupling Kit (Bio-Rad, 171-406001) according to manufacturer’s instructions. When necessary, pre-processing with Micro Bio-Spin 6 columns (Bio-Rad) was undertaken to ensure antibodies were in PBS buffer containing no additives prior to the attachment procedure. Assays were initially developed and optimised individually for target protein before being combined step-by-step to identify any problems due to reagent cross-reactivity that may have occurred.
Luminex procedure
Designated wells of a filter plate were pre-wetted with wash buffer (PBS-0.05% Tween20 (PBST)). A bead suspension containing 5000 of each antibody-conjugated bead set was added to each well, washed twice with PBST and resuspended in incubation buffer (PBS-1% BSA). To this, a sample of appropriately diluted clinical sample or protein standard was added, and the plate was incubated for 2 hr at 25°C on a rotating plate shaker (600 rpm). Wells were then washed twice with PBST, and incubated with a cocktail of biotinylated detection antibodies (containing each antibody at a pre-determined optimal concentration). The plate was incubated for 1 hr at 25°C on a rotating plate shaker after which wells were again washed twice with PBST and the reporter molecule (Streptavidin-RPE) was added to the appropriate wells. Finally, following a 30 min incubation at 25°C on a rotating plate shaker and two washes with PBST, PBST was added to each well in preparation for analysis. Data were acquired on a Bio-Plex 200 system and analysed with associated software (Bio-Rad). All multiplex assays were performed in duplicate. In each well, a minimum of 100 beads per target molecule were analysed for both bead designation and R-phycoerythrin fluorescence. Clinical sample concentrations of each target protein were determined from standard curves generated using recombinant proteins.
Human cytokine 30-plex Luminex assay
Cytokine levels in both plasma and CSF were measured using the Luminex human cytokine 30-plex panel (Biosource, Camarillo, CA). This kit is able to simultaneously measure human IL-1β, IL-1RA, IL-2, IL-2R, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12p40/p70, IL-13, IL-15, IL-17, TNF-α, IFN-α, IFN-γ, GM-CSF, MIP-1α, MIP-1β, IP-10, MIG, Eotaxin-1, RANTES, MCP-1, VEGF, G-CSF, EGF, FGF-basic and HGF. Samples were analysed according to manufacturer’s instructions. Briefly, incubation buffer and 1:2 diluted plasma samples were pipetted into wells and incubated with the beads for 2 hr. Wells were washed, incubated with biotinylated detector antibody for 1 hr, washed again and incubated for 30 mins with streptavidin-RPE. Wells were washed to remove unbound streptavidin-RPE prior to analysis. All samples and standard curves were performed in duplicate. Data was acquired on a Bio-Plex 200 system and analysed with associated software (Bio-Rad). Standard curves for each cytokine were generated using the pre-mixed lyophilised standards provided in the kits and the cytokine concentrations in samples were determined from the appropriate standard curve.
Individual biomarker analysis
Data were analysed using GraphPad Prism Version 5.02. Concentrations of the markers in the cohorts were compared using the Mann-Whitney test and Spearman correlation coefficients were determined as appropriate. A p-value ≤ 0.05 was considered to be a practical level of clinical significance.
Biomarker panel analysis
To determine the effectiveness of each individual marker, the statistical data was used to calculate the proportion of both the healthy and the diseased population that would be incorrectly diagnosed. It was assumed that both the healthy and the diseased population are normally distributed, but the principle is valid for other distributions.
Considering first the healthy population, take Ph(c) as the probability distribution with respect to marker concentration c. The first step involves calculating the range of marker concentrations where the diseased population overlaps with the healthy population. This is entirely covered by the range of concentrations for the diseased population, so the range can be set to μd ± nσd, where μd and σd are the mean and standard deviation of the diseased population respectively, and n is the number of standard deviations from the mean. In this work, n was chosen as 3. We define the proportion of the healthy population that lies within this range to be Oh. An example can be seen in Figure 1, where the healthy population that cannot be correctly diagnosed with the single example marker is shown in grey.
Figure 1.
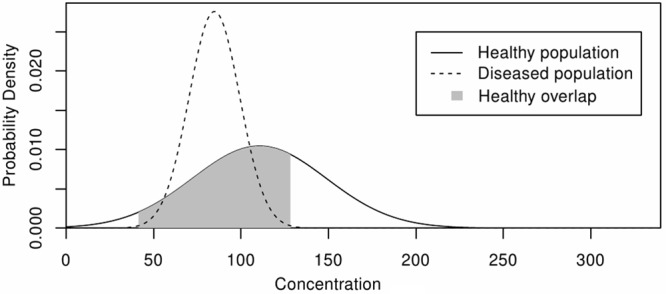
Determining Population Distribution Overlaps. The overlap (grey) in population distributions of a healthy (black line) and disease (dashed line) cohort for a single disease biomarker.
Individuals with marker concentrations in the overlap range are at risk of being misdiagnosed. Determining Oh for a marker gives a measure of its usefulness. Oh can be calculated as the cumulative distribution of the healthy population, Ph, that lies within the overlap range. This can be found by integrating the probability density function for the healthy population, Ph, over the overlap range, as shown in Equation 1.
 |
In some cases the lower integration limit will be negative, implying a negative marker concentration which is of course not possible, so the general case above must be modified. The probability density function, Ph(c), is multiplied by the step function H(c) as defined in Equation 2.
 |
This removes the possibility of negative marker concentrations. It also means that the total area of Ph(c) that is under consideration, defined as Th, is less than 1. Th must be found so that Oh can be calculated as a proportion. Th is calculated in Equation 3.
 |
Th may also be calculated using Equation 4, where the upper integration limit is set based on the healthy population mean and standard deviation to give a more practical limit. The parameter m = 6 was chosen in this work.
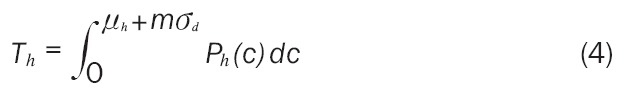 |
The final result for Oh is given in Equation 5 with the same result for the diseased population in Equation 6.
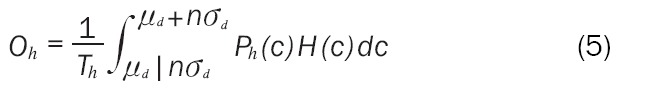 |
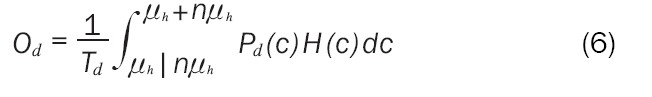 |
Supervised learning techniques
The parameters Oh and Od give an indication of how well each individual marker performs as a diagnostic tool, but to get the full power they must be considered together as a panel of markers. To validate the data without preconception of how to classify the subject groupings, three data analysis techniques based on supervised learning techniques were employed; a decision tree classifier (C4.5), a Bayesian classifier (Naïve Bayes) and a Multilayer Perceptron artificial neural network (ANN).
In the C4.5 algorithm each attribute of the data can be used to make a decision that splits the data into smaller subsets. It examines the normalised information gain that results from choosing an attribute for splitting the data. The attribute with the highest normalised information gain is used to make the decision. The algorithm then recurs on the smaller sublists. The system outputs as a decision tree, or a set of if-then rules, which can be used to classify new cases, with an emphasis on making the model understandable and accurate [21].
The Naïve Bayes is a probabilistic classifier based on Bayes’ theorem with strong independence assumptions that can deal with large numbers of cases and variables. It aims to predict the class of test instances as accurately as possible and is termed naive because it is based on two simplifying assumptions: (1) That the predictive attributes are conditionally independent given the class and (2) The values of numeric attributes are normally distributed within each class [22].
The Multilayer Perceptron is a feed-forward ANN model that maps sets of input data onto a set of appropriate output. It has three distinctive characteristics: (1) The model of each neuron in the network includes a nonlinear activation function. (2) The network contains one or more layers of hidden neurons (not part of the input or output of the network) which enable the network to learn complex tasks by extracting progressively more meaningful features from the input patterns. (3) The network exhibits a high degree of connectivity, determined by the synapses of the network. A change in the connectivity of the network requires a change in the population of synaptic connections or their weights. The Multilayer Perceptron derives its computing power from these characteristics together with the ability to learn from experience through training [23]. This system is trained to solve problems using the error back-propagation algorithm.
The classification algorithms were applied to the two data sets (CSF/Plasma) to examine whether the classifications into Healthy/AD could be reproduced. Each technique was implemented using a ‘leave-one-out’ cross-validation, to estimate how accurately the predictive model performs in practice. At each step, the whole data but one point are used for training and the remaining point is used for testing. The process is then repeated n-times, where n is the total number of patients.
Results
The concentrations of eight putative AD protein biomarkers were determined in plasma and CSF samples collected simultaneously from individual ‘donors’. The ‘matched’ nature of the samples makes it feasible for comparisons to be made between the protein constituents of each sample, but this also led to restrictions of the overall cohort sizes. All of the proteins examined (Aβ40, Aβ42, ApoE, Clusterin, Fibrinogen alpha chain, Keratin 9, SPARCL1, Tau, Tetranectin) could be detected in CSF and plasma using immunoassays (Figure 2). The mean concentrations of the proteins measured in both sample sets are shown in Table 2. SPARCL1 (p = 0.035) and Aβ42 (p = 0.049) were found to be significantly decreased in CSF from AD patients in comparison to healthy controls. Keratin 9 was only detectable in CSF samples from three AD patients (Mean concentration ± SD; 3.75 ± 5.9 pg/ml) and not any healthy individuals. In plasma from AD patients, the concentrations of Aβ42 (p = 0.0008) and Tau (p = 0.047) were significantly decreased in comparison to those observed in healthy controls. Significant correlations between the plasma and CSF concentrations of ApoE, SPARCL1, Tau and Tetranectin were determined in healthy individuals but were found to be weaker in the AD patient cohort (Table 2).
Figure 2.
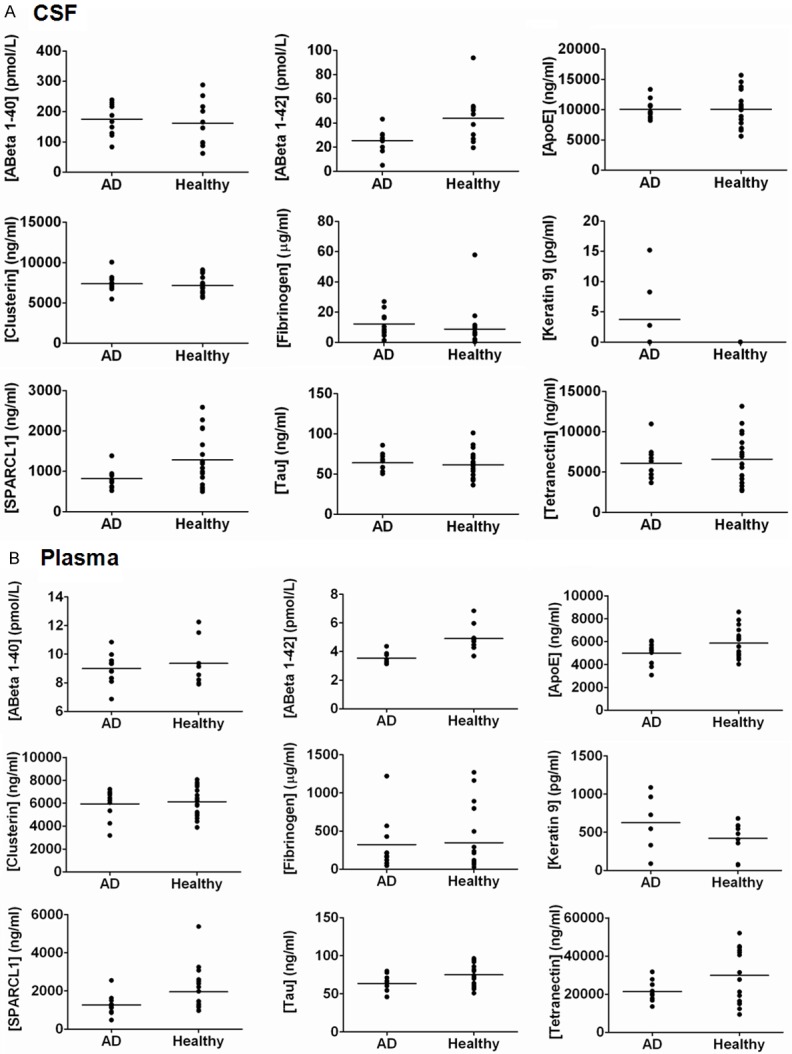
Immunoassays of Biomarker Panel Components. Quantification of components of the biomarker panel in AD patients and healthy individuals in (A) CSF and (B) plasma samples. Samples were interrogated using either Luminex or ELISA immunoassays as described in the Methods section.
Table 2.
Protein Biomarkers in CSF and Plasma
| Target Marker | Mean concentrations (± SD) | P-value (change due to AD) | Plasma/CSF correlation | |||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Plasma | CSF | |||||||
|
| ||||||||
| Healthy | AD | Healthy | AD | Plasma | CSF | Healthy | AD | |
| Aβ40 (pmol/L) | 9.36 ±1.65 | 9.00 ±1.10 | 161 ±76.6 | 175 ±53.4 | 0.8968 | 0.5787 | 0.93 | 0.66 |
| Aβ42 (pmol/L) | 4.91 ±0.90 | 3.54 ±0.41 | 43.8 ±21.6 | 25.5 ±10.1 | 0.0008* | 0.0493* | 0.61 | 0.14 |
| ApoE (µg/ml) | 5.86 ±1.28 | 4.98 ±0.99 | 10.08 ±2.88 | 10.05 ±1.62 | 0.1718 | 0.9809 | 0.02 | 0.54 |
| Clusterin (µg/ml) | 6.12 ±1.31 | 5.93 ±1.31 | 7.14 ±1.02 | 7.38 ±1.19 | 0.9046 | 0.5490 | 0.14 | 0.18 |
| Fibrinogen α-chain (µg/ml) | 348 ±401 | 323 ±354 | 8.77 ±13.08 | 12.2 ±8.36 | 0.7191 | 0.0887 | 0.30 | 0.87 |
| Keratin 9 (pg/ml) | 422 ±219 | 625 ±379 | 0.0 ±0.0 | 3.75 ±5.9 | 0.2721 | - | - | 0.24 |
| SPARCL1 (ng/ml) | 1957 ±1136 | 1271 ±557 | 1286 ±620 | 815 ±245 | 0.0887 | 0.0349* | 0.05 | 0.89 |
| Tau (ng/ml) | 75.1 ±14.3 | 63.4 ±11.9 | 61.5 ±17.6 | 63.9 ±11.7 | 0.0466* | 0.5490 | ≤ 0.0001 | 0.47 |
| Tetranectin (µg/ml) | 29.9 ±14.3 | 21.4 ±5.4 | 6.59 ±3.09 | 6.08 ±2.18 | 0.3498 | 0.9046 | ≤ 0.0001 | 0.58 |
Expression levels of the biomarker panel components were quantified in CSF and plasma samples using immunoassays. Variations between the cohorts were evaluated using the Mann-Whitney test. Spearman correlation coefficients were determined for protein values between the two sample types. A p-value 0.05 was considered statistically significant.
p ≤ 0.05.
To investigate the inflammatory component of AD, samples were examined using Luminex technology, allowing simultaneous assessment of 30 cytokines. In the plasma and CSF samples, 11 and 12 respectively of the cytokines examined were not present at concentrations that could be detected using this technology and are omitted from Table 3. Furthermore, some samples contained levels of RANTES that exceeded the maximum range of the standard curve so these values have also been omitted from Table 3. Significant differences (p-value ≤ 0.05) in concentration between AD and healthy samples were observed for FGF-basic (p = 0.05), IL-1RA (p = 0.02), MCP-1 (p = 0.01) and MIP-1β (p = 0.04) in plasma samples as illustrated in Figure 3. These differences were not replicated in the matched CSF samples in which only IL-12 (p = 0.04) demonstrated a significant change.
Table 3.
Inflammatory Proteins in CSF and Plasma
| Marker | Mean concentrations (± SD) | P-value (change due to AD) | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Plasma | CSF | |||||
|
| ||||||
| Healthy | AD | Healthy | AD | Plasma | CSF | |
| EGF | 8.08 ±5.60 | 9.54 ±8.04 | 5.24 ±6.91 | 3.93 ±8.68 | 0.82 | 0.07 |
| Eotaxin | 54.7 ± 44.1 | 39.8 ± 19.0 | 25.2 ± 26.2 | 10.1 ± 21.1 | 0.47 | 0.26 |
| FGF-basic | 2.30 ± 3.44 | 0.124 ± 0.392 | 3.80 ± 3.00 | 2.62 ± 1.11 | 0.05* | 0.29 |
| G-CSF | 10.6 ± 10.4 | 17.8 ± 14.1 | 11.8 ± 24.8 | 2.97 ± 4.28 | 0.42 | 0.32 |
| HGF | 111 ± 38.2 | 84.9 ± 14.5 | 89.6 ± 30.2 | 77.7 ± 40.9 | 0.10 | 0.18 |
| IFN-α | 256 ± 163 | 165 ± 116 | 93.3 ± 143 | 59.4 ± 111 | 0.27 | 0.73 |
| IL-1RA | 41.0 ± 53.5 | 2.98 ± 9.43 | 7.82 ± 22.5 | 1.65 ± 3.53 | 0.02* | 0.95 |
| IL-2R | 143 ± 132 | 64.9 ± 80.2 | 40.6 ± 50.7 | 38.1 ± 78.4 | 0.14 | 0.57 |
| IL-6 | 0.293 ± 0.927 | 0.647 ± 2.05 | 0.833 ± 1.52 | 0.266 ± 0.841 | 1.00 | 0.11 |
| IL-8 | 3.22 ± 5.82 | 0.576 ± 1.82 | 11.6 ± 11.6 | 6.99 ± 8.45 | 0.23 | 0.58 |
| IL-10 | 34.8 ± 110 | 3.68 ± 7.76 | 0.0 ± 0.0 | 0.084 ± 0.266 | 0.67 | - |
| IL-12 | 116 ± 99.2 | 109 ± 52.7 | 52.1 ± 48.8 | 25.6 ± 52.0 | 0.97 | 0.04* |
| IP-10 | 13.8 ± 9.23 | 14.0 ± 6.83 | 8.32 ± 5.22 | 6.46 ± 6.00 | 0.91 | 0.36 |
| MCP-1 | 235 ± 102 | 150 ± 62.9 | 237 ± 54.4 | 240 ± 96.7 | 0.01* | 0.72 |
| MIG | 45.5 ± 34.5 | 39.3 ± 24.1 | 25.3 ± 35.0 | 11.4 ± 19.5 | 0.45 | 0.45 |
| MIP-1α | 13.4 ± 12.2 | 6.23 ± 5.71 | 9.70 ± 11.9 | 1.81 ± 3.94 | 0.15 | 0.15 |
| MIP-1β | 62.2 ± 48.0 | 152 ± 88.1 | 47.7 ± 77.1 | 49.7 ± 131 | 0.04* | 0.36 |
| VEGF | 0.477 ± 0.485 | 0.205 ± 0.261 | 0.730 ± 0.513 | 0.533 ± 0.276 | 0.21 | 0.21 |
Expression levels of inflammatory proteins were quantified in CSF and plasma samples using the Luminex human cytokine 30-plex panel (Biosource). Variations between the cohorts were evaluated using the Mann-Whitney test. A p-value 0.05 was considered statistically significant.
p ≤ 0.05.
Figure 3.
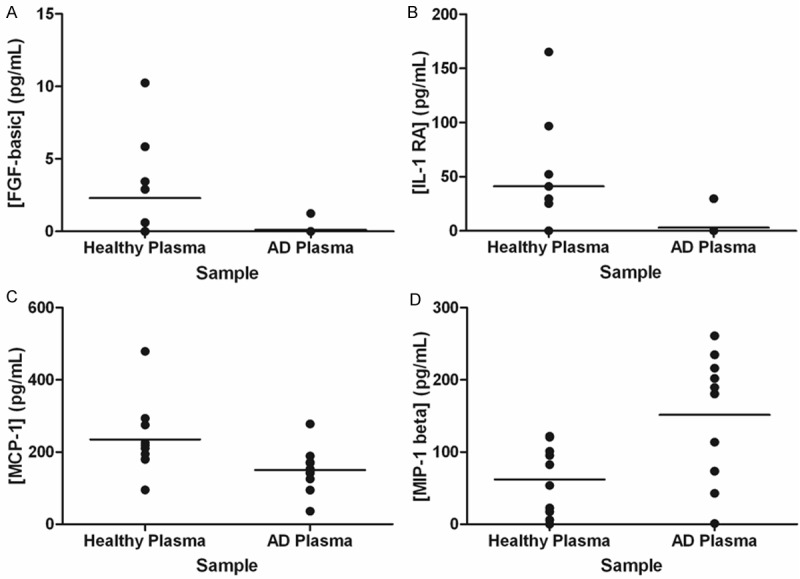
Inflammatory Proteins in AD. Inflammatory proteins were examined in blood plasma samples from healthy and AD cohorts. Samples were interrogated using the Luminex human cytokine 30-plex panel (Biosource). Data was collected using a Bio-Plex 200 system and analysed with associated software (Bio-Rad). Only those proteins demonstrated to differ significantly between the two patient cohorts are displayed: (A) FGF-basic; (B) IL-1RA; (C) MCP-1 and (D) MIP-1β.
In order to gain more insight into the mechanisms of disease and potential pathways implicated, we sought to identify any correlations that exist between components of the targeted biomarker panel and the inflammatory proteins shown to be significantly altered in AD (FGF-basic, IL-12, IL-1RA, MCP-1 and MIP-1β). The correlation of Keratin 9 with the other proteins could only be established in the disease cohort as it was not detectable in samples from the healthy cohort. Whilst all potential combinations of proteins were analysed, only those shown to be significantly correlated are displayed in Table 4 along with the p-value of the correlation and the effect of disease on the correlation. All of the targeted biomarker panel and inflammatory proteins were found to be significantly correlated (p ≤ 0.05) with at least one other protein (Table 4). Identification of a correlation in CSF samples did not automatically infer a correlation between that protein pair in the corresponding plasma samples as only the Aβ40/Aβ42 and MCP-1/Tetranectin pairings demonstrated the same patterns of correlations in CSF and plasma samples. In these cases a correlation that existed between the proteins in healthy samples was disrupted in the disease cohorts. Interestingly, some of the protein targets that were not significantly altered in the disease state (Figure 2 and Table 2) were found to correlate with some of those that exhibited significant changes. For example, clusterin, which had p-values of 0.90 and 0.55 in plasma and CSF respectively, was found to correlate with Aβ40, Aβ42, FGF-basic, MCP-1 and Tau demonstrating its potential importance in the disease network.
Table 4.
Correlations Between Proteins Concentrations in CSF and Plasma
| CSF Correlations | Plasma Correlations | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| Target 1 | Target 2 | Healthy | AD | Effect due to AD | Target 1 | Target 2 | Healthy | AD | Effect due to AD |
| Target 1 | Target 2 | Healthy | AD | Effect due to AD | Target 1 | Target 2 | Healthy | AD | Effect due to AD |
| Aβ40 | Aβ42 | 0.0368* | 0.5364 | Disrupted correlation | Aβ40 | Aβ42 | 0.0167* | 0.9768 | Disrupted correlation |
| Aβ40 | ApoE | 0.4279 | 0.0154* | Correlation | Aβ40 | Clusterin | 0.0333* | 0.7033 | Disrupted correlation |
| Aβ40 | SPARCL1 | 0.0279* | 0.7033 | Disrupted correlation | Aβ40 | Fibrinogen | 0.3556 | 0.0368* | Correlation |
| Aβ40 | Tau | 0.1710 | 0.0458* | Correlation | Aβ40 | IL-12 | 0.0167* | 0.0962 | Disrupted correlation |
| Aβ40 | Tetranectin | 0.0831 | 0.0072** | Correlation | Aβ40 | Tau | 0.0333* | 0.2675 | Disrupted correlation |
| Aβ42 | MCP-1 | 0.4976 | 0.0072** | Correlation | Aβ42 | Clusterin | 0.0072** | 0.0458* | Weaker correlation |
| ApoE | MCP-1 | 0.0067** | 0.0831 | Disrupted correlation | Aβ42 | Tau | 0.0154* | 0.1323 | Disrupted correlation |
| ApoE | Tau | 0.0368* | 0.0458* | No change in correlation | ApoE | Fibrinogen | 0.7033 | 0.0368* | Correlation |
| ApoE | Tetranectin | 0.0458* | 0.0368* | No change in correlation | ApoE | IL-12 | 0.0154* | 0.7930 | Disrupted correlation |
| Clusterin | FGF-basic | 0.5560 | 0.0107* | Correlation | ApoE | Tau | 0.1710 | 0.0046** | Correlation |
| Clusterin | MCP-1 | 0.0341* | 0.9349 | Disrupted correlation | ApoE | Tetranectin | 0.4618 | 0.0368* | Correlation |
| FGF-basic | MIP-1beta | 0.0480* | 0.3268 | Disrupted correlation | Clusterin | Tau | 0.0046** | 0.2162 | Disrupted correlation |
| MCP-1 | Tau | 0.0123* | 0.3599 | Disrupted correlation | Fibrinogen | Tau | 0.9768 | 0.0154* | Correlation |
| MCP-1 | Tetranectin | 0.0480* | 0.2992 | Disrupted correlation | Fibrinogen | Tetranectin | 0.0831 | 0.0107* | Correlation |
| Tau | Tetranectin | 0.0022** | 0.0107* | Weaker correlation | IL-12 | Tau | 0.0107* | 0.7520 | Disrupted correlation |
| MCP-1 | Tetranectin | 0.0458* | 0.3268 | Disrupted correlation | |||||
| Tau | Tetranectin | 0.5364 | 0.0072** | Correlation | |||||
Spearman correlation coefficients were determined for all protein pairings in CSF and plasma. A p-value 0.05 was considered statistically significant. Target markers showing significant correlations either in the healthy or disease state are shown.
p ≤ 0.05;
p ≤ 0.01.
Despite having identified significant differences in the concentrations of several proteins in AD samples, it would still not be possible to accurately differentiate an AD sample from a healthy sample using purely individual protein biomarkers as there is still significant overlap present between the ranges of concentrations of the two separate cohorts (Figure 2). Examination of this overlapping region allowed numerical assessment of the diagnostic power of each individual marker in the healthy and disease states for both CSF and plasma samples (Table 5). Keratin 9 values were determined using the detection limits of the ELISA kit used during sample analysis. When analysed in this manner, the individual markers that provided the most power in identifying healthy CSF, AD CSF, healthy plasma and AD plasma were IL-1RA (66.43%), Keratin 9 (98.76%), FGF-basic (82.13%) and MIP-1β (28.05%) respectively. It is important, however, that these single protein markers are investigated as a combined panel of biomarkers to determine whether this can provide improved diagnostic ability and permit identification of a subject’s disease state. Data analysis techniques based on supervised learning techniques were, therefore, employed in order to validate our data without preconception of how to classify the subject groupings. The biomarker panel examined comprised the original biomarker panel plus all detectable inflammatory markers. Due to the nature of the analysis, i.e. analysis of a complete biomarker panel, only samples where measurements had been obtained for all of these markers could be utilised.
Table 5.
The Power of Individual Proteins Within the Biomarkers Panels
| Marker | Healthy CSF | AD CSF | Healthy Plasma | AD Plasma | ||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Rank | Power (%) | Rank | Power (%) | Rank | Power (%) | Rank | Power (%) | |
| Aβ40 | 4 | 40.58 | 21 | 0 | 14 | 5.13 | 12 | 0.002 |
| Aβ42 | 5 | 29.68 | 17 | 0 | 3 | 56.67 | 10 | 0.07 |
| ApoE | 11 | 10.48 | 6 | 4.50 | 20 | 0.002 | 4 | 4.52 |
| Clusterin | 17 | 0.57 | 8 | 2.71 | 18 | 0.03 | 8 | 1.97 |
| EGF | 21 | 0.02 | 9 | 0.82 | 22 | 0.0003 | 5 | 3.21 |
| Eotaxin | 14 | 4.00 | 14 | 0.0006 | 7 | 19.08 | 18 | 0 |
| FGF-basic | 6 | 26.37 | 19 | 0 | 1 | 82.13 | 15 | 0 |
| Fibrinogen | 7 | 18.41 | 18 | 0 | 21 | 0.002 | 7 | 2.41 |
| G-CSF | 2 | 52.30 | 22 | 0 | 23 | 0.0001 | 3 | 5.02 |
| HGF | 22 | 0.013 | 10 | 0.62 | 5 | 35.51 | 19 | 0 |
| IFN-α | 16 | 2.45 | 13 | 0.002 | 13 | 6.19 | 20 | 0 |
| IL-12 | 19 | 0.46 | 12 | 0.06 | 12 | 7.33 | 23 | 0 |
| IL-1RA | 1 | 66.43 | 20 | 0 | 2 | 73.54 | 16 | 0 |
| IL-2R | 23 | 0.0003 | 7 | 3.54 | 10 | 12.74 | 21 | 0 |
| IL-6 | 8 | 13.92 | 23 | 0 | - | - | - | - |
| IL-8 | 13 | 4.34 | 15 | 0.0002 | 4 | 44.19 | 22 | 0 |
| IP-10 | 20 | 0.10 | 11 | 0.21 | 17 | 1.37 | 11 | 0.003 |
| Keratin 9 | 24 | 0 | 1 | 98.76 | 24 | 0 | 2 | 7.96 |
| MCP-1 | 24 | 0 | 4 | 8.57 | 9 | 15.56 | 17 | 0 |
| MIG | 9 | 13.14 | 24 | 0 | 16 | 3.06 | 13 | 0.0003 |
| MIP-1α | 3 | 46.84 | 24 | 0 | 6 | 23.96 | 24 | 0 |
| MIP-1β | 24 | 0 | 5 | 6.23 | 24 | 0 | 1 | 28.05 |
| SPARCL1 | 12 | 6.66 | 16 | 0 | 11 | 7.68 | 14 | 0 |
| Tau | 15 | 3.92 | 2 | 15.68 | 19 | 0.02 | 6 | 2.48 |
| Tetranectin | 18 | 0.56 | 3 | 14.95 | 15 | 3.86 | 9 | 1.49 |
| VEGF | 10 | 11.85 | 24 | 0 | 8 | 17.48 | 24 | 0 |
Contribution of each individual marker to the overall power of the biomarker panels. Values are presented as the percentage of a cohort that can be accurately identified using that marker. Each marker is ranked according to its contribution to the strength of the biomarker panel.
In the CSF samples, the C4.5 algorithm did not perform well, resulting in only seven (46.7%) patients being correctly classified, whilst the remaining eight were misclassified. The naïve Bayes classifier performed slightly better, returning 60% (i.e. nine patients) accuracy. The remaining six patients were not correctly assigned to their category. The most accurate results were obtained with the neural network, with 13 patients been correctly classified (86.7%). When the whole data set was used for both training and testing, all three classification techniques reached an accuracy of 100% (all patients correctly classified). For the measurements made in plasma samples, the C4.5 algorithm classified 14 out of 16 (87.5%) correctly. The C4.5 algorithm shows that, based upon this dataset, it is enough to check the values of the M7 variable (Aβ42) to establish whether a patient should be classified as Healthy or as AD. However, when the whole data were used for both training and testing, not all patients were correctly classified (15/16). In plasma samples the naïve Bayes algorithm reached an accuracy of 75% (i.e. 12 patients correctly classified). This method was also able to classify correctly all patients when both training and testing were performed using the whole data. Finally, the ANN was less accurate than for the CSF samples, assigning only 13 out of 16 patients (81.25%) to the correct group, but it reached 100% accuracy when the whole data was used for training and testing. It is evident that the classification techniques performed better on the plasma samples with more data available. The naïve Bayes classifier was unable to outperform the other two techniques; this is likely to be due to the cohort size as this method is known to perform well with big datasets and if data are normally distributed. For the CSF samples the ANN was the best classifier, but unfortunately it is difficult to access the ‘rules’ used for classification by this method as they are not ‘visible’.
Discussion
Confirmation of the presence of a disease to acceptable levels of clinical confidence from a single molecular indicator (i.e. a biomarker) is not a trivial task. Practical examples of such markers do, however, exist. Prostate-specific antigen (PSA) and Human Epidermal Growth Factor Receptor 2 (HER2), for example, are employed diagnostically for prostate and breast cancers respectively. Whilst even these exemplars display limitations (HER2 is not definitive of all breast cancer subtypes [24] and the diagnostic assay for PSA produces large numbers of false positives [25]), they provide useful diagnostic tools and, particularly in the case of HER2, have lead to improved therapeutic intervention [26]. Investigations into biomarkers for AD have, to date, failed to identify a single molecule capable of fully depicting the disease state due in part to the complex nature of the pathways underlying the disease [27]. An alternative approach whereby panels of biomarkers are investigated (as opposed to a singular marker) has, in comparison, heralded some success an example of which being the recent identification of a panel of CSF biomarkers capable of differentiating between AD and healthy individuals. This panel comprised some novel protein biomarkers not previously associated with AD and, encouragingly, was applied in the identification of a cohort of individuals with MCI [18].
The initial aim of this study was to demonstrate that the components of this biomarker panel could be detected and quantified in blood plasma. Further to this our aim was to determine the concentrations of these proteins in CSF and to examine their utility as individual biomarkers in their own rights in both types of clinical sample (CSF and plasma). It is well documented that albumin is the most abundant protein component of both blood plasma [28] and CSF [29]. It also has limited utility as a biomarker due to its physical properties, the inter-individual variation in ranges that it exhibits and the vastly higher concentrations that it exists at compared to the other biomarker components of the panel under consideration [28,30]. Instead of providing an accurate measure of disease onset and/or progression, we now consider that it is more likely that albumin is acting as a marker of damage to the blood-CSF barrier [31] and therefore it was omitted from our current study. As an association between inflammatory genes/proteins and AD has been previously demonstrated [32-34], the inflammatory profiles of the samples were also assessed to investigate whether they would positively augment the diagnosis obtained using the original biomarker panel.
As illustrated in Figure 2, all of the components of the biomarker panel that were under consideration (Aβ40, Aβ42, ApoE, Clusterin, Fibrinogen alpha chain, Keratin 9, SPARCL1, Tau and Tetranectin) could be measured in blood plasma samples from both healthy and diseased individuals using immunoassay. All proteins could also be detected in CSF samples from both healthy and diseased individuals with the exception of Keratin 9 which was not detectable in CSF from healthy individuals. To our knowledge, this is only the second demonstration of the presence of Keratin 9, a protein normally considered to be associated with skin, in blood. Previous data suggests that Keratin 9 may act as a serum marker of metastasis of hepatocellular carcinoma [35]. It is also the first immunoassay validation of the presence of Keratin 9 in CSF; the only previous association between CSF Keratin 9 and AD was reported in a study by Vafadar-Isfahani and colleagues [18].
The cohort sizes under investigation in this study were modest, but are in keeping with other similar studies [36]. Perhaps unexpectedly given these relatively small cohorts, significant differences in the mean concentrations of some of the biomarker panel components and inflammatory proteins were found in both CSF and plasma samples (Figures 2 and 3; Tables 2 and 3) suggesting that these proteins may have potential utility as individual biomarkers in AD. The most notable potential AD biomarker identified in this study is CSF Keratin 9 as this was found to only be present in CSF samples collected from AD patients, not healthy individuals. Another promising protein marker is SPARCL1, levels of which were significantly altered in CSF samples of AD patients when compared to healthy controls (p = 0.035) and approaching significance (p = 0.089) in plasma samples. SPARCL1 has been linked to several diseases including various forms of cancer [37-43], uterine leiomyomas [44] and multiple sclerosis [45]. An association between SPARCL1 and AD has previously been demonstrated by Yin et al using 1D electrophoresis followed by LC-MS/MS [46]. The study also identified SPARCL1 as a potential target for Parkinson’s disease [46]. The other proteins found to be significantly altered in the disease state during this study, Aβ42 and Tau have extensively been linked to AD and thus this study provides further validation of their potential utility in AD diagnostics.
When looking at the significance of the association of the proteins with AD in CSF, the most powerful (excluding Keratin 9 for which a p value cannot be determined) are SPARCL1, Aβ42 and Fibrinogen. These three proteins were also identified as the three most important components of the AD biomarker panel derived in the mass spectrometry study that preceded this study [18]. The levels of correlation between plasma and CSF protein concentrations were found to be variable (Table 2). The blood brain barrier restricts molecular diffusion (including capillary walls among other structures) and associated biochemical and physiological processes to maintain a dynamical blood/CSF concentration gradient. The observed differences that we found seem most likely due to compromises in this barrier function which have been demonstrated to be compromised in neurological conditions including AD [47].
Evidence of an inflammatory component to AD has been emerging in the literature over the last few years, however the nature and extent of the inflammatory response has yet to be fully elucidated. A variety of cytokines and chemokines have been implicated in the disease process [36,48,49]. Indeed, the first suggestion of a biomolecular fingerprint for AD included a large number of inflammatory markers [34]. The majority of the cytokines examined in this study were detectable in both plasma and CSF which is in contrast to a previous study undertaken using Luminex in which only one cytokine out of the 22 examined was detectable [50]. These differences could be due to the sensitivity of the detection kit being utilised or the disease stage of the patients being studied. Correlation between cytokine levels and disease progression have been previously identified [51]. Motta and colleagues demonstrated an initial large increase in immune responsiveness for some cytokines in mild-stage AD with respect to comparative healthy individuals. As the disease progressed from the early stages a gradual decline in cytokine levels were observed until concentrations in individuals with late-stage disease had dropped below those observed in the healthy cohort [51]. When taking the findings of this previous study into consideration, the decrease in levels of many of the inflammatory markers observed within this study may have been expected as the samples investigated represent late-stage disease.
Of the detectable cytokines, we observed significant decreases in CSF levels of IL-12 (p = 0.04) and plasma levels of FGF-basic (p = 0.05), IL-1RA (p = 0.02), MCP-1 (p = 0.01) and a significant increase in plasma MIP-1β (p = 0.04) (Figure 3 and Table 3). All five of these proteins have previously been linked to AD but the extent and strength of these associations is varied. Whilst FGF basic has been linked to AD processes [52] and MIP-1β (CCL4) has been identified as a component of some Aβ1-42 pathways [53,54], to our knowledge this is one of the first instances of them being highlighted as potential AD biomarkers. In contrast, several studies have examined expression patterns of MCP-1 (CCL2) in AD with slightly conflicting findings, but generally it appears that it increases in AD CSF and decreases in AD plasma [55-58] which is in accordance with the findings of this study. The IL-1 cytokine family has been widely implicated in AD pathology [59-61] and decreased expression of IL-1RA in CSF has previously been linked to AD [62]. Levels of IL-12 have previously been shown to decrease in CSF from AD patients [63] corroborating the findings of this study. Expression of IL-12 in plasma samples has also been tracked throughout AD progression with an increased concentration being observed during mild stage disease which gradually decreases as the health of the individual declines [51]. However these results were not replicated in this study as we observed comparable levels of IL-12 in plasma from AD patients and healthy individuals.
In addition to the foregoing points, Figures 2 and 3 highlight the difficulties encountered whilst trying to define a singular biomarker indicative of the presence of a complex disease condition such as AD. Even if, as demonstrated by some of the data presented here, a prospective biomarker is shown to be significantly different in the disease condition compared to that in the healthy, there still remains uncertainty as to the accuracy of the clinical decision based on the marker measurement from a patient. This uncertainty has its origins in the overlap between the marker distributions (Figure 1). Only those concentration values falling outside of the overlapping ranges can be considered as an affirmative diagnosis, thus extensive levels of overlap may render the practical application of a respective marker untenable.
One solution to the foregoing marker screening problem involves combination of the measurements of separate molecular markers into a singular analytical protocol to yield a more reliable clinical decision. During this study this approach was investigated through application of established supervised learning techniques to the datasets. Using the C4.5 algorithm, a diagnostic accuracy (in correctly classifying either an AD or Healthy sample) of 87.5% was obtained for the plasma data sets whilst application of the neural network algorithm to the CSF dataset yielded a diagnostic accuracy of 86.7%. It is not appropriate to assign sensitivity or specificity values to the results obtained within this study due to the nature of the analytical methods used which were implemented using a ‘leave-one-out’ cross validation of the entire healthy and AD cohorts. The values of diagnostic accuracy obtained are promising, particularly for the plasma samples, but need to be validated further using larger sample cohorts. We envisage that this type of multiparametric biomarker testing would be used as a screening tool to inform decisions about further diagnostic requirements e.g. whether a patient should be referred for an MRI scan. When used in conjunction with clinical and neuropsychometric evaluation, the ability to define a healthy individual from a blood test would enable a subset of the population to avoid the need for referral for imaging tests such as MRI [64] which are expensive and tend to be of limited availability outside specialist centres.
Acknowledgements
This work was funded by a Medical Research Council Developmental Pathways Grant, grant number 1001500 (http://www.mrc.ac.uk/index.htm). The work of OPTIMA has been supported over 20 years by major grants from Bristol-Myers Squibb, Merck, USA Inc, MRC, Charles Wolfson Charitable Trust. GW was partly funded by the NIHR Biomedical Research Centre Programme, Oxford. No funding bodies had any role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure of conflict of interest
There are no actual or potential conflicts of interest to disclose.
Abbreviations
- AD
Alzheimer’s disease
- CSF
Cerebrospinal fluid
References
- 1.Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer’s disease. Lancet. 2011;377:1019–1031. doi: 10.1016/S0140-6736(10)61349-9. [DOI] [PubMed] [Google Scholar]
- 2.Herrmann N, Chau SA, Kircanski I, Lanctot KL. Current and emerging drug treatment options for Alzheimer’s disease: a systematic review. Drugs. 2011;71:2031–2065. doi: 10.2165/11595870-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 3.Salomone S, Caraci F, Leggio GM, Fedotova J, Drago F. New pharmacological strategies for treatment of Alzheimer’s disease: focus on disease modifying drugs. Br J Clin Pharmacol. 2012;73:504–517. doi: 10.1111/j.1365-2125.2011.04134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer disease: back to the future. Neuron. 2010;68:270–281. doi: 10.1016/j.neuron.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 5.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Hull M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O’Donovan M, Owen MJ, Williams J. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, Thomas C, Richards A, Ivanov D, Widdowson C, Chapman J, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Beaumont H, Warden D, Wilcock G, Love S, Kehoe PG, Hooper NM, Vardy ER, Hardy J, Mead S, Fox NC, Rossor M, Collinge J, Maier W, Jessen F, Ruther E, Schurmann B, Heun R, Kolsch H, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Gallacher J, Hull M, Rujescu D, Giegling I, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Pankratz VS, Sando SB, Aasly JO, Barcikowska M, Wszolek ZK, Dickson DW, Graff-Radford NR, Petersen RC, van Duijn CM, Breteler MM, Ikram MA, Destefano AL, Fitzpatrick AL, Lopez O, Launer LJ, Seshadri S, Berr C, Campion D, Epelbaum J, Dartigues JF, Tzourio C, Alperovitch A, Lathrop M, Feulner TM, Friedrich P, Riehle C, Krawczak M, Schreiber S, Mayhaus M, Nicolhaus S, Wagenpfeil S, Steinberg S, Stefansson H, Stefansson K, Snaedal J, Bjornsson S, Jonsson PV, Chouraki V, Genier-Boley B, Hiltunen M, Soininen H, Combarros O, Zelenika D, Delepine M, Bullido MJ, Pasquier F, Mateo I, Frank-Garcia A, Porcellini E, Hanon O, Coto E, Alvarez V, Bosco P, Siciliano G, Mancuso M, Panza F, Solfrizzi V, Nacmias B, Sorbi S, Bossu P, Piccardi P, Arosio B, Annoni G, Seripa D, Pilotto A, Scarpini E, Galimberti D, Brice A, Hannequin D, Licastro F, Jones L, Holmans PA, Jonsson T, Riemenschneider M, Morgan K, Younkin SG, Owen MJ, O’Donovan M, Amouyel P, Williams J. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fievet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O, de Pancorbo MM, Lendon C, Dufouil C, Jaillard C, Leveillard T, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossu P, Piccardi P, Annoni G, Seripa D, Galimberti D, Hannequin D, Licastro F, Soininen H, Ritchie K, Blanche H, Dartigues JF, Tzourio C, Gut I, Van Broeckhoven C, Alperovitch A, Lathrop M, Amouyel P. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 8.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JS, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, George-Hyslop PS, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, Decarli C, Dekosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, Bis JC, Smith AV, Carassquillo MM, Lambert JC, Harold D, Schrijvers EM, Ramirez-Lorca R, Debette S, Longstreth WT Jr, Janssens AC, Pankratz VS, Dartigues JF, Hollingworth P, Aspelund T, Hernandez I, Beiser A, Kuller LH, Koudstaal PJ, Dickson DW, Tzourio C, Abraham R, Antunez C, Du Y, Rotter JI, Aulchenko YS, Harris TB, Petersen RC, Berr C, Owen MJ, Lopez-Arrieta J, Varadarajan BN, Becker JT, Rivadeneira F, Nalls MA, Graff-Radford NR, Campion D, Auerbach S, Rice K, Hofman A, Jonsson PV, Schmidt H, Lathrop M, Mosley TH, Au R, Psaty BM, Uitterlinden AG, Farrer LA, Lumley T, Ruiz A, Williams J, Amouyel P, Younkin SG, Wolf PA, Launer LJ, Lopez OL, van Duijn CM, Breteler MM. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303:1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee SH, Harold D, Nyholt DR, Goddard ME, Zondervan KT, Williams J, Montgomery GW, Wray NR, Visscher PM. Estimation and partitioning of polygenic variation captured by common SNPs for Alzheimer’s disease, multiple sclerosis and endometriosis. Hum Mol Genet. 2013;22:832–841. doi: 10.1093/hmg/dds491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ward A, Crean S, Mercaldi CJ, Collins JM, Boyd D, Cook MN, Arrighi HM. Prevalence of apolipoprotein E4 genotype and homozygotes (APOE e4/4) among patients diagnosed with Alzheimer’s disease: a systematic review and meta-analysis. Neuroepidemiology. 2012;38:1–17. doi: 10.1159/000334607. [DOI] [PubMed] [Google Scholar]
- 12.Prvulovic D, Hampel H. Amyloid beta (Abeta) and phospho-tau (p-tau) as diagnostic biomarkers in Alzheimer’s disease. Clin Chem Lab Med. 2011;49:367–374. doi: 10.1515/CCLM.2011.087. [DOI] [PubMed] [Google Scholar]
- 13.Ewers M, Mielke MM, Hampel H. Blood-based biomarkers of microvascular pathology in Alzheimer’s disease. Exp Gerontol. 2010;45:75–79. doi: 10.1016/j.exger.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hampel H, Burger K, Teipel SJ, Bokde AL, Zetterberg H, Blennow K. Core candidate neurochemical and imaging biomarkers of Alzheimer’s disease. Alzheimers Dement. 2008;4:38–48. doi: 10.1016/j.jalz.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 15.Mattsson N, Zetterberg H, Hansson O, Andreasen N, Parnetti L, Jonsson M, Herukka SK, van der Flier WM, Blankenstein MA, Ewers M, Rich K, Kaiser E, Verbeek M, Tsolaki M, Mulugeta E, Rosen E, Aarsland D, Visser PJ, Schroder J, Marcusson J, de Leon M, Hampel H, Scheltens P, Pirttila T, Wallin A, Jonhagen ME, Minthon L, Winblad B, Blennow K. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA. 2009;302:385–393. doi: 10.1001/jama.2009.1064. [DOI] [PubMed] [Google Scholar]
- 16.Schneider P, Hampel H, Buerger K. Biological marker candidates of Alzheimer’s disease in blood, plasma, and serum. CNS Neurosci Ther. 2009;15:358–374. doi: 10.1111/j.1755-5949.2009.00104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buchhave P, Minthon L, Zetterberg H, Wallin AK, Blennow K, Hansson O. Cerebrospinal fluid levels of beta-amyloid 1-42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch Gen Psychiatry. 2012;69:98–106. doi: 10.1001/archgenpsychiatry.2011.155. [DOI] [PubMed] [Google Scholar]
- 18.Vafadar-Isfahani B, Ball G, Coveney C, Lemetre C, Boocock D, Minthon L, Hansson O, Miles AK, Janciauskiene SM, Warden D, Smith AD, Wilcock G, Kalsheker N, Rees R, Matharoo-Ball B, Morgan K. Identification of SPARC-like 1 Protein as Part of a Biomarker Panel for Alzheimer’s Disease in Cerebrospinal Fluid. J Alzheimers Dis. 2012;28:625–636. doi: 10.3233/JAD-2011-111505. [DOI] [PubMed] [Google Scholar]
- 19.Thambisetty M, Simmons A, Velayudhan L, Hye A, Campbell J, Zhang Y, Wahlund LO, Westman E, Kinsey A, Guntert A, Proitsi P, Powell J, Causevic M, Killick R, Lunnon K, Lynham S, Broadstock M, Choudhry F, Howlett DR, Williams RJ, Sharp SI, Mitchelmore C, Tunnard C, Leung R, Foy C, O’Brien D, Breen G, Furney SJ, Ward M, Kloszewska I, Mecocci P, Soininen H, Tsolaki M, Vellas B, Hodges A, Murphy DG, Parkins S, Richardson JC, Resnick SM, Ferrucci L, Wong DF, Zhou Y, Muehlboeck S, Evans A, Francis PT, Spenger C, Lovestone S. Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch Gen Psychiatry. 2010;67:739–748. doi: 10.1001/archgenpsychiatry.2010.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams JH, Wilcock GK, Seeburger J, Dallob A, Laterza O, Potter W, Smith AD. Non-linear relationships of cerebrospinal fluid biomarker levels with cognitive function: an observational study. Alzheimers Res Ther. 2011;3:5. doi: 10.1186/alzrt64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quinlan JR. C4.5: programs for machine learning. Morgan Kaufmann Publishers Inc.; 1993. [Google Scholar]
- 22.John GH, Langley P. Estimating continuous distributions in Bayesian classifiers. Proceedings of the Eleventh conference on Uncertainty in artificial intelligence. 1995:338–345. [Google Scholar]
- 23.Haykin S. Neural Networks: A Comprehensive Foundation. Prentice Hall PTR; 1998. [Google Scholar]
- 24.Gutierrez C, Schiff R. HER2: biology, detection, and clinical implications. Arch Pathol Lab Med. 2011;135:55–62. doi: 10.1043/2010-0454-RAR.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lim LS, Sherin K. Screening for prostate cancer in U. S. men ACPM position statement on preventive practice. Am J Prev Med. 2008;34:164–170. doi: 10.1016/j.amepre.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 26.de Bono JS, Ashworth A. Translating cancer research into targeted therapeutics. Nature. 2010;467:543–549. doi: 10.1038/nature09339. [DOI] [PubMed] [Google Scholar]
- 27.Richens JL, Morgan K, O’Shea P. Reverse engineering of Alzheimer’s disease based on biomarker pathways analysis. Neurobiol Aging. 2014 doi: 10.1016/j.neurobiolaging.2014.02.024. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 28.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics. 2002;1:845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 29.Yuan X, Russell T, Wood G, Desiderio DM. Analysis of the human lumbar cerebrospinal fluid proteome. Electrophoresis. 2002;23:1185–1196. doi: 10.1002/1522-2683(200204)23:7/8<1185::AID-ELPS1185>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 30.Richens JL, Lunt EA, Sanger D, McKenzie G, O’Shea P. Avoiding nonspecific interactions in studies of the plasma proteome: practical solutions to prevention of nonspecific interactions for label-free detection of low-abundance plasma proteins. J Proteome Res. 2009;8:5103–5110. doi: 10.1021/pr900487y. [DOI] [PubMed] [Google Scholar]
- 31.Chalbot S, Zetterberg H, Blennow K, Fladby T, Andreasen N, Grundke-Iqbal I, Iqbal K. Blood-cerebrospinal fluid barrier permeability in Alzheimer’s disease. J Alzheimers Dis. 2011;25:505–515. doi: 10.3233/JAD-2011-101959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morgan K. The three new pathways leading to Alzheimer’s disease. Neuropathol Appl Neurobiol. 2011;37:353–357. doi: 10.1111/j.1365-2990.2011.01181.x. [DOI] [PubMed] [Google Scholar]
- 33.Jones L, Holmans PA, Hamshere ML, Harold D, Moskvina V, Ivanov D, Pocklington A, Abraham R, Hollingworth P, Sims R, Gerrish A, Pahwa JS, Jones N, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, Heun R, Kolsch H, van den Bussche H, Heuser I, Peters O, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Hull M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Ruther E, Carrasquillo MM, Pankratz VS, Younkin SG, Hardy J, O’Donovan MC, Owen MJ, Williams J. Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer’s disease. PLoS One. 2010;5:e13950. doi: 10.1371/journal.pone.0013950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ray S, Britschgi M, Herbert C, Takeda-Uchimura Y, Boxer A, Blennow K, Friedman LF, Galasko DR, Jutel M, Karydas A, Kaye JA, Leszek J, Miller BL, Minthon L, Quinn JF, Rabinovici GD, Robinson WH, Sabbagh MN, So YT, Sparks DL, Tabaton M, Tinklenberg J, Yesavage JA, Tibshirani R, Wyss-Coray T. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat Med. 2007;13:1359–1362. doi: 10.1038/nm1653. [DOI] [PubMed] [Google Scholar]
- 35.Fu BS, Liu W, Zhang JW, Zhang T, Li H, Chen GH. Serum proteomic analysis on metastasis-associated proteins of hepatocellular carcinoma. Nan Fang Yi Ke Da Xue Xue Bao. 2009;29:1775–1778. [PubMed] [Google Scholar]
- 36.Lee KS, Chung JH, Lee KH, Shin MJ, Oh BH, Hong CH. Bioplex analysis of plasma cytokines in Alzheimer’s disease and mild cognitive impairment. Immunol Lett. 2008;121:105–109. doi: 10.1016/j.imlet.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 37.Hu H, Zhang H, Ge W, Liu X, Loera S, Chu P, Chen H, Peng J, Zhou L, Yu S, Yuan Y, Zhang S, Lai L, Yen Y, Zheng S. Secreted Protein Acidic and Rich in Cysteines-like 1 Suppresses Aggressiveness and Predicts Better Survival in Colorectal Cancers. Clin Cancer Res. 2012;18:5438–5448. doi: 10.1158/1078-0432.CCR-12-0124. [DOI] [PubMed] [Google Scholar]
- 38.Zhang H, Widegren E, Wang DW, Sun XF. SPARCL1: a potential molecule associated with tumor diagnosis, progression and prognosis of colorectal cancer. Tumour Biol. 2011;32:1225–1231. doi: 10.1007/s13277-011-0226-x. [DOI] [PubMed] [Google Scholar]
- 39.Yu SJ, Yu JK, Ge WT, Hu HG, Yuan Y, Zheng S. SPARCL1, Shp2, MSH2, E-cadherin, p53, ADCY-2 and MAPK are prognosis-related in colorectal cancer. World J Gastroenterol. 2011;17:2028–2036. doi: 10.3748/wjg.v17.i15.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hurley PJ, Marchionni L, Simons BW, Ross AE, Peskoe SB, Miller RM, Erho N, Vergara IA, Ghadessi M, Huang Z, Gurel B, Park BH, Davicioni E, Jenkins RB, Platz EA, Berman DM, Schaeffer EM. Secreted protein, acidic and rich in cysteine-like 1 (SPARCL1) is down regulated in aggressive prostate cancers and is prognostic for poor clinical outcome. Proc Natl Acad Sci U S A. 2012;109:14977–14982. doi: 10.1073/pnas.1203525109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Esposito I, Kayed H, Keleg S, Giese T, Sage EH, Schirmacher P, Friess H, Kleeff J. Tumor-suppressor function of SPARC-like protein 1/Hevin in pancreatic cancer. Neoplasia. 2007;9:8–17. doi: 10.1593/neo.06646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li P, Qian J, Yu G, Chen Y, Liu K, Li J, Wang J. Down-regulated SPARCL1 is associated with clinical significance in human gastric cancer. J Surg Oncol. 2012;105:31–37. doi: 10.1002/jso.22025. [DOI] [PubMed] [Google Scholar]
- 43.Lau CP, Poon RT, Cheung ST, Yu WC, Fan ST. SPARC and Hevin expression correlate with tumour angiogenesis in hepatocellular carcinoma. J Pathol. 2006;210:459–468. doi: 10.1002/path.2068. [DOI] [PubMed] [Google Scholar]
- 44.Mencalha AL, Levinsphul A, Deterling LC, Pizzatti L, Abdelhay E. SPARC-like1 mRNA is overexpressed in human uterine leiomyoma. Mol Med Report. 2008;1:571–574. [PubMed] [Google Scholar]
- 45.Hammack BN, Fung KY, Hunsucker SW, Duncan MW, Burgoon MP, Owens GP, Gilden DH. Proteomic analysis of multiple sclerosis cerebrospinal fluid. Mult Scler. 2004;10:245–260. doi: 10.1191/1352458504ms1023oa. [DOI] [PubMed] [Google Scholar]
- 46.Yin GN, Lee HW, Cho JY, Suk K. Neuronal pentraxin receptor in cerebrospinal fluid as a potential biomarker for neurodegenerative diseases. Brain Res. 2009;1265:158–170. doi: 10.1016/j.brainres.2009.01.058. [DOI] [PubMed] [Google Scholar]
- 47.Carmeliet P, De Strooper B. Alzheimer’s disease: A breach in the blood-brain barrier. Nature. 2012;485:451–452. doi: 10.1038/485451a. [DOI] [PubMed] [Google Scholar]
- 48.Lee KS, Chung JH, Choi TK, Suh SY, Oh BH, Hong CH. Peripheral cytokines and chemokines in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2009;28:281–287. doi: 10.1159/000245156. [DOI] [PubMed] [Google Scholar]
- 49.Hu WT, Chen-Plotkin A, Arnold SE, Grossman M, Clark CM, Shaw LM, Pickering E, Kuhn M, Chen Y, McCluskey L, Elman L, Karlawish J, Hurtig HI, Siderowf A, Lee VM, Soares H, Trojanowski JQ. Novel CSF biomarkers for Alzheimer’s disease and mild cognitive impairment. Acta Neuropathol. 2010;119:669–678. doi: 10.1007/s00401-010-0667-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choi C, Jeong JH, Jang JS, Choi K, Lee J, Kwon J, Choi KG, Lee JS, Kang SW. Multiplex analysis of cytokines in the serum and cerebrospinal fluid of patients with Alzheimer’s disease by color-coded bead technology. J Clin Neurol. 2008;4:84–88. doi: 10.3988/jcn.2008.4.2.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Motta M, Imbesi R, Di Rosa M, Stivala F, Malaguarnera L. Altered plasma cytokine levels in Alzheimer’s disease: correlation with the disease progression. Immunol Lett. 2007;114:46–51. doi: 10.1016/j.imlet.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 52.Stopa EG, Gonzalez AM, Chorsky R, Corona RJ, Alvarez J, Bird ED, Baird A. Basic fibroblast growth factor in Alzheimer’s disease. Biochem Biophys Res Commun. 1990;171:690–696. doi: 10.1016/0006-291x(90)91201-3. [DOI] [PubMed] [Google Scholar]
- 53.Ito S, Sawada M, Haneda M, Ishida Y, Isobe K. Amyloid-beta peptides induce several chemokine mRNA expressions in the primary microglia and Ra2 cell line via the PI3K/Akt and/or ERK pathway. Neurosci Res. 2006;56:294–299. doi: 10.1016/j.neures.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 54.Smits HA, Rijsmus A, van Loon JH, Wat JW, Verhoef J, Boven LA, Nottet HS. Amyloid-beta-induced chemokine production in primary human macrophages and astrocytes. J Neuroimmunol. 2002;127:160–168. doi: 10.1016/s0165-5728(02)00112-1. [DOI] [PubMed] [Google Scholar]
- 55.Conductier G, Blondeau N, Guyon A, Nahon JL, Rovère C. The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J Neuroimmunol. 2010;224:93–100. doi: 10.1016/j.jneuroim.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 56.Galimberti D, Schoonenboom N, Scheltens P, Fenoglio C, Bouwman F, Venturelli E, Guidi I, Blankenstein MA, Bresolin N, Scarpini E. Intrathecal chemokine synthesis in mild cognitive impairment and Alzheimer disease. Arch Neurol. 2006;63:538–543. doi: 10.1001/archneur.63.4.538. [DOI] [PubMed] [Google Scholar]
- 57.Galimberti D, Fenoglio C, Lovati C, Venturelli E, Guidi I, Corra B, Scalabrini D, Clerici F, Mariani C, Bresolin N, Scarpini E. Serum MCP-1 levels are increased in mild cognitive impairment and mild Alzheimer’s disease. Neurobiol Aging. 2006;27:1763–1768. doi: 10.1016/j.neurobiolaging.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 58.Sun YX, Minthon L, Wallmark A, Warkentin S, Blennow K, Janciauskiene S. Inflammatory markers in matched plasma and cerebrospinal fluid from patients with Alzheimer’s disease. Dement Geriatr Cogn Disord. 2003;16:136–144. doi: 10.1159/000071001. [DOI] [PubMed] [Google Scholar]
- 59.Payao SL, Goncalves GM, de Labio RW, Horiguchi L, Mizumoto I, Rasmussen LT, de Souza Pinhel MA, Silva Souza DR, Bechara MD, Chen E, Mazzotti DR, Ferreira Bertolucci PH, Cardoso Smith Mde A. Association of interleukin 1beta polymorphisms and haplotypes with Alzheimer’s disease. J Neuroimmunol. 2012;247:59–62. doi: 10.1016/j.jneuroim.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 60.Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, Cribbs DH, LaFerla FM. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J Immunol. 2011;187:6539–6549. doi: 10.4049/jimmunol.1100620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Forlenza OV, Diniz BS, Talib LL, Mendonca VA, Ojopi EB, Gattaz WF, Teixeira AL. Increased serum IL-1beta level in Alzheimer’s disease and mild cognitive impairment. Dement Geriatr Cogn Disord. 2009;28:507–512. doi: 10.1159/000255051. [DOI] [PubMed] [Google Scholar]
- 62.Tarkowski E, Liljeroth AM, Nilsson A, Minthon L, Blennow K. Decreased levels of intrathecal interleukin 1 receptor antagonist in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2001;12:314–317. doi: 10.1159/000051276. [DOI] [PubMed] [Google Scholar]
- 63.Rentzos M, Paraskevas GP, Kapaki E, Nikolaou C, Zoga M, Rombos A, Tsoutsou A, Vassilopoulos DD. Interleukin-12 is reduced in cerebrospinal fluid of patients with Alzheimer’s disease and frontotemporal dementia. J Neurol Sci. 2006;249:110–114. doi: 10.1016/j.jns.2006.05.063. [DOI] [PubMed] [Google Scholar]
- 64.Hampel H, Frank R, Broich K, Teipel SJ, Katz RG, Hardy J, Herholz K, Bokde AL, Jessen F, Hoessler YC, Sanhai WR, Zetterberg H, Woodcock J, Blennow K. Biomarkers for Alzheimer’s disease: academic, industry and regulatory perspectives. Nat Rev Drug Discov. 2010;9:560–574. doi: 10.1038/nrd3115. [DOI] [PubMed] [Google Scholar]