

Abstract
The deacetylase SIRT1 regulates multiple biological processes including cellular metabolism and aging. Importantly, SIRT1 can also inactivate the p53 tumor suppressor via deacetylation, suggesting a role in oncogenesis. Recently, SIRT1 was shown to be released from its endogenous inhibitor DBC1 by a process requiring AMPK and the phosphorylation of SIRT1 by yet undefined kinase(s). Here we provide further evidence that AMPK directly phosphorylates SIRT1 on T344, releasing it from DBC1. Furthermore, a phospho-mimetic SIRT1 (T334E) showed decreased binding to DBC1, supporting the importance of this phosphorylation in AMPK-mediated regulation of SIRT1 activity. In addition, inhibition of AMPK by Compound C led to increased p53 acetylation, suggesting a role for the AMPK/SIRT1 pathway in regulating p53 signaling. Together, our results support a hypothesis that AMPK negatively regulates p53 acetylation via phosphorylation of SIRT1 on T344. Furthermore, our findings also define the AMPK/SIRT1 axis as a possible targetable pathway to regulate p53 function.
Keywords: AMPK, SIRT1 deacetylase, DBC1, p53, phosphorylation, acetylation
Introduction
SIRT1, a mammalian ortholog of the yeast Sir2, is a NAD+-dependent deacetylase that plays a critical role in multiple biological processes including apoptosis [1], aging [2], metabolism [3] and various stress responses [4]. It does so by deacetylating a diverse group of substrates such as NF-ĸB [5], PGC-1α [3], Ku70 [1], MyoD [6], FOXO [7] and histones [8]. Interestingly, it was recently shown by two independent groups that SIRT1 could also deacetylate the p53 tumor suppressor in vitro, inhibiting p53 function and promoting cell survival under various stresses [9,10]. SIRT1 regulation of p53 was further corroborated in vivo as SIRT1-deficient thymocytes, harvested from SIRT1 knockout mice, displayed enhanced apoptosis upon γ-irradiation [11]. Based on these findings, SIRT1 function could have important implications in cancer development and progression.
As SIRT1 can regulate a wide range of cellular pathways, and may further play an intricate role in tumorigenesis, fully understanding the mechanisms of SIRT1 regulation is of extreme importance. Despite the extensive studies of SIRT1 downstream effectors, the upstream regulatory network is relatively less understood. To date, some work has examined the various inhibitors and activators of SIRT1 at both the transcriptional and translational levels. Transcriptionally, SIRT1 is under the control of at least two negative feedback loops. While the transcription factor E2F1 can induce SIRT1 expression, deacetylation of E2F1 by SIRT1 can also inhibit its activity [12]. Furthermore, while being a well-characterized target of SIRT1 deacetylation, p53 can also binds the SIRT1 promoter region to repress SIRT1 transcription [4]. Translationally, the microRNA miR-34a can bind SIRT1 mRNA preventing its translation [13], while the tumor suppressor HUR can similarly interact with SIRT1 mRNA but rather stabilizing the transcript [14]. While these previous studies have clearly begun to elucidate the multi-layered regulation of SIRT1 function, more recent studies have examined post-translational regulation of this deactylase by various proteinprotein interactions.
Activate Regulator of SIRT1 (AROS, #123) is one such protein that is known to directly interact and regulate SIRT1 activity. AROS was initially indicated to stimulate SIRT1 function and attenuate p53-dependent transcriptional activation. Furthermore, depletion of AROS, using anti-sense technology, increased cell susceptibility to apoptosis following DNA damage, consistent with the capacity of SIRT1 to regulate p53 [15]. Another molecule that can be found in complex with SIRT1 is the neuronal protein Necdin. Located primarily in post-mitotic neurons, Necdin can negatively control p53 activity by potentiating SIRT1-mediated deacetylation of p53 [16]. The Necdin-SIRT1-p53 signaling axis was further identified to prevent p53-mediated apoptosis in response to DNA damage [16]. Finally, multiple studies have recently reported that Deleted in Breast Cancer 1 (DBC1) can bind SIRT1 in an inhibitory complex to prevent the activation of SIRT1 [17,18]. Mechanistically, DBC1 and SIRT1 form a dynamic inhibitory complex both in vitro and in vivo [19]. Furthermore, the binding between these two proteins is dependent on the energetic state of the cell [19]. Consistent with metabolic stresses mediating the dynamic interaction between DBC1 and SIRT1, activation of AMP-activated Protein Kinase (AMPK), a crucial cellular energy sensor, was recently identified to promote SIRT1-DBC1 dissociation, leading to activation of SIRT1 [20]. Interestingly, while this group indicated that AMPK activity was crucial for SIRT1 to detach from DBC1, they were unable to identify a direct molecular mechanism from AMPK activation to subsequent SIRT1 activation. Furthermore, while phosphorylation of SIRT1 was shown to be important for its dissociation from DBC1, the kinase directly involved in this regulatory axis remains uncharacterized.
Here, we report that AMPK can directly phosphorylate both SIRT1 and DBC1 in vitro. We also identify T344 as the critical phosphorylation site on human SIRT1 required for AMPK regulation of DBC1-SIRT1 dissociation. Unfortunately, we currently do not understand the significance of DBC1 phosphorylation by AMPK. It may suggest that the upstream signaling pathways regulating DBC1-SIRT1 dissociation are far more complicated than previously thought, and that multi-subunit phosphorylation events may have a role in the integrity of this inhibitory complex.
Materials and methods
Cell culture
Cell culture was performed as previously described [21,22]. Lenti-viral shRNA packaging and infection were also performed as previously described [23]. The AMPK inhibitor Compound C (Calbiochem) and the AMPK activator AICAR (Cell Signaling Technology) were used as instructed by the manufacturers.
Plasmids
HA-DBC1 was a kind gift from Dr. Zhenkun Lou. Flag-SIRT1-7 was obtained from Addgene. To generate HA-SIRT1, pGEX-DBC1 and pGEX-SIRT1, DBC1 and SIRT1 cDNA were amplified by PCR from HA-DBC1 and Flag-SIRT1, respectively, and then the PCR product was subcloned into HA-pcDNA and pGEX-4T-1. Various versions of HA-SIRT1 and HA-DBC1 mutation constructs were generated using the QuikChange XL Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer’s instructions, with specific primer sequences available upon request.
Antibodies and reagents
Anti-K382-p53 (2525), anti-SIRT1 (9475), anti-pT172-AMPK (2535), anti-pS79-ACC1 (3661), anti-AMPKα (2603), and anti-AMPKβ (4150) were purchased from Cell Signaling Technology. Anti-DBC1 (A300-432A) was purchased from Bethyl Laboratories. Anti-Tubulin (T-5168), anti-Vinculin (V-4504), polyclonal anti-Flag (F-2425), monoclonal anti-Flag (F-3165), anti-HA agarose beads (A-2095), peroxidase-conjugated anti-mouse secondary (A-4416), and peroxidase-conjugated anti-rabbit-secondary antibodies (A4914) were purchased from Sigma. Monoclonal anti-HA antibody (MMS-101P) was purchased from Covance. Polyclonal anti-HA antibody (SC-805) and monoclonal anti-p53 (SC-126) was purchased from Santa Cruz Biotechnology. Lipofectamine and Plus reagents were purchased from Invitrogen.
Immunoblots and immunoprecipitation
Cells were lysed in EBC buffer (50 mM Tris pH 7.5, 120 mM NaCl, 0.5% NP-40) supplemented with protease inhibitors (Complete Mini, Roche) and phosphatase inhibitors (phosphatase inhibitor cocktail set I and II, Calbiochem). Protein concentrations were then measured using a Bio-Rad protein assay reagent on a Beckman Coulter DU-800 spectrophotometer. Finally, the lysates were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. For immunoprecipitation experiments, 1 mg of lysate was incubated with either anti-HA or anti-Flag conjugated agarose beads overnight at 4°C. Immuno-complexes were then washed five times with NETN buffer (20 mM Tris pH 8.0, 100 mM NaCl, 1 mM EDTA, and 0.5% NP-40) and resolved by SDS-PAGE and immunoblotted with the indicated antibodies.
In vitro kinase assay
For use in in vitro kinase assays, active AMPK was purchased from Millipore. The protocol followed was previously described by Inuzuka et al. [24]. Briefly, 2.5 μg of the indicated GST fusion proteins were incubated with active AMPK in the presence of 0.5 μCi [γ-32P] ATP and 200 μM cold ATP in AMPK kinase buffer supplemented with 1 mM AMP. The reaction was allowed to proceed for 30 minutes before being stopped by the addition of SDS-containing loading buffer. The samples were then resolved by SDS-PAGE.
Mass spectrometry analysis
Mass spectrometry analysis to identify novel phosphorylation sites on both SIRT1 and DBC1 was performed similarly as previously described [25].
Results
AMPK regulates p53 acetylation via SIRT1
It was previously shown that AMPK could activate SIRT1 by promoting its phosphorylation and subsequent dissociation from the inhibitory molecule, DBC1 [20]. Active SIRT1 has also been identified to deacetylate p53, attenuating p53 transcriptional activity [9,10]. Therefore, we first explored whether AMPK regulation of SIRT1 function was capable of affecting p53 acetylation on Lysine 382 (K382). K382 was examined as readout of p53 activity as p300 was previously shown to acetylate this residue, enhancing the ability of p53 to bind DNA [26,27]. Interestingly, in U2OS cells treated with the AMPK inhibitor Compound C [28], we observed that p53 displayed elevated acetylation levels on K382 compared to control treatment (Figure 1A). Furthermore, in support of a regulatory role for AMPK in p53 acetylation, deletion of AMPK-α in MEFs also led to increased acetylated p53, while Compound C was unable to further elevate K382 acetylation (Figure 1B). Next, we wanted to examine whether AMPK was functioning via SIRT1 to affect p53 acetylation. Consistent with SIRT1 deacylating p53, depletion of SIRT1 led to elevated levels of acetylated p53 (Figure 1C). More importantly, to insure that the p53 acetylation phenotype was indeed occurring through the AMPK-SIRT1 signaling axis, we treated SIRT1-depleted cells with either Compound C or the AMPK activator AICAR [29]. Notably, while WT-SIRT1 cells were sensitive to both Compound C and AICAR treatments, with Compound C promoting p53 acetylation and AICAR causing a subtle decrease, cells depleted of SIRT1 showed no changes in p53 acetylation status with either treatment (Figure 1D). Together, these findings support the notion that AMPK might regulate p53 acetylation largely via SIRT1 (Figure 1E).
Figure 1.

AMPK regulates p53 acetylation via SIRT1. A. Immunoblot (IB) analysis of whole cell lysates harvested from U2OS cells treated with the AMPK inhibitor Compound C (20 μM). B. Immunoblot (IB) analysis of whole cell lysates harvested from AMPK-α-/- MEFs either treated with the AMPK inhibitor Compound C (20 μM) or vehicle. C. U2OS cells were infected with multiple shRNA constructs targeting Sirt1, or GFP as a control, followed by selection with 1 μg/ml puromycin for three days to eliminate non-infected cells. The cells were then harvested and the whole cell lysate was subjected to immunoblot (IB) analysis as indicated. D. Immunoblot (IB) analysis of whole cell lysates harvested from Sirt1-/- MEFs either treated with the AMPK inhibitor Compound C (20 μM), the AMPK activator AICAR (1 mM), or vehicle. E. Schematic of how AMPK may directly affect p53 acetylation via direct regulation of SIRT1 activity.
AMPK phosphorylates DBC1 in vitro and in vivo
We next wanted to further characterize the molecular mechanism(s) by which AMPK may be regulating p53 acetylation via SIRT1. As SIRT1 binds DBC1 in an inhibitory complex [17,18], and SIRT1 phosphorylation could promote its dissociation from DBC1 [20], we were curious to explore whether DBC1 could similarly be phosphorylated by AMPK. Interestingly, there are two putative AMPK sites, S808 and T897, present within DBC1 (Figure 2A and 2B). Consistent with these sites being possible AMPK phosphorylation acceptors, only a C-terminal truncation mutant, containing S808 and T897, was phosphorylated by recombinant AMPK in vitro (Figure 2A and 2C). Furthermore, mass spectrometry analysis identified only S808 phosphorylation on DBC1 treated with recombinant AMPK in vitro (Figure 2D). This phosphorylation was further confirmed in vivo with mass spectrometry analysis on DBC1 isolated from cells treated with the AMPK activator AICAR (Figure 2E). In order to test the importance of this site to AMPK-mediated phosphorylation of DBC1, we mutated S808 to an alanine and performed an in vitro kinase assay. As seen in Figure 2F, mutation of S808, but not T897, was capable of abolishing DBC1 phosphorylation by AMPK. In summary, DBC1 can be directly phosphorylated by AMPK on S808 both in vivo and in vitro.
Figure 2.
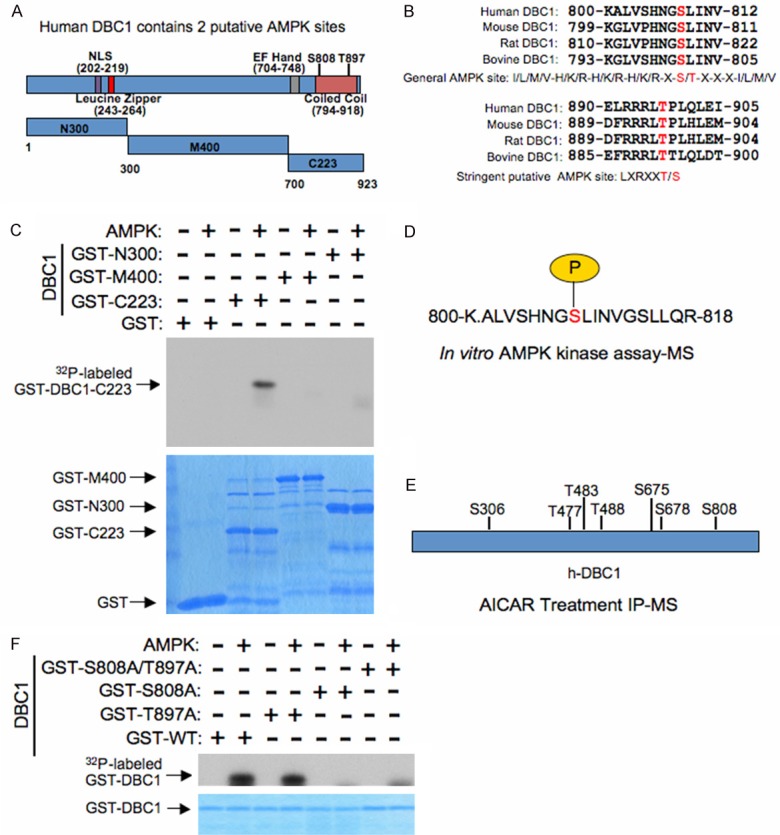
AMPK directly phosphorylates the Sirt1 inhibitory molecule, DBC1. A. Schematic illustrating the location of possible putative AMPK phosphorylation sites on human DBC1 (h-DBC1) as well as truncation mutants used in Figure 2C. B. Alignment of candidate AMPK sequences, both general and stringent, in human DBC1 across several species. C. Purified AMPK was incubated with the indicated GST-DBC1 truncation mutants in the presence of γ-32P-ATP. The kinase reaction products were resolved by SDS-PAGE and phosphorylation was detected by autoradiography. D. Schematic representation of S808 phosphorylation identified in human DBC1 via an in vitro AMPK kinase assay followed by mass spectrometry analysis. E. Schematic representation of in vivo phosphorylation sites identified in human DBC1 via immunoprecipitation of DBC1 from 293T cells treated with the AMPK activator AICAR (1 mM) followed by mass spectrometry analysis. F. Purified AMPK was incubated with the indicated GST-DBC1 phosphorylation mutants, or WT-DBC1, in the presence of γ-32P-ATP. The kinase reaction products were resolved by SDS-PAGE and phosphorylation was detected by autoradiography.
AMPK phosphorylates SIRT1 in vitro
While a previous study had shown that AMPK activation could promote the phosphorylation of SIRT1, leading to its dissociation from DBC1, the kinase directly responsible for this phosphorylation-dependent mechanism remains elusive [20]. Furthermore, while this group was able to ascertain sites involved in this regulation (S47, S605, and S615), they were unable to provide evidence showing that these sites were indeed phosphorylated upon AMPK activation [20]. As we were able to use multiple methods to identify DBC1 as a substrate of AMPK (Figure 2), we continued to examine if direct phosphorylation of SIRT1 by AMPK could also be playing a role. Interestingly, human SIRT1 contains a putative AMPK site at T344 (Figure 3A and 3B) that was previously identified [30]. Furthermore, consistent with SIRT1 being a direct substrate of AMPK, we were able to observe phosphorylation of GST-SIRT1 upon treatment with AMPK using an in vitro kinase assay (Figure 3C). Moreover, mass spectrometry analysis confirmed that T344 was phosphorylated upon recombinant AMPK treatment in vitro (Figure 3D). To further identify the importance of T344 in AMPK-mediated phosphorylation of SIRT1, we next mutated T344 to an alanine and performed an in vitro kinase assay. As illustrated in Figure 3E, GST-SIRT1-344A was no longer phosphorylated by AMPK, further supporting T344 is the major AMPK site present in SIRT1. In summary, these findings further support that AMPK can directly phosphorylate SIRT1 on T344.
Figure 3.

AMPK directly phosphorylates Sirt1. A. Schematic illustrating the location of a possible putative AMPK phosphorylation site on human SIRT1 (h-SIRT1). B. Alignment of the candidate AMPK sequence in human SIRT1 across several species. C. Purified AMPK was incubated with GST-SIRT1 or GST-DBC1 in the presence of γ-32P-ATP. The kinase reaction products were resolved by SDS-PAGE and phosphorylation was detected by autoradiography. D. Schematic representation of T344 phosphorylation identified in human SIRT1 via an in vitro AMPK kinase assay followed by mass spectrometry analysis. E. Purified AMPK was incubated with the indicated GST-SIRT1 phosphorylation mutant (GST-SIRT1-344A), or WT-SIRT1, in the presence of γ-32P-ATP. The kinase reaction products were resolved by SDS-PAGE and phosphorylation was detected by autoradiography.
AMPK phosphorylation of SIRT1, but not DBC1, is crucial for AMPK-mediated dissociation of the SIRT1-DBC1 inhibitory complex
After identifying an AMPK phosphorylation site on DBC1, as well reconfirming T344 as an AMPK site on SIRT1, we next attempted to determine the physiological significance of these modifications. Consistent with SIRT1 forming an inhibitory complex with DBC1 [17,18], we observed that DBC1 bound only SIRT1 but not other members of the Sirtuin family (Figure 4A). As this inhibitory complex is mainly regulated through phosphorylation, we first examined whether a phospho-mimetic mutant of SIRT1 (T344E) had differential binding to DBC1. Surprisingly, we observed that SIRT1-T344E could no longer bind DBC1, suggesting that the direct phosphorylation of SIRT1 by AMPK could regulate the dissociation of SIRT1 from DBC1 (Figure 4B). On the other hand, the phospho-mimetic mutant of DBC1 (S808E and S897E) showed no depreciation of binding to SIRT1 (Figure 4C). These results suggest that AMPK-mediated phosphorylation of SIRT1, but not DBC1, might signal the “OFF” switch to dissociate DBC1, thereby activating SIRT1.
Figure 4.
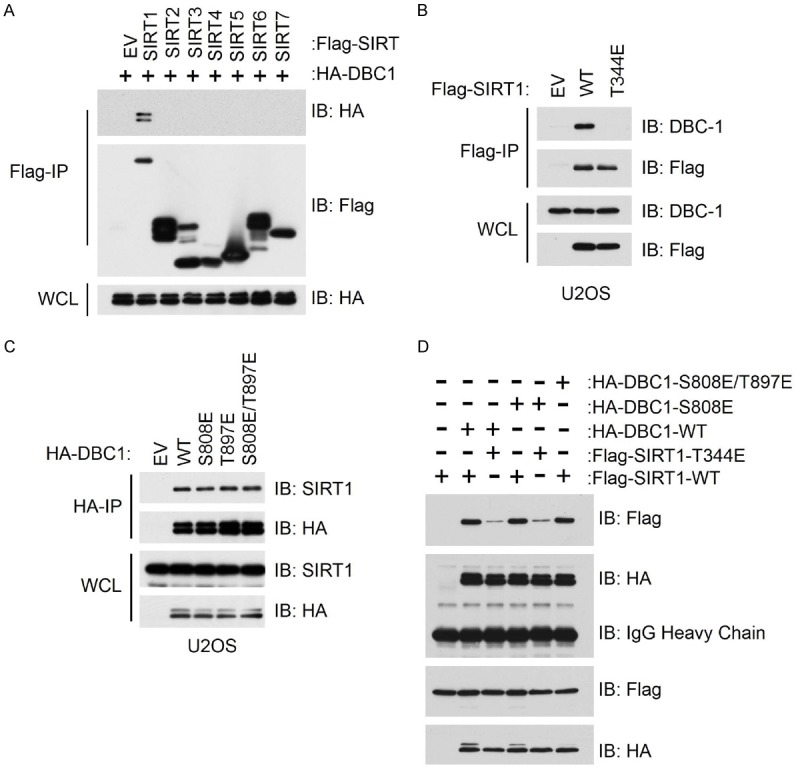
Phosphorylation of SIRT1 by AMPK disrupts the interaction between SIRT1 and its inhibitor molecule DBC1. A. Immunoblot (IB) analysis of whole cell lysate (WCL) and immunoprecipitates (IP) derived from 293T cells transfected with HA-DBC1 and the indicated Flag-SIRT constructs. B. Immunoblot (IB) analysis of whole cell lysate (WCL) and immunoprecipitates (IP) derived from U2OS cells transfected with a Flag-tagged phospho-mimetic SIRT1 mutant (Flag-Sirt1-344E) or wild-type SIRT1 (Flag-Sirt1-WT). C. Immunoblot (IB) analysis of whole cell lysate (WCL) and immunoprecipitates (IP) derived from U2OS cells transfected with the indicated HA-tagged phospho-mimetic DBC1 mutants or wild-type DBC1. D. Immunoblot (IB) analysis of whole cell lysate (WCL) and immunoprecipitates (IP) derived from U2OS cells co-transfected with the indicated HA-tagged phospho-mimetic DBC1 mutants and Flag-tagged phopho-mimetic SIRT1 mutant.
Notably, previous work has also shown that phosphorylation of DBC1 by ATM/ATR could promote the formation of the SIRT1-DBC1 inhibitory complex, which serves to further inactivate SIRT1 [31]. To determine the potential significance of AMPK-mediated phosphorylation of both SIRT1 and DBC1 in regulating SIRT1-DBC1 binding, we performed binding assays utilizing the various generated phospho-mimetic mutants of SIRT1 and DBC1. Interestingly, regardless of which DBC1 construct used (WT or S808E), SIRT1 (T344E) always exhibited depreciated binding (Figure 4D). These results would suggest that AMPK-mediated phosphorylation of SIRT1 is the major regulatory modification which mediates binding of SIRT1 to its inhibitory protein DBC1.
Discussion
In the present study, we provide further biochemical evidence showing that both SIRT1 and DBC1 are direct substrates for AMPK phosphorylation. Furthermore, we also offer evidence that AMPK may regulate p53 acetylation via this direct SIRT1 phosphorylation mechanism involving DBC1 dissociation. In support of these claims, we observed that pharmacological inhibition of AMPK activity by Compound C led to a marked increase in acetylated p53 (Figure 1A). Furthermore, this regulation by AMPK was largely dependent on SIRT1, as depletion of this deacetylase could adversely affect the ability of AMPK to mediate p53 acetylation (Figure 1C and 1D). More importantly, our results also gave further insight into the molecular mechanism by which AMPK could possibly trigger changes in SIRT1 activity. In particular, we identified T344 as the residue directly phosphorylated by AMPK (Figure 3). In support of T344 mediating the ability of AMPK to regulate SIRT1, we showed that a phospho-mimetic version of SIRT1 (T344E) was no longer capable of binding DBC1 (Figure 4B and 4D), a protein well documented to inhibit SIRT1 activity [17].
It is important to further discuss that while it has already been shown that AMPK can regulate SIRT1 activity by promoting its phosphorylation and dissociation from DBC1, this work did not completely verify the molecular mechanism [20]. It was identified by Nin et al. that AMPK activation was required for PKA-mediated SIRT1 activation. This group went on to surmise that phosphorylation of SIRT1 was also critical, and further identified that three residues on SIRT1 seemed to play an important role in this regulatory mechanism (S47, S605, and S615) [20]. However, only phosphorylation of S47 has been previously described [32,33] and it remains unknown whether S605 or S615 are actually modified. Therefore, the work presented here brings new light to the molecular mechanism by which AMPK mediates SIRT1 phosphorylation via DBC1 dissociation. Briefly, our work supports a hypothesis that AMPK can directly phosphorylate SIRT1 at T344. Phosphorylation of this residue may stimulate SIRT1 dissociation from its inhibitory subunit DBC1, subsequently promoting the deacetylation of SIRT1 downstream substrates such as p53 (Figure 5). While our mass spectrometry analysis did not identify S47, S605 and S615 as AMPK sites, it is still possible that these residues may play crucial roles in regulating SIRT1 activity. Consistent with this notion, S47 had been shown to be a target of JNK1 [33]. More importantly, JNK1 activation led to SIRT1 phosphorylation and an increase in SIRT1 activity [34]. In addition to JNK1, multiple other kinases have also been identified to target SIRT1 including CK2 [35], DYRK1A [36] and DYRK3 [36]. Therefore, multiple kinases may feed into SIRT1 regulation and we are only beginning to understand the intricate regulatory mechanisms behind this deacetylase. It is also important to note that it was recently reported that AMPK directly phosphorylates SIRT1 on T344 leading to changes in p53 acetylation status [30]. However, contrary to our findings, this group showed that AMPK phosphorylation of SIRT1 actually led to its inactivation in liver cancer cells. Furthermore, this group did not determine if DBC1 was involved. We, on the other hand, showed that AMPK inactivation increased p53 acetylation in bone tumor cells (Figure 1A) and the mechanism most likely involves DBC1 dissociation (Figure 5). While it would require further in-depth studies to truly identify the differences between our work and the work from Lee et al [30], one possible explanation may stem from the different cancers examined. It is possible that AMPK may have differing roles in regulating SIRT1 activity in liver cancer cells versus bone tumor cells. It is also possible that SIRT1 phosphorylation on T344 may lead to the dissociation of DBC1 in both cancer types, but may subsequently lead to the binding of SIRT1 to a yet unidentified inhibitor in liver cancer cells that is not present in bone tumors.
Figure 5.
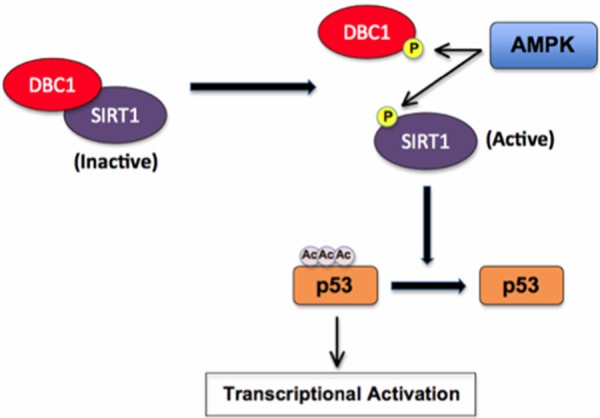
Proposed model for how AMPK directly affects the acetylation status of p53 by phosphorylating SIRT1, leading to its dissociation from the inhibitory protein DBC1 and subsequent activation.
Interestingly, in addition to identifying T344 as a key regulatory phosphorylation site on SIRT1, we also uncovered a novel AMPK site on DBC1 as well (Figure 2). While we did not see any decrease in binding between a phospho-mimetic mutant of DBC1 (S808E) and SIRT1 (Figure 4C and 4D), this may simply have been due to our experimental conditions. It is also possible that phosphorylation of DBC1 by AMPK may stabilize the DBC1/SIRT1 complex. As such, we would not expect to see any change in binding between DBC1 (S808E) and wild type SIRT1. As a matter of fact, it was recently shown that ATM/ATR phosphorylation of DBC1 after DNA damage led to increased binding of DBC1 to SIRT1 [31]. Such a possibility would suggest that the integrity of the DBC1/SIRT1 complex is regulated by multiple upstream phosphorylation signals such that SIRT1 activity may be dependent on the net, rather than single, phosphorylation status of the complex. With greater DBC1 phosphorylation, SIRT1 would stay bound to DBC1, effectively inhibiting its deacetylase activity. On the other hand, with greater SIRT1 phosphorylation, SIRT1 would dissociate with DBC1 and thus be more active. Nevertheless, this is mere speculation and additional experiments are still required to fully elucidate the functional significance of AMPK-mediated phosphorylation of DBC1.
In summary, our study expands on the recent observations that AMPK can regulate SIRT1 deacetylase activity [20]. It further suggests that targeting AMPK may be a novel means to regulate p53 activity in tumorigenic situations.
Acknowledgements
This work was supported in part by a grant from the National Natural Science Foundation of China (Grant No.81372602) to D.G. This work was supported by grants from National Institute on Aging, NIH (H.I., AG041218). We thank Shavali Shaik, Lixin Wan and other members for critical reading and discussion of this manuscript.
Disclosure of conflicting of interest
The authors have no conflicting interests.
References
- 1.Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 2.Cha YI, Kim HS. Emerging role of sirtuins on tumorigenesis: possible link between aging and cancer. BMB Rep. 2013;46:429–438. doi: 10.5483/BMBRep.2013.46.9.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 4.Brooks CL, Gu W. How does SIRT1 affect metabolism, senescence and cancer? Nat Rev Cancer. 2009;9:123–128. doi: 10.1038/nrc2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fulco M, Schiltz RL, Iezzi S, King MT, Zhao P, Kashiwaya Y, Hoffman E, Veech RL, Sartorelli V. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell. 2003;12:51–62. doi: 10.1016/s1097-2765(03)00226-0. [DOI] [PubMed] [Google Scholar]
- 7.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 8.Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell. 2004;16:93–105. doi: 10.1016/j.molcel.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 9.Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 10.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 11.Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003;100:10794–10799. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang C, Chen L, Hou X, Li Z, Kabra N, Ma Y, Nemoto S, Finkel T, Gu W, Cress WD, Chen J. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat Cell Biol. 2006;8:1025–1031. doi: 10.1038/ncb1468. [DOI] [PubMed] [Google Scholar]
- 13.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A. 2008;105:13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abdelmohsen K, Pullmann R Jr, Lal A, Kim HH, Galban S, Yang X, Blethrow JD, Walker M, Shubert J, Gillespie DA, Furneaux H, Gorospe M. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol Cell. 2007;25:543–557. doi: 10.1016/j.molcel.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim EJ, Kho JH, Kang MR, Um SJ. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol Cell. 2007;28:277–290. doi: 10.1016/j.molcel.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 16.Hasegawa K, Yoshikawa K. Necdin regulates p53 acetylation via Sirtuin1 to modulate DNA damage response in cortical neurons. J Neurosci. 2008;28:8772–8784. doi: 10.1523/JNEUROSCI.3052-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583–586. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
- 18.Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008;451:587–590. doi: 10.1038/nature06515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Escande C, Chini CC, Nin V, Dykhouse KM, Novak CM, Levine J, van Deursen J, Gores GJ, Chen J, Lou Z, Chini EN. Deleted in breast cancer-1 regulates SIRT1 activity and contributes to high-fat diet-induced liver steatosis in mice. J Clin Invest. 2010;120:545–558. doi: 10.1172/JCI39319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nin V, Escande C, Chini CC, Giri S, Camacho-Pereira J, Matalonga J, Lou Z, Chini EN. Role of deleted in breast cancer 1 (DBC1) protein in SIRT1 deacetylase activation induced by protein kinase A and AMP-activated protein kinase. J Biol Chem. 2012;287:23489–23501. doi: 10.1074/jbc.M112.365874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao D, Inuzuka H, Tseng A, Chin RY, Toker A, Wei W. Phosphorylation by Akt1 promotes cytoplasmic localization of Skp2 and impairs APCCdh1-mediated Skp2 destruction. Nat Cell Biol. 2009;11:397–408. doi: 10.1038/ncb1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG Jr. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature. 2004;428:194–198. doi: 10.1038/nature02381. [DOI] [PubMed] [Google Scholar]
- 23.Boehm JS, Hession MT, Bulmer SE, Hahn WC. Transformation of human and murine fibroblasts without viral oncoproteins. Mol Cell Biol. 2005;25:6464–6474. doi: 10.1128/MCB.25.15.6464-6474.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inuzuka H, Tseng A, Gao D, Zhai B, Zhang Q, Shaik S, Wan L, Ang XL, Mock C, Yin H, Stommel JM, Gygi S, Lahav G, Asara J, Xiao ZX, Kaelin WG Jr, Harper JW, Wei W. Phosphorylation by casein kinase I promotes the turnover of the Mdm2 oncoprotein via the SCF(beta-TRCP) ubiquitin ligase. Cancer Cell. 2010;18:147–159. doi: 10.1016/j.ccr.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inuzuka H, Gao D, Finley LW, Yang W, Wan L, Fukushima H, Chin YR, Zhai B, Shaik S, Lau AW, Wang Z, Gygi SP, Nakayama K, Teruya-Feldstein J, Toker A, Haigis MC, Pandolfi PP, Wei W. Acetylation dependent regulation of Skp2 function. Cell. 2012;150:179–193. doi: 10.1016/j.cell.2012.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 27.Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW, Appella E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998;12:2831–2841. doi: 10.1101/gad.12.18.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang WL, Perillo W, Liou D, Marambaud P, Wang P. AMPK inhibitor compound C suppresses cell proliferation by induction of apoptosis and autophagy in human colorectal cancer cells. J Surg Oncol. 2012;106:680–688. doi: 10.1002/jso.23184. [DOI] [PubMed] [Google Scholar]
- 29.Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H, Kang H, Shaw RJ, Evans RM. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134:405–415. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee CW, Wong LL, Tse EY, Liu HF, Leong VY, Lee JM, Hardie DG, Ng IO, Ching YP. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res. 2012;72:4394–4404. doi: 10.1158/0008-5472.CAN-12-0429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zannini L, Buscemi G, Kim JE, Fontanella E, Delia D. DBC1 phosphorylation by ATM/ATR inhibits SIRT1 deacetylase in response to DNA damage. J Mol Cell Biol. 2012;4:294–303. doi: 10.1093/jmcb/mjs035. [DOI] [PubMed] [Google Scholar]
- 32.Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279:50754–50763. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- 33.Nasrin N, Kaushik VK, Fortier E, Wall D, Pearson KJ, de Cabo R, Bordone L. JNK1 phosphorylates SIRT1 and promotes its enzymatic activity. PLoS One. 2009;4:e8414. doi: 10.1371/journal.pone.0008414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao Z, Zhang J, Kheterpal I, Kennedy N, Davis RJ, Ye J. Sirtuin 1 (SIRT1) protein degradation in response to persistent c-Jun N-terminal kinase 1 (JNK1) activation contributes to hepatic steatosis in obesity. J Biol Chem. 2011;286:22227–22234. doi: 10.1074/jbc.M111.228874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zschoernig B, Mahlknecht U. Carboxy-terminal phosphorylation of SIRT1 by protein kinase CK2. Biochem Biophys Res Commun. 2009;381:372–377. doi: 10.1016/j.bbrc.2009.02.085. [DOI] [PubMed] [Google Scholar]
- 36.Guo X, Williams JG, Schug TT, Li X. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J Biol Chem. 2010;285:13223–13232. doi: 10.1074/jbc.M110.102574. [DOI] [PMC free article] [PubMed] [Google Scholar]