

Abstract
Cell adhesion proteins that connect each cell to neighboring cells and the extracellular matrix are an important determinant of cell morphology and metastasis. Migfilin is a member of LIM-containing protein family, mediated the linkage between cytoskeleton and focal adhesion through interacting with other proteins and involved in cell adhesion and motility. The present study used immunohistochemistry and Western blot analysis of migfilin in clinical specimens of esophageal squamous cell carcinoma (ESCC) to identify that the expression of migfilin was significantly upregulated in ESCC in concomitance with a nuclear-cytoplasm translocation compared to normal adjacent tissue. Alternately, using the published datasets we identified that the expression of β-catenin was upregulated in esophageal cancer cells with focal invasion, while inversely correlated with migfilin in esophageal cancer cell lines. We demonstrated that the reduced expression of β-catenin by migfilin was through the inhibition of Akt activation. In conclusion, these results illustrated that migfilin upregulated in ESCCs and repressed β-catenin in an Akt-GSK3β signaling dependent manner.
Keywords: Migfilin, β-catenin, Akt, GSK3β, cell adhesion, esophageal cancer
Introduction
Esophageal cancer is one of the most aggressive cancers occurring with the eighth of incidence and sixth of cancer-related mortality worldwide [1]. The focal invasion and distant metastasis are the cardinal cause of death in esophageal cancer and more than 50 percent of patients have either unresectable tumors or radiographically visible metastases at the time of the diagnosis [2]. In China and other Asian countries, esophageal squamous cell carcinoma (ESCC) is the most prevalent distribution [1]. Cumulative evidences illustrated that a variety of genetic and molecular abnormalities including gene dysregulation, biochemical modifications and aberrant signaling pathways underlying the development and progression of ESCCs [3]. Among them, particular attention has been attracted to LIM domain containing protein as the expression of them altered and involved in pathogenesis or represented properties specific to ESCC [4-8].
LIM domains are cysteine-rich sequences of approximately 50 residues that fold into a zincfinger structure serving as modular protein-binding interfaces [9]. Zyxin family is one group of the LIM domain proteins which are indentified in the cytoplasm, and can translocate into the nucleus and facilitate transcription of target genes, involved in tissue-specific gene regulation, cell fate determination and cytoskeleton organization [10]. Migfilin is an adaptor protein of the zyxin family, regulates cell physiology and drives carcinogenesis. Previous study had defined that migfilin mediated a connection between focal adhesion and the actin cytoskeleton through interacting with mig-2 and filamin, participated in the orchestration of actin assembly and cell shape modulation [11]. Migfilin competed with integrin-β tails for common binding sites on filamin provided a mechanism for switching between different integrin-cytoskeleton linkages [12-16]. Besides, migfilin regulated cell migration and anoikis resistance through interacting with VASP and Src [17,18]. Alternatively, migfilin could also shuttle form cytoplasm to nuclear to induce cardiomyocyte differentiation through associating with cardiac transcriptional factor CSX/NKX2-5 [19]. These findings provided additional support for the role of migfilin in tumorigenesis and implicated that the function of migfilin was in a genetic and tissues context specific dependent manner.
Our previous study had exhibited that migfilin inhibited esophageal cancer cell motility through elevating GSK3β-mediated degradation of β-catenin [20]. Although the important role of migfilin and β-catenin in cell adhesion, cytoskeleton organization and migration via cultured cells has been delineated, the expression and functional roles of migfilin and β-catenin in esophageal tissue, either in normal or diseased conditions and the relationship between migfilin, β-catenin levels and the clinicopathologic parameters or survival in ESCCs needs to be further elucidated. In the present study, we evaluated the expression of migfilin in clinical ESCC samples and their matched adjacent normal esophageal epithelia and analyzed the relationships between migfilin, β-catenin expression and the clinicopathologic parameters of ESCCs. Additionally, we verified that the expression of β-catenin was inversely correlated with migfilin in esophageal cancer cells and provided a new perspective about the underlying mechanism of migfilin-mediated repression of β-catenin.
Materials and methods
Tissue specimens
Tissue microarray comprised of 105 patients with ESCCs was obtained at the First Affiliated Hospital of Anhui Medical University (Anhui province, China). Patients received no treatment before surgery and signed informed consents in accordance with Institutional Review Board of the Chinese Academy of Medical Sciences Cancer Institute and Anhui Medical University standards and guidelines. Representative primary tumor regions and the corresponding histologically normal esophageal mucosa from each patient were snap-frozen in liquid nitrogen and stored at -80°C [21]. Additional sections were collected and embedded in paraffin for histological examination [20,22].
Cell culture
Human ESCC cell lines (KYSE series) were generous gifts from Dr. Shimada Y of Kyoto University (Kyoto, Japan; [23]). Cells were maintained in RPMI-1640 supplemented with 10% FBS, 100 U/mL streptomycin, and 100 U/mL penicillin.
Antibodies
Monoclonal anti-migfilin Ab (clone 43) was kindly provided by Prof. Wu C as described previously [11]. Antibodies against β-catenin, p-Ser473-Akt (Santa Cruz), p-β-catenin, p-Ser9-GSK3β, GSK3β, Akt (Cell Signaling), FLAG, β-actin (Sigma) and cyclin D1 (Protein innovation) were also used.
Immunohistochemistry
The human ESCC tissue microarray was subjected to immunohistochemistry using monoclonal antibody to migfilin. Briefly, deparaffinized tissue sections were treated by 3% hydrogen peroxide and processed for antigen retrieval (by heating in a microwave oven at 96°C, in 0.01 M citrate buffer, at pH 6, for 15 min), after cooled at room temperature, they were blocked by normal goat serum for 30 min and incubated with anti-migfilin antibody at 4°C overnight. The reaction products were colorized with PV9003 immunohistochemistry (IHC) kit (Zhong Shan Goldenbridge) and DAB substrate-chromogen kit (Zhong Shan Goldenbridge), resulting in a brown signal. The evaluation of immunohistochemical staining was described as previously [24]. Briefly, specimens were reviewed with staining intensity and staining extent. Staining intensity was rated as follows: 0, negative; 1, faint; 2, moderate; 3, strong. The expression extent were graded on a scale from 0 to 3 as follows: 0, no positive staining or only a few scattered positive cells; 1, cluster(s) of positively stained cells, but accounting for < 25% of the cells within a visual field; 2, cluster(s) of positively stained cells that accounted for 25% to 50% of the cells within a visual field; 3, cluster(s) of positively stained cells that accounted for > 50% of the cells within a visual field. The results of intensity and extent gave an overall score. The samples of staining where the score was less than 3 were marked as low expression; those scored more than 3 (including 3) were marked as high expression.
Western blot analysis
Proteins were separated by SDS-PAGE, transferred to PVDF membranes (Amersham), blocked with milk, and then probed with antibodies as indicated. Protein bands were detected by chemiluminescence using the ECL system (Vigorous) according to the manufacture’s protocol.
SiRNA transfection and generation of stable cell lines
SiRNA transfection and establishment of stable cell lines were performed as previously described [20].
Statistical analysis
Association of migfilin expression level with clinicopathologic features of ESCCs and the statistical difference of migfilin expression in ESCC and its corresponding normal epithelium were validated by χ2 test and 2-tailed paired Student t test with the SPSS17.0 statistical program for windows. A value of P < 0.05 was considered to be statistically significant.
Results
Increased expression and nuclear-to-cytoplasmic translocation of migfilin in human esophageal squamous cell carcinoma
Our previously results showed that the expression of migfilin in ESCCs is potentially relevant for the clinical outcome in patients and played a role in suppressing esophageal cancer cells malignant phenotype and nodal metastasis [20]. To determine the significance of migfilin in esophageal squamous cell carcinoma progression, we conducted immunohistochemistry analysis for migfilin in 95 pairs of human ESCC tissue specimens and their normal counterparts from the 105 ESCCs tissue array which lost 10 adjacent normal tissue specimens. As shown in Figure 1A, the staining of migfilin protein was predominantly localized in the nucleus of normal esophageal epithelial cells, while in neoplastic cells, the immunoreactivity shifted to the cytoplasm of the tumor nest. A statistically significant overexpression of migfilin was observed in ESCC samples (Table 1). Western blot analysis on eight pairs of ESCC and their matched normal esophageal adjacent tissues validated the increased expression of migfilin in ESCC (Figure 1B). To further confirm this result, we analyzed a public data set in ESCC [25] and found migfilin mRNA was significantly overexpressed in ESCCs as compared to matched adjacent normal esophageal mucosa (Figure 2). These results showed that the expression of migfilin was upregulated in concomitance with a dynamic intracellular shuttling in ESCCs.
Figure 1.
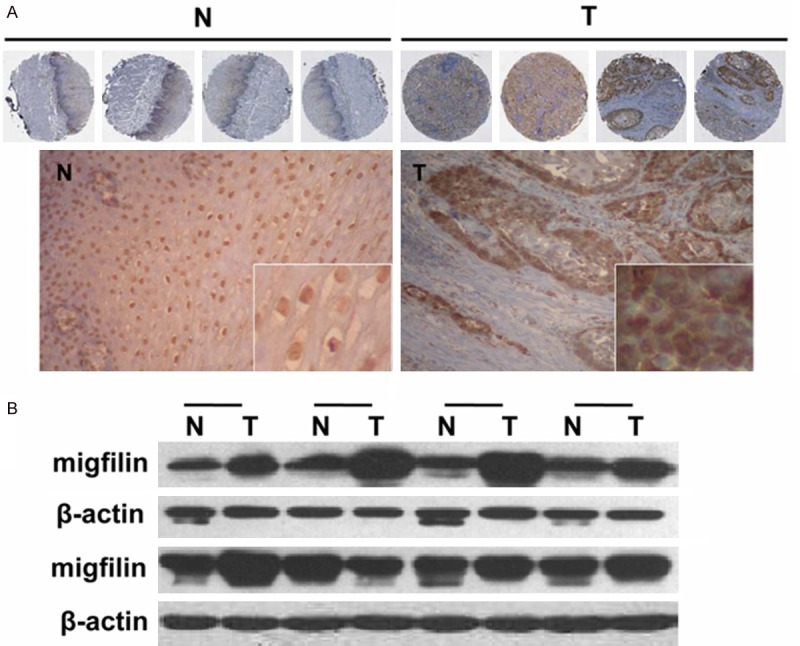
The protein expression of migfilin in esophageal cancer samples. A. Representative immunohistochemical staining showing the expression and localization of migfilin in four cases of paired esophageal normal and cancer tissues. B. Western blot analysis showing the expression of migfilin in paired esophageal normal and cancer tissues. β-actin served as an internal control. T: ESCC tissue, N: corresponding normal adjacent mucosa.
Table 1.
The different expression of migfilin between ESCCs and normal adjacent tissues
| Characteristics | Low (%) | High (%) | Total | P |
|---|---|---|---|---|
| Normal | 94 (98.9) | 1 (1.1) | 95 | < 0.001*** |
| Tumor | 52 (54.7) | 43 (45.3) | 95 |
NOTE: The result was analyzed by the Pearson χ2 test. P values with significance are shown as superscripts.
p < 0.001.
Figure 2.
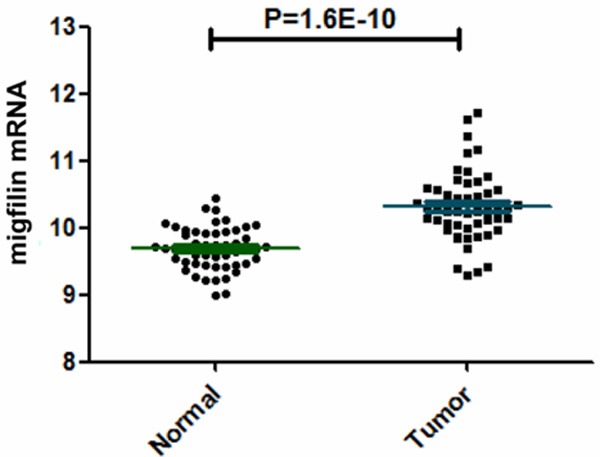
The mRNA levels of migfilin in esophageal cancer samples. Scatter plot displaying migfilin mRNA levels between ESCCs and matched adjacent normal esophageal mucosa in published microarray data sets (GSE23400).
The association between migfilin expression and ESCC clinicopathologic parameters
To further validate the alteration of migfilin expression in ESCCs and analyze the relationship between migfilin and clinicopathologic features, we assessed the correlation between migfilin protein levels and clinicopathologic parameters in the tissue microarray comprised of 105 esophageal cancer by IHC analysis. The immunostaining results for migfilin in ESCCs showed that the protein expression of migfilin was not correlated with ESCC depth of tumor invasion, differentiation or clinical stage (Table 2).
Table 2.
The correlation between migfilin expression and clinicopathologic features in ESCCs
| Characteristics | Low (%) | High (%) | Total | P |
|---|---|---|---|---|
| Overall | 17 | 88 | 105 | |
| TNM classification | ||||
| pT | ||||
| pT1 | 1 (33.3) | 2 (66.7) | 3 | 0.684 |
| pT2 | 5 (13.5) | 32 (86.5) | 37 | |
| pT3 | 11 (16.9) | 54 (83.1) | 65 | |
| Clinical stage | ||||
| I | 1 (50) | 1 (50) | 2 | 0.723 |
| II | 11 (15.9) | 58 (84.1) | 69 | |
| III | 1 (12.5) | 7 (87.5) | 8 | |
| IV | 4 (15.4) | 22 (84.6) | 26 | |
| Differentiation | ||||
| Well | 1 (4.8) | 20 (95.2) | 21 | 0.201 |
| Moderately | 12 (18.5) | 53 (81.5) | 65 | |
| Poorly | 4 (21.1) | 15 (78.9) | 19 |
Inverse correlation between β-catenin and migfilin mRNA expression in esophageal cancer cells
Since our previous results demonstrated that the inhibition of cell motility by migfilin was conferred partially by β-catenin-mediated signaling pathway [20], invigorated us to further investigate the difference of β-catenin level in esophageal cancer cells with clear invasion status and statistic result showed that in the published data sets [26], β-catenin mRNA was significantly overexpressed in invading cells grown in organotypic culture, as compared to noninvading cells (Figure 3A). To confirm the repressive effect of migfilin on β-catenin expression in cell lines, we examined their mRNA levels in a subset of esophageal cancer cell lines [27]. Pairwise correlation analysis indicated that a statistically significant inverse correlation between migfilin and β-catenin (Figure 3B). The negative correlation between migfilin and β-catenin suggested that migfilin participated in regulating the transcription of β-catenin.
Figure 3.
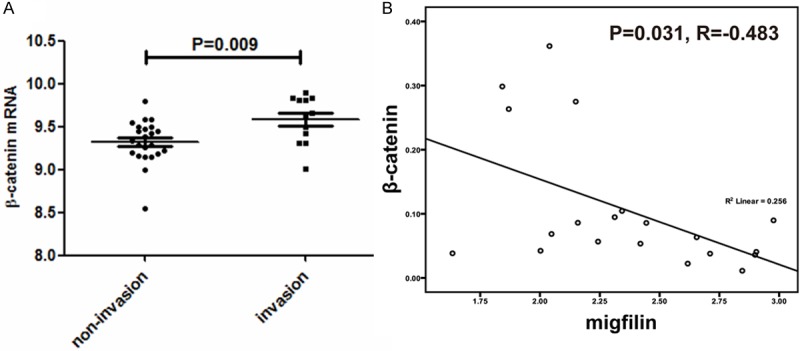
The expression of β-catenin was upregualted in invading esophageal cancer cells and inversely correlated with migfilin in esophageal cancer cell lines. A. Scatter plot displaying β-catenin mRNA levels in 35 esophageal carcinoma cell samples (GSE21293) with (12 cases) or without (23 cases) invasion. B. Scatter plot displaying migfilin and β-catenin mRNA levels in 20 esophageal cancer cell lines (GSE9982) with pairwise correlation analysis.
Migfilin-induced reduction of β-catenin is mediated through PI3K/Akt/GSK3β signaling pathway
Since we have recently reported that GSK3β is required for migfilin-mediated β-catenin degradation [20], and it has been demonstrated that phosphorylation of GSK-3β at Ser9 by Akt/PKB results in its inactivation and subsequently accumulation of cytoplasmic β-catenin [28], we hypothesized that the PI3K/Akt signaling pathway might be involved in the destabilization of β-catenin induced by migfilin. Results in Figure 4A showed that overexpression of migfilin was accompanied with a decreased phosphorylation of Akt at Ser473 (the predominant site for Akt activation) and its downstream target GSK3β in migfilin stable transfectants, although the total protein levels of each protein remained unchanged compared with the vector control. Conversely, transfection of KYSE150 cells with migfilin siRNA substantially increased the activity of Akt and phosphorylation of GSK3β as well as the expression of cyclinD1, a classic downstream target of β-catenin, compared with the control cells transfected with an irrelevant siRNA, but treatment with the LY294002, a reversible PI3K inhibitor, attenuated knockdown of migfilin induced phosphorylation of Akt and GSK3β (Figure 4B). Taken together, these observations suggested that migfilin functions as an upstream regulator of β-catenin through modulation of the PI3K/Akt signaling pathway.
Figure 4.
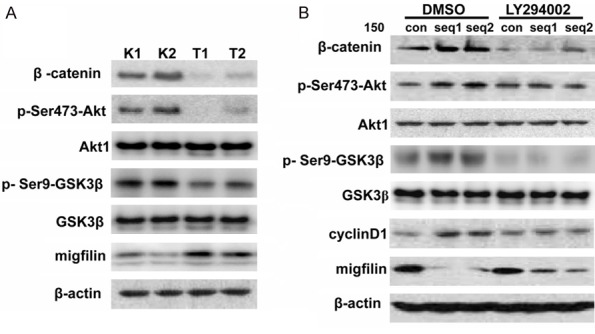
Migfilin regulated β-catenin expression via the PI3K/Akt/GSK3β pathway. A. Lysates of KYSE450 stable transfectants (T1 and T2) were prepared and immunoblots were performed with antibodies as indicated. K1 and K2 denote stable transfectants with vector controls. B. KYSE150 cells were transiently transfected with migfilin siRNAs (seq1 and seq2) or non-targeting siRNA (con). LY294002 (50 μM) or dimethyl sulfoxide (DMSO) was added to the cell culture medium 1 hour prior to harvesting the cells. Cells were lysed and immunoblotting analysis was performed with antibodies as indicated.
Discussion
Cell-extra cellular matrix and cell-cell adhesion maintain tissue integrity and orchestrate cell morphology and involve in accumulate fundamental cellular processes. Consequently, aberration of the expression or subcellular distribution of cell adhesion molecules is frequently amenable to dissociation of cells from the original tumor and invasion of surrounding tissues. Migfilin, the adhesion protein connects the focal adhesion with the actin cytoskeleton, are associated with a number of proteins that can serve as a docking sites for protein-protein interactions [29,30]. Distinctively, a recent publication by Moik and colleagues considered that loss of migfilin expression has no overt consequences on murine development and homeostasis [31]. However, another study with migfilin-knockout mice model identified migfilin as an important modulator of bone homeostasis through regulating both the intrinsic properties of bone marrow stromal cells and the communication between osteoblast and bone marrow monocytes which controls osteoclastogenesis [32]. One possible explanation for the contradictory evidence acquired by using the same animal model and experiment in vivo is that the tissue and genetic context have a decisive role in switching migfilin between its multiple functions. In carcinogenesis, it had been reported that migfilin was significantly reduced in the majority of the breast cancer tissues compared to normal tissues [33]. However, in human leiomyosarcoma, the cytoplasmic level of migfilin was strongly associated with higher tumor grades [34]. Our immunohistochemical and Western blot analysis using clinical ESCC samples revealed an increased expression of migfilin in ESCC (Figure 1A and 1B). The result was verified by detecting the migfilin levels in ESCCs through using an independent published dataset (Figure 2). Whereas we previously observed that migfilin overexpression was associated with a less aggressive biological behavior and promoted the GSK3β-mediated degradation of β-catenin [20], which reminisced us that migfilin played a negative regulatory role in ESCC metastasis. The contrary results mentioned above are compatible with the expression of migfilin was upregulated in osteoarthritis, but silencing of migfilin rather exacerbated than ameliorated the osteoarthritic phenotype via inducing β-catenin expression [35]. Hence, we extrapolated that migfilin may facilitate tumorgenicity of early-stage cancers, whereas it acted cytostatically in more malignant lesions, which was conceivably related to changes in its genetic context during tumor progressions. Recently, an increasing amount of evidences indicated that migfilin could translocate to the nucleus and facilitate transcription of tissue-specific gene and determination of cell fate [10,29]. In myocardial cells, migfilin could shuttle from the cytoplasm to the nucleus in response to calcium and induce cardiomyocyte differentiation [19]. Migfilin also contained a nuclear export sequence in its proline-rich domain, which mediated its dynamic intracellular shuttling [19,36]. Our present study observed that migfilin was restricted to the nucleus in the adjacent normal epithelium, whereas increased levels of migfilin were observed in both the cytoplasm and the nucleus in ESCC. The mechanism for nuclear translocation of migfilin in ESCC is currently unknown and requires further investigation.
The different functions of migfilin in the specific tissue context not only depend on its different subcellular localization but also effect on different signal pathways. It had been reported that migfilin acted as a key regulator in sensitizing glioma cells to cisplatin-induced apoptosis through modulation of apoptosis-related proteins, such as PARP, caspase-3 and Bcl-xL [37]. Reciprocally, upregulation of migfilin was associated with poor prognosis of glioma and mediated metastasis depended on activating the epithermal growth factor receptor-induced PLC-γ and STAT3-signaling pathways [38]. Our previous study revealed that migfilin promoted GSK3β-mediated phosphorylation and degradation of β-catenin by enhancing the interaction between β-catenin and GSK3β [20]. In the present study, we further substantiated that the expression of β-catenin was dramatically negative correlated with migfilin in esophageal cancer cell lines by using the existing GEO database (Figure 3B). Besides, overexpression of migfilin significantly abrogated the function of β-catenin by decreasing phosphosrylation of Akt and GSK3β (Figure 4A) and inhibition of PI3K-Akt signaling by LY294002 blunted the increase of β-catenin protein expression induced by migfilin knockdown (Figure 4B). GSK-3β had been reported to be inactivated by phosphorylation at Ser9 by serine/ threonine kinases [28], here we showed that the phosphorylation and activity of GSK-3β regulated by migfilin via the PI3K/Akt pathway and the increased GSK-3β activity leading to phoshporylation of β-catenin at sites which are critical for its recognition and degradation by β-Trcp [39,40]. We also found that down-regulation of migfilin promoted the activation of Akt which resulted in the up-regulation of β-catenin and augmentation in TCF activity by inhibiting GSK-3β (Figure 4B). Since the regulation of Akt activity is PI3K dependent, we have shown, with the use of pharmacologic PI3K inhibitor LY294002, that knockdown of migfilin-induced Akt activation is PI3K dependent (Figure 4B).
Currently, the precise mechanism by which PI3K is targeted by migfilin action remains to be determined. Ras, one of the PI3K upstream regulators can directly bind and stimulate p110, which is the catalytic subunit of PI3K [41]. And another recent paper validated that active Ras co-localized with Filamin A (FLNa) coordinately increased cell migration by enhancing integrin activation and fibronectin matrix assembly [42]. Previous studies had revealed that FLNa is a migfilin-binding protein [11]. Therefore we speculate that the competition between migfilin and Ras, both of which can bind FLNa, may dissociate FLNa from Ras, thereby regulate Ras activity, inhibit the activation of PI3K/Akt signaling pathway, and facilitate β-catenin phosphorylation by GSK3β. Clearly, future studies are required to test this hypothesis.
In conclusion, we have shown that the expression of migfilin significantly increased in the majority of the ESCCs compared to adjacent normal tissues regardless of differentiation and disease stage and illuminated that migfilin modulated β-catenin levels by affecting the activity/phosphorylation of GSK-3β through the PI3K/Akt pathway. Identification of the underlying molecular mechanisms will shed light on the understanding of cell adhesion-mediated carcinogenesis and spread to target organs.
Acknowledgements
This work was supported by National Basic Research Program of China Grant 2013CB911004 and National Natural Science Foundation of China Grants 81130043 and 81302329.
Disclosure of conflict of interest
None.
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003;349:2241–2252. doi: 10.1056/NEJMra035010. [DOI] [PubMed] [Google Scholar]
- 3.Denlinger CE, Thompson RK. Molecular basis of esophageal cancer development and progression. Surg Clin North Am. 2012;92:1089–1103. doi: 10.1016/j.suc.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 4.Kashyap MK, Pawar HA, Keerthikumar S, Sharma J, Goel R, Mahmood R, Kumar MV, Kumar KV, Pandey A, Kumar RV, Prasad TS, Harsha HC. Evaluation of protein expression pattern of stanniocalcin 2, insulin-like growth factor-binding protein 7, inhibin beta A and four and a half LIM domains 1 in esophageal squamous cell carcinoma. Cancer Biomark. 2012;12:1–9. doi: 10.3233/CBM-120289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lo PH, Ko JM, Yu ZY, Law S, Wang LD, Li JL, Srivastava G, Tsao SW, Stanbridge EJ, Lung ML. The LIM domain protein, CRIP2, promotes apoptosis in esophageal squamous cell carcinoma. Cancer Lett. 2012;316:39–45. doi: 10.1016/j.canlet.2011.10.020. [DOI] [PubMed] [Google Scholar]
- 6.Takeshita N, Mori M, Kano M, Hoshino I, Akutsu Y, Hanari N, Yoneyama Y, Ikeda N, Isozaki Y, Maruyama T, Akanuma N, Miyazawa Y, Matsubara H. miR-203 inhibits the migration and invasion of esophageal squamous cell carcinoma by regulating LASP1. Int J Oncol. 2012;41:1653–1661. doi: 10.3892/ijo.2012.1614. [DOI] [PubMed] [Google Scholar]
- 7.Tao Y, Chai D, Ma L, Zhang T, Feng Z, Cheng Z, Wu S, Qin Y, Lai M. Identification of distinct gene expression profiles between esophageal squamous cell carcinoma and adjacent normal epithelial tissues. Tohoku J Exp Med. 2012;226:301–311. doi: 10.1620/tjem.226.301. [DOI] [PubMed] [Google Scholar]
- 8.He B, Yin B, Wang B, Chen C, Xia Z, Tang J, Yuan Y, Feng X, Yin N. Overexpression of LASP1 is associated with proliferation, migration and invasion in esophageal squamous cell carcinoma. Oncol Rep. 2013;29:1115–1123. doi: 10.3892/or.2012.2199. [DOI] [PubMed] [Google Scholar]
- 9.Willier S, Butt E, Richter GH, Burdach S, Grunewald TG. Defining the role of TRIP6 in cell physiology and cancer. Biol Cell. 2011;103:573–591. doi: 10.1042/BC20110077. [DOI] [PubMed] [Google Scholar]
- 10.Zheng Q, Zhao Y. The diverse biofunctions of LIM domain proteins: determined by subcellular localization and protein-protein interaction. Biol Cell. 2007;99:489–502. doi: 10.1042/BC20060126. [DOI] [PubMed] [Google Scholar]
- 11.Tu Y, Wu S, Shi X, Chen K, Wu C. Migfilin and Mig-2 link focal adhesions to filamin and the actin cytoskeleton and function in cell shape modulation. Cell. 2003;113:37–47. doi: 10.1016/s0092-8674(03)00163-6. [DOI] [PubMed] [Google Scholar]
- 12.Das M, Ithychanda SS, Qin J, Plow EF. Migfilin and filamin as regulators of integrin activation in endothelial cells and neutrophils. PLoS One. 2011;6:e26355. doi: 10.1371/journal.pone.0026355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ithychanda SS, Das M, Ma YQ, Ding K, Wang X, Gupta S, Wu C, Plow EF, Qin J. Migfilin, a molecular switch in regulation of integrin activation. J Biol Chem. 2009;284:4713–4722. doi: 10.1074/jbc.M807719200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ithychanda SS, Hsu D, Li H, Yan L, Liu DD, Das M, Plow EF, Qin J. Identification and characterization of multiple similar ligand-binding repeats in filamin: implication on filamin-mediated receptor clustering and cross-talk. J Biol Chem. 2009;284:35113–35121. doi: 10.1074/jbc.M109.060954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ithychanda SS, Qin J. Evidence for multisite ligand binding and stretching of filamin by integrin and migfilin. Biochemistry. 2011;50:4229–4231. doi: 10.1021/bi2003229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lad Y, Jiang P, Ruskamo S, Harburger DS, Ylanne J, Campbell ID, Calderwood DA. Structural basis of the migfilin-filamin interaction and competition with integrin beta tails. J Biol Chem. 2008;283:35154–35163. doi: 10.1074/jbc.M802592200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Tu Y, Gkretsi V, Wu C. Migfilin interacts with vasodilator-stimulated phosphoprotein (VASP) and regulates VASP localization to cell-matrix adhesions and migration. J Biol Chem. 2006;281:12397–12407. doi: 10.1074/jbc.M512107200. [DOI] [PubMed] [Google Scholar]
- 18.Zhao J, Zhang Y, Ithychanda SS, Tu Y, Chen K, Qin J, Wu C. Migfilin interacts with Src and contributes to cell-matrix adhesion-mediated survival signaling. J Biol Chem. 2009;284:34308–34320. doi: 10.1074/jbc.M109.045021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akazawa H, Kudoh S, Mochizuki N, Takekoshi N, Takano H, Nagai T, Komuro I. A novel LIM protein Cal promotes cardiac differentiation by association with CSX/NKX2-5. J Cell Biol. 2004;164:395–405. doi: 10.1083/jcb.200309159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He H, Ding F, Li Y, Luo A, Chen H, Wu C, Liu Z. Migfilin regulates esophageal cancer cell motility through promoting GSK-3beta-mediated degradation of beta-catenin. Mol Cancer Res. 2012;10:273–281. doi: 10.1158/1541-7786.MCR-11-0419. [DOI] [PubMed] [Google Scholar]
- 21.Zhang L, Ding F, Cao W, Liu Z, Liu W, Yu Z, Wu Y, Li W, Li Y. Stomatin-like protein 2 is overexpressed in cancer and involved in regulating cell growth and cell adhesion in human esophageal squamous cell carcinoma. Clin Cancer Res. 2006;12:1639–1646. doi: 10.1158/1078-0432.CCR-05-1858. [DOI] [PubMed] [Google Scholar]
- 22.Chen H, Ma J, Sunkel B, Luo A, Ding F, Li Y, He H, Zhang S, Xu C, Jin Q, Wang Q, Liu Z. S100A14: Novel Modulator of Terminal Differentiation in Esophageal Cancer. Mol Cancer Res. 2013;11:1542–1553. doi: 10.1158/1541-7786.MCR-13-0317. [DOI] [PubMed] [Google Scholar]
- 23.Shimada Y, Imamura M, Wagata T, Yamaguchi N, Tobe T. Characterization of 21 newly established esophageal cancer cell lines. Cancer. 1992;69:277–284. doi: 10.1002/1097-0142(19920115)69:2<277::aid-cncr2820690202>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 24.Tong T, Zhong Y, Kong J, Dong L, Song Y, Fu M, Liu Z, Wang M, Guo L, Lu S, Wu M, Zhan Q. Overexpression of Aurora-A contributes to malignant development of human esophageal squamous cell carcinoma. Clin Cancer Res. 2004;10:7304–7310. doi: 10.1158/1078-0432.CCR-04-0806. [DOI] [PubMed] [Google Scholar]
- 25.Su H, Hu N, Yang HH, Wang C, Takikita M, Wang QH, Giffen C, Clifford R, Hewitt SM, Shou JZ, Goldstein AM, Lee MP, Taylor PR. Global gene expression profiling and validation in esophageal squamous cell carcinoma and its association with clinical phenotypes. Clin Cancer Res. 2011;17:2955–2966. doi: 10.1158/1078-0432.CCR-10-2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michaylira CZ, Wong GS, Miller CG, Gutierrez CM, Nakagawa H, Hammond R, Klein-Szanto AJ, Lee JS, Kim SB, Herlyn M, Diehl JA, Gimotty P, Rustgi AK. Periostin, a cell adhesion molecule, facilitates invasion in the tumor microenvironment and annotates a novel tumor-invasive signature in esophageal cancer. Cancer Res. 2010;70:5281–5292. doi: 10.1158/0008-5472.CAN-10-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimokuni T, Tanimoto K, Hiyama K, Otani K, Ohtaki M, Hihara J, Yoshida K, Noguchi T, Kawahara K, Natsugoe S, Aikou T, Okazaki Y, Hayashizaki Y, Sato Y, Todo S, Hiyama E, Nishiyama M. Chemosensitivity prediction in esophageal squamous cell carcinoma: novel marker genes and efficacy-prediction formulae using their expression data. Int J Oncol. 2006;28:1153–1162. [PubMed] [Google Scholar]
- 28.Sharma M, Chuang WW, Sun Z. Phosphatidylinositol 3-kinase/Akt stimulates androgen pathway through GSK3beta inhibition and nuclear beta-catenin accumulation. J Biol Chem. 2002;277:30935–30941. doi: 10.1074/jbc.M201919200. [DOI] [PubMed] [Google Scholar]
- 29.Gkretsi V, Zhang Y, Tu Y, Chen K, Stolz DB, Yang Y, Watkins SC, Wu C. Physical and functional association of migfilin with cell-cell adhesions. J Cell Sci. 2005;118:697–710. doi: 10.1242/jcs.01638. [DOI] [PubMed] [Google Scholar]
- 30.Wu C. Migfilin and its binding partners: from cell biology to human diseases. J Cell Sci. 2005;118:659–664. doi: 10.1242/jcs.01639. [DOI] [PubMed] [Google Scholar]
- 31.Moik DV, Janbandhu VC, Fassler R. Loss of migfilin expression has no overt consequences on murine development and homeostasis. J Cell Sci. 2011;124:414–421. doi: 10.1242/jcs.075960. [DOI] [PubMed] [Google Scholar]
- 32.Xiao G, Cheng H, Cao H, Chen K, Tu Y, Yu S, Jiao H, Yang S, Im HJ, Chen D, Chen J, Wu C. Critical role of filamin-binding LIM protein 1 (FBLP-1)/migfilin in regulation of bone remodeling. J Biol Chem. 2012;287:21450–21460. doi: 10.1074/jbc.M111.331249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gkretsi V, Papanikolaou V, Zacharia LC, Athanassiou E, Wu C, Tsezou A. Mitogen-inducible Gene-2 (MIG2) and migfilin expression is reduced in samples of human breast cancer. Anticancer Res. 2013;33:1977–1981. [PubMed] [Google Scholar]
- 34.Papachristou DJ, Gkretsi V, Tu Y, Shi X, Chen K, Larjava H, Rao UN, Wu C. Increased cytoplasmic level of migfilin is associated with higher grades of human leiomyosarcoma. Histopathology. 2007;51:499–508. doi: 10.1111/j.1365-2559.2007.02791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gkretsi V, Papanikolaou V, Dubos S, Papathanasiou I, Giotopoulou N, Valiakou V, Wu C, Malizos KN, Tsezou A. Migfilin’s elimination from osteoarthritic chondrocytes further promotes the osteoarthritic phenotype via beta-catenin upregulation. Biochem Biophys Res Commun. 2013;430:494–499. doi: 10.1016/j.bbrc.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takafuta T, Saeki M, Fujimoto TT, Fujimura K, Shapiro SS. A new member of the LIM protein family binds to filamin B and localizes at stress fibers. J Biol Chem. 2003;278:12175–12181. doi: 10.1074/jbc.M209339200. [DOI] [PubMed] [Google Scholar]
- 37.Fan J, Ou YW, Wu CY, Yu CJ, Song YM, Zhan QM. Migfilin sensitizes cisplatin-induced apoptosis in human glioma cells in vitro. Acta Pharmacol Sin. 2012;33:1301–1310. doi: 10.1038/aps.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ou Y, Ma L, Dong L, Zhao Z, Zhou W, Fan J, Wu C, Yu C, Zhan Q, Song Y. Migfilin protein promotes migration and invasion in human glioma through epidermal growth factor receptor-mediated phospholipase C-gamma and STAT3 protein signaling pathways. J Biol Chem. 2012;287:32394–32405. doi: 10.1074/jbc.M112.393900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma S, Bao JY, Kwan PS, Chan YP, Tong CM, Fu L, Zhang N, Tong AH, Qin YR, Tsao SW, Chan KW, Lok S, Guan XY. Identification of PTK6, via RNA sequencing analysis, as a suppressor of esophageal squamous cell carcinoma. Gastroenterology. 2012;143:675–86. doi: 10.1053/j.gastro.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 40.Song S, Mazurek N, Liu C, Sun Y, Ding QQ, Liu K, Hung MC, Bresalier RS. Galectin-3 mediates nuclear beta-catenin accumulation and Wnt signaling in human colon cancer cells by regulation of glycogen synthase kinase-3beta activity. Cancer Res. 2009;69:1343–1349. doi: 10.1158/0008-5472.CAN-08-4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rodriguez-Viciana P, Warne PH, Vanhaesebroeck B, Waterfield MD, Downward J. Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J. 1996;15:2442–2451. [PMC free article] [PubMed] [Google Scholar]
- 42.Gawecka JE, Griffiths GS, Ek-Rylander B, Ramos JW, Matter ML. R-Ras regulates migration through an interaction with filamin A in melanoma cells. PLoS One. 2010;5:e11269. doi: 10.1371/journal.pone.0011269. [DOI] [PMC free article] [PubMed] [Google Scholar]