

Abstract
Squamous cell carcinoma is the predominant type of oral malignancy and is a result of oral carcinogenesis. Oral carcinogenesis is a mutifactorial and complex process related to the sequential occurrence of alterations in genetic structures, promoting inhibitory or excitatory effects of the tumor oncogenes and gene suppressors, compromising the histophysiology of the division, differentiation and cell death; and therefore, methods to prevent, detect, or treat it in the best way is constantly being searched for. Biomarkers reveal the genetic and molecular changes related to early, intermediate and late endpoints in the process of oral carcinogenesis. Thereby, they are likely to not only refine our ability to predict the biologic course of oral cancer and distinguish individuals at high and/or low risk of oral cancer development; but, also they will also reveal the genetic and molecular changes related to various endpoints of oral carcinogenesis. Chemopreventives are chemicals of natural or synthetic origin, which reduce the incidence of fatal diseases such as cancer before clinical symptoms occur. Chemopreventives are agents whose curative capacity is defined with help of biomarkers, as the later determine the effectiveness and safety of chemopreventives.
Keywords: β-carotene, δλ-alpha-tocopherol, biomarkers, chemopreventives, retinoid
INTRODUCTION
The predominant type of cancer found in the oral cavity is squamous cell carcinoma. Oral carcinogenesis is a mutifactorial and complex process related to the sequential occurrence of alterations in genetic structures, promoting inhibitory or excitatory effects of the tumor oncogenes and gene suppressors, compromising the histophysiology of the division, differentiation and cell death. Biomarkers help in the evaluation of prevention or use of therapies and the detection of the earliest stages of oral mucosal malignant transformation [Figure 1]. Biomarkers reveal the genetic and molecular changes related to early, intermediate and late endpoints in the process of oral carcinogenesis. These markers will refine our ability in predicting the biologic course of oral cancer and thereby, distinguishing individuals at high and/or low risk of oral cancer development. Genetic and molecular biomarkers will also determine the effectiveness and safety of chemopreventives. Biomarkers will also reduce the number of patients and the time for long-term follow-up required to define a significant clinical response to a chemopreventive agent, thereby, clarifying the types, doses, frequencies and regimens to achieve the maximum level of benefit from chemopreventives. This review will focus on the development of genetic and molecular biomarkers and the use of chemopreventives to intervene in the oral carcinogenesis [Figure 2] process.[1]
Figure 1.
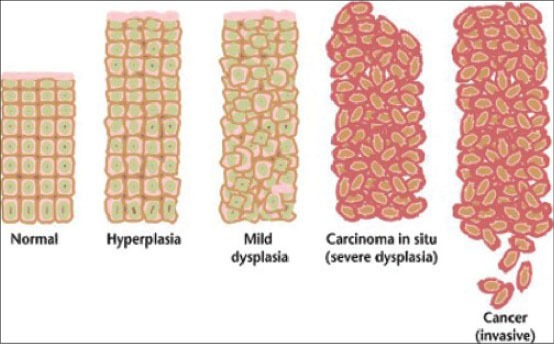
Cancers are caused by a series of mutations. Each mutation alters the behavior of the cell somewhat (multistep process) (Courtesy: Wikimedia Commons)
Figure 2.
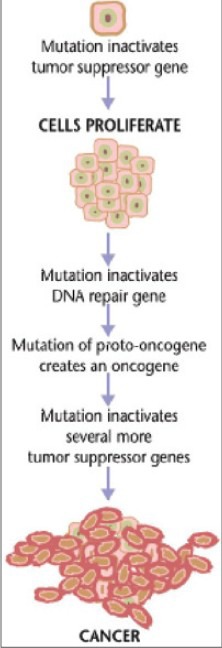
Steps of carcinogenesis (initiation, promotion and progression) (Courtesy: En.wikipedia)
ROLES OF CELLULAR BIOMARKERS
Indicators of deoxyribonucleic acid (DNA) repair mechanisms
Indicators of programmed cell death (PCD)
Indicators of tumor development and growth
Indicators of genetic markers of oral cancer.
Indicators of DNA repair mechanisms
PCD functions not by itself, but in concert with systems that facilitates DNA repair. Present evidence indicates that cancer cells require a high level of DNA repair [Figure 3]. In general, these include repair of the telomeric ends of chromosomes produced through the action of telomerase and repair of nucleotide sequences, exemplified by mismatch repair and nucleotide excision repair (NER). Telomeres are heterochromatic structures at the ends of eukaryotic chromosomes and consist of simple, highly conserved, repeated DNA sequences (e.g., TTAGGG, as observed in humans and mice). Every species has a characteristic average of telomeric subunits. In human oral carcinomas, telomerase is elevated in the proliferative areas of the carcinoma. Defective repair processes and checkpoints are also linked to cancer genomic instability. The types of genomic instability seen in most cases of sporadic cancer suggest a commonality of break types and DNA repair processes. Examples of the more common DNA repair sites are approximately 20 genes known to be involved in the process of NER or the repair/transcription factors, such as Transcription factor II Human (TFIIH), that are required to orchestrate the function of incisional proteins, that is, DNA polymerases and ligases. These changes may become additional markers for the aggressive and metastatic characteristics of oral carcinoma. DNA repair influences the progression of oral carcinogenesis through the regulation of various growth factors, for example, transforming growth factor (TGF)-β3. The development of a DNA repair defect and the presentation of a mutated TGF-β3 receptor gene (e.g. glutamine and proline.
Figure 3.
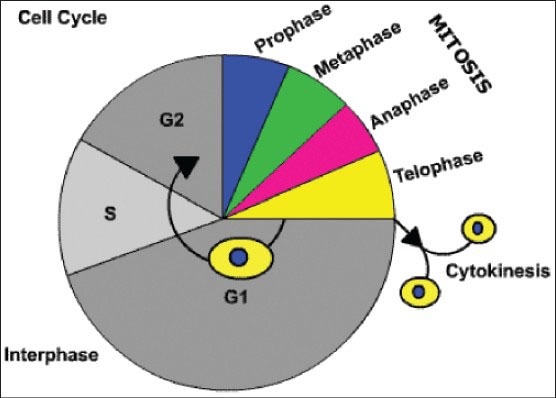
The cell cycle. It consists of: (i) interphase - G1 = growth and preparation of the chromosomes occurs for replication after G1 checkpoint; S = synthesis of deoxyribonucleic acid (DNA) and duplication of the centrosome; G2 = preparation for mitoses of cell size and content after G2 checkpoint. (ii) M = mitosis (prophase, metaphase, anaphase and telophase). (iii) Cytokinesis = separation of cell membrane to form two new cells. (iv) G0 = Resting Phase when cell leaves cell cycle temporarily/permanently (Courtesy: http://click4biology.info/)
Indicators of PCD
There is a considerable body of work that has identified several hundred cellular changes or biomarkers associated with the growth of oral carcinoma and carcinomas of the aerodigestive tract. Many of these indicators are also markers for PCD. PCD or gene-directed apoptosis is a common means used in nature to remove unwanted cells. It is characterized by the lack of an inflammation-driven necrosis of the tissue and the histologic appearance of apoptotic cells. PCD is also an important feature of oral keratinocytes undergoing differentiation or transformation during oral cancer development. PCD may also play an important screening role in cancer formation [Figure 4]. PCD results in the modification of the surviving cell population in transforming clones by altering the numbers and types of cells in a tumor. The surviving transforming cells appear to have suppressed PCD and they have a high rate of proliferation, enhanced levels of resistance to different antitumor therapies and elevated levels of DNA repair. There are numerous genetic and molecular changes that are used to identify PCD[3] [Figure 5].
Figure 4.
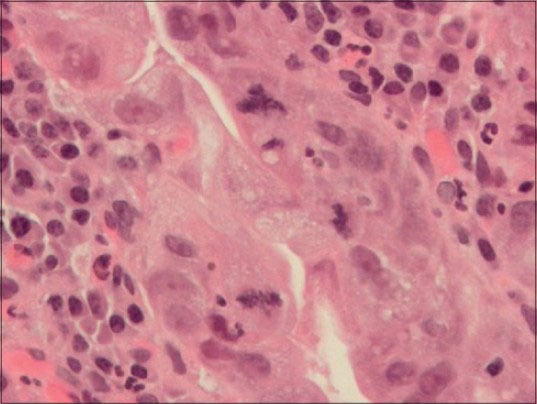
Photomicrograph of gastric mucosa with high grade dysplasia (H&E stain, ×400) Three clearly identifiable mitoses are seen. The one at the 12:00 o’clock position is tripolar, that is, atypical. The mitoses at 3:00 and 6:00 o’clock are normal. Acute (neutrophils) and chronic (plasma cells and lymphocytes) inflammatory cells are in the lamina propria immediately underneath of the dysplastic epithelium. Atypical mitoses are a characteristic of precancerous lesions, that is, dysplasia and malignancy, that is, cancer. (Coutesy: En.wikipedia)
Figure 5.
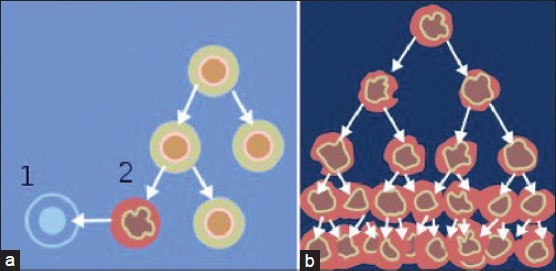
When normal cells are damaged beyond repair, they are eliminated by apoptosis (a). Cancer cells avoid apoptosis and continue to multiply in an unregulated manner (b). (courtesy: Normal_cancer_ cell_division_from_NIH.png)
Oxidation and the effect on PCD
Oral malignant transformation could be a product of oxidative state change. The oxidative state change would then result in changes in DNA repair and PCD. The observed cellular manifestations of these processes are losses of cell growth control and modifications in cell-cell interactions, which could enhance the potential for tumor metastases. Nutrients that act as chemopreventives alter the oxidative state of the oral transforming cell by acting as reducing agents (e.g., antioxidant) and/or oxidizing agents (e.g., pro-oxidant).[4]
Chemopreventives and PCD
The treatment with chemopreventives, during malignant transformation or in fully transformed malignant oral mucosal cells, results in the induction of PCD and the observed inhibition of oral carcinogenesis and malignant tumor growth. Chemopreventives induce PCD because of their oxygen-responsive characteristics, which trigger inducers such as the tumor suppressor gene p53, modifiers of PCD such as the bcl-2 family and immune-derived cytokines, for example, tumor necrosis factor (TNF). Chemopreventives, as exemplified by retinoids, carotenoids, tocopherols, bioflavonoids, isothiocyanates, indoles and polyphenols induce PCD.[5,6]
Indicators of tumor development and growth
Biomarkers establish the level of risk for individuals in a target group of patients and they may provide information concerning the etiology and the process of carcinogenesis. The primary goal for the use of early, intermediate and late biomarkers is to identify individuals at risk of developing malignancy and indicate their level of risk.[1,7]
Indicators of genetic markers of oral cancer
A developing solid clone of transforming cells found in an oral carcinoma arrives at the state of malignancy by proceeding through stages of transformation [Figure 1]. The transformation rate is dependent on the location of the clone in the spherical tumor mass and the oxygen states of the cells.[8]
Chemopreventives
Chemoprevention is an appealing strategy and its success has been demonstrated in breast cancer and familial adenomatous polyposis. High-dose retinoids have been shown to be active against oral premalignant lesions and in prevention of second primary tumors in the head and neck.[9]
The rationale for pharmacologic chemoprevention in patients at risk for the development of invasive cancer is based upon two factors:
Field cancerization - Patients with head and neck cancer have a predilection for cancer development throughout the oropharyngeal mucosa. Whether this is also true for human papillomavirus (HPV)-associated oropharyngeal tumors is not clear
Multistep carcinogenesis - Squamous cell cancers of the head and neck result from a multistep process with defined intermediate stages; leading to fully transformed, invasive and metastatic cancer.[10]
Thus, chemopreventives are chemicals of natural or synthetic origin which, unlike other drugs, do not prevent disease but instead, chemopreventives reduce the incidence of diseases such as cancer before clinical symptoms occur. This development is critical for the understanding of early oral mucosal transformation.
Classification of Chemopreventive agents (Pharmacological and chemical structural classification)
-
Antimutagens/Carcinogen Blocking Agents
- Phase II metabolic enzyme inducers
- N-acetyl L-cysteine
- Polyphenols
- Curcumin and dehydroepiendrosterone (DHEA).
-
Antiproliferatives
- Retinoids/caretinoids: β-carotene, 13-cis-retinoic acid, vitamin-A
- Glucose-6-phosphate dehydrogenase inhibitors
- Aspirin.[11]
Antioxidants.
Commonly tried chemopreventive agents in oral cancer
Vitamin A and other retinoids
β-carotene
Vitamin E
Dietary agents
Other agents[11]
Overview
A concept common to chemoprevention is the ability of chemical agents to function as reducing agents, antioxidants or oxidizing agents (pro-oxidants). The chemical character of the chemopreventive will depend on the partial pressure of oxygen and the level of oxidative metabolites produced or derived in the cell.
β-carotene, a carotenoid, acts as an antioxidant, but can also act as a pro-oxidant, depending on the oxygen state of the cell. β-carotene as well as ellagic acid (from garlic) are carcinogen-blocking agents that either suppress promotion or act as antioxidants, which are reducing agents. Their specific mechanism of action is unknown, but blocking agents prevent carcinogenic compounds from reacting with critical target sites of DNA by inhibiting the metabolic activation of carcinogens catalyzed by cytochrome P450 (Phase I enzymes).
Other chemopreventive agents, such as dλ-alpha-tocopherol (vitamin E), are strong antioxidants that enhance the cellular detoxification system by increasing the levels of glutathione-S-transferases (GSTs; Phase II enzymes). The activation of these enzymes may also lead to the trapping of reactive carcinogen metabolites or the triggering of apoptosis.
Some chemopreventives may suppress the promotion of cancer and prevent the transformation of premalignant cells by altering differentiation. For example, retinoids enhance differentiation while affecting housekeeping genes and oncogene expression.
Another class of chemopreventives, the terpenes, may also inhibit oncogenic expression and cell proliferation, reducing dedifferentiation.
Indomethacin, an anti-inflammatory drug, blocks prostaglandin synthesis and reduces tumor development, resulting in a normal differentiation pattern.
Cells also protect themselves from reactive oxygen substances (ROSs) by activating antioxidant pathways and molecular systems that use enzymes. Examples are superoxide dismutase, which controls the level of the superoxide anion (e.g., O2−); whereas, catalase modifies the levels of hydroxyl radicals (e.g., OH−) and glutathione- S-transferase (GSTs) alters the level of the intracellular antioxidant glutathione.
Less obvious cellular antioxidants are proteins such as Bcl-2, which is comprised of a family of proteins that modifies PCD.
There are also several protein families that function as redox, antioxidant/pro-oxidant molecules that also regulate PCD (e.g., p53).
Perhaps the most important common feature of chemopreventives is their ability to trigger PCD in transformed cells. The retinoids, carotenoids, tocopherols, isothiocyanates and polyphenols induce PCD in various cell types. Clinically, there is an advantage to the combination of chemopreventives with chemotherapeutics, especially with the alkylating agents [Figure 6].[6]
Figure 6.
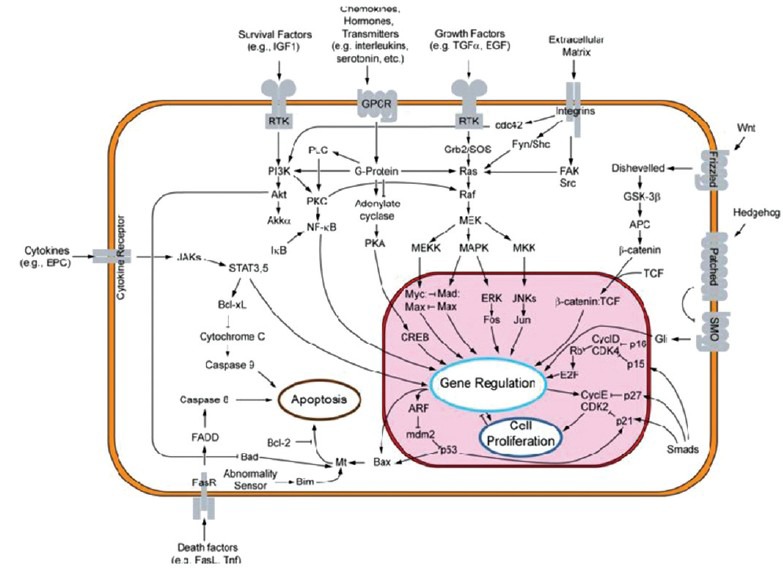
An overview of major signal transduction pathways. In each signal transduction system, an activation/inhibition signal from a biologically active molecule (hormone and neurotransmitter) is mediated via the coupling of a receptor/enzyme to a second messenger system or to an ion channel. Signal transduction plays an important role in activating cellular functions, cell differentiation and cell proliferation. Many tumor suppressor genes effect signal transduction pathways which regulate apoptosis, also known as “programmed cell death” (courtesy: http://en.wikipedia.org/wiki/File:Signal_transduction_v1.png)
Genetic, molecular and biochemical activities of chemopreventives
Retinoid chemopreventives
The retinoid molecule family consists of vitamin A and its derivatives. The primary source of retinoids in the diet is retinyl esters from animal tissues and retinol from the conversion of carotenoids (e.g., 3-carotene) derived from a vegetable source. Retinol tends to be the biologically active form, but retinoic acid (RA) may substitute for retinol for many functions and both can be found in the serum linked to proteins. Retinol binds to proteins designated as retinol-binding proteins, while RA is associated with albumin. The normal plasma level for retinol is approximately 2 mmol/L, while that of RA is 10-20 nmol/L, about 150-fold lower than retinol. RA is metabolized by hydroxylation of the cyclohexenyl ring, a reaction mediated by cytochrome P450, producing a 4-hydroxy metabolite which can undergo further oxidation. These oxygen reactive molecules could enhance genetic instability and result in abnormal cell growth, differentiation and cell-cell interactions. In cells, retinol and RA bind to specific cytoplasmic binding proteins, which include cytoplasmic retinol binding proteins I and II (CRABP-I/II). The cellular binding proteins mediate transfer of retinol and RA from the cytoplasm to the nucleus. The nuclear RA receptors (RAR) are members of the steroid-thyroid superfamily of nuclear receptors. Retinoid modulation of gene expression is the result of the activation of one of four possible receptors. The RARs are found in many different isoforms, as shown by mRNA, which indicates that the isoforms arise from different promoters or by alternative RNA splicing in a region at the 5’ end of the mRNA. Isoforms appear to be tissue-specific and their expression is stage-specific, suggesting distinctive roles for each isoform. The expression of mRNA for RAR-; -β, -γ and -X has been found in normal oral mucosa. The development of premalignant change and smoking or alcohol use do not appear to change the general distribution of these receptors in oral tissue. However, recent studies have indicated that with retinoid treatment there is an increase in the level of RAR-β in premalignant lesions. Lotan et al., (1995)[12] presented a studywhere RAR-1 was depressed during premalignant change and retinoid treatment and remission of oral leukoplakia was coincident with an increase in RAR-β. It appears that RAR-β may be an important indicator for a retinoid response in premalignant or malignant oral mucosa. The aberrant response to RA, as reflected by RAR-β modulation, is also echoed by changes in related growth factor receptors such as epidermal growth factor. Retinoid ligand specificities are found in recombinant RAR-α and -β receptors, with at least two retinoid analogs binding preferentially to either RAR-α or -β. Initial studies suggested that RAR-β was approximately 10-fold more sensitive to all-trans RA than RAR-α. The results of these studies indicated that the blockage of the RAR changed tissue oxidation of retinol to RA, but other oxidation processes, perhaps linked to cytochrome P450 activity, may also be altered. Recent studies have also indicated that the conversion of retinol to RA might be mediated by the RAR. Therefore, the ability of retinoids to inhibit or increase the growth of cancer cells could be dependent on the accumulation and rates of conversion and/or non-conversion of RA in the cell. It is well-known that head and neck squamous cell carcinoma (HNSCC) patients, particularly those who smoke tobacco, have low levels of serum retinoids. Their low intake of fruits and vegetables may play a role in the development of oral cancer, but the cellular state of the oral mucosa must also be considered. The concentration of any retinoid metabolite may be initially dependent on the type, number and affinity of the RARs present on the cell. Changes in the binding and concentration of the cytoplasmic retinal binding protein (CRBP) or CRABPs might also influence the triggering of RA response elements (RARE) and associated genes. Exogenous influences affect the conversion of retinol to RA by altering the activity of alcohol dehydrogenase and the cytochrome P450 system noted above. In addition, RARs appear to be involved in the differentiation of myeloid cells and keratinocytes. RA binding to its receptors has also been shown to induce apoptosis in the differentiating cell population.[12,13,14]
The finding that PCD occurred following treatment with RA indicates that a cascade of definitive genetic changes was set into motion. RA induction of PCD may be different from other chemopreventive inductions of PCD by triggering BAG-1 gene and internal phosphorylation signals that trigger the Bcl-2 system of proteins. Other chemopreventives, such as β-carotene or vitamin E, appear to activate the oncogene-inductive process (e.g., p53) and/or the immune-inductive process {e.g., the tumor necrosis factor I (TNFI) receptor family, FAS/FASL} for PCD. Retinoid-induced genetic changes will not only inhibit the growth of developing cancer cells or established cancer but, at times may possibly promote the growth of premalignant or malignant cancers. These results again indicate that the responses of tissues to retinoids are more complex than is apparent from studies that emphasize RAR expression.[12,14,15]
Genetic and molecular activities of carotenoid and tocopherol
The carotenoids and tocopherols produce genetic and molecular responses similar to those observed with the retinoids. The specificities of these responses have been previously reviewed. Animal studies with the hamster buccal pouch tumor model have further confirmed some of the similarities between these chemopreventive groups and the retinoids. These include the overexpression and half-life of p53, enhanced differentiation of the oral mucosa, reduction in neovascularization of developing oral cancers and the induction of apoptosis. The major cellular difference between the retinoid response and the carotenoids or tocopherols is lack of a complex receptor system described above for retinoids. In general, the major biochemical similarity among these groups (e.g., carotenoids, retinoids and tocopherols) is their isoprenoid structure and biochemical sterol effects. Their sterol chemistry is derived from their hydrophobic cyclic structures that contain unsaturated double bonds. Chemopreventives, as a group, respond to changes in oxygen partial pressure that influences their ability to act as oxygen free radical quenchers or reactive oxygen molecules.[16] In a well-oxygenated environment, the carotenoid, β-carotene, can inhibit the growth of oral cancer cells, because the carotenoid induces an oxidative stress in the tumor cell. At the identical partial pressure of oxygen, tocopherol tends to have an antioxidant activity and reduces the oxidative stress. These oxygen-handling characteristics could affect the triggering of PCD, the cell cycle, by modifying kinase activities and eventually lead to an alteration in activities such as c-fos and other cellular processes linked to differentiation or DNA repair. The responses of the oral mucosal cells to nutritional agents such as retinoids, carotenoids or tocopherols will also depend on the degree of transformation that has occurred in the cell. Normal cells appear to react to these agents by differentiating, while transforming cells or fully transformed cells may also differentiate, but they continue towards PCD. Some of the more common membrane-related responses to β-carotene and/or tocopherols are the reductions in membrane-associated enzymes such as serine and threonine kinases, connexin 43 and a reduction in ras proteins. Other previously discussed proteins, such as proto-oncogenes, c-fos, c-myb, N-myc and the tumor suppressor p53, have been shown to be affected by treatment with these chemopreventives. Stress proteins, acting as cellular chaperonins, can complex to p53 and following treatment of oral carcinoma cells with β-carotene, their levels of expression increase and affect the movement of proteins. In addition, growth factors and immune regulatory factors such as epidermal growth factor (EGF) and TGF-α have a reduced expression, while TGF-β1 and TNF-α expression are elevated during the inhibition of oral carcinogenesis. The EGF receptor's (EGFR's) affinity, number and protein expression were reduced following β-carotene treatment and suppression of oral cancer growth. Studies reveal the broad effects of β-carotene and other similar chemopreventives on many different cellular functions. Importantly, the carotenoids and tocopherols do not appear to produce the toxicity seen with retinoids, but their effectiveness at preventing or reversing premalignant change is unclear.[16,17,18]
Oxidative biochemistry of chemopreventives
Chemopreventive agents can be defined by their suppression or blocking of mutagenic activity, initiation and/or promotion during oral carcinogenesis. Agents that alter the mutagenic process are generally enzymes that enhance the solubilization or degradation of mutagenic or carcinogenic agents. This process usually activates the intracellular antioxidant, glutathione and its associated system. The glutathione pathway regulates free radical activity by producing reducing agents, such as thiol-containing proteins, with cysteine-glycine residues. The primary antioxidant in this pathway is glutathione, which can react with electrophilic sites produced on a carcinogen molecule through the action of cytochrome P450 hydrolyses. Glutathione also acts to block the nucleophilic attack of the carcinogen on DNA. Chemopreventive agents, such as glutathione, may also function by enhancing the elimination of the genotoxic agent from the liver and other tissues. In these tissues, the hydroxylation of the cytochrome P450 system and mixed-function oxidases (MFO) eventually terminates with conjugates of reduced protein salts containing glucuronate and sulfates.[14,19,20]
Unfortunately, the activation of the cytochrome P450 system by the chemopreventive agents may sometimes increase carcinogenesis by producing more highly electrophilic epoxides. The result could be the conversion of procarcinogens to their genotoxic form, which could generate oxygen free radicals. Many of the chemopreventives may work not only through this pathway but also by altering the level of GSTs which mediates electrophile scavenging. The carotenoids, tocopherols and retinoids all appear to be capable of modifying GSTs. The carotenoid, β-carotene, has been shown to depress GST levels, while other chemopreventives, such as alpha-tocopherol acid succinate or dithiolthione oltipaz, elevate the level of GST.[21,22]
Chemopreventives have been shown to inhibit both initiation and promotion during oral carcinogenesis. The inhibition of initiation may occur by preventing the carcinogen from becoming fully active by enhancing DNA repair and/or the activation of tumor suppressor genes. The inhibition of promotion could result by triggering differentiation. Initiation and promotion may also be affected by the elimination of transformed malignant clones of cells. The induction of an inflammatory cytotoxic immune reaction or the generation of apoptosis could accomplish this.[23,24,25] Examples of chemopreventives that have exhibited anti-promotional activity include tamoxifen (an anti-estrogen), retinoids and carotenoids, which are also inhibitors of proliferation. In addition, alpha-tocopherol acid succinate and piroxicam are known to suppress arachidonic acid metabolism, which, in turn, could affect apoptosis or tumor development by reducing prostaglandins. Alpha-tocopherol can inhibit the activity of prostaglandins by blocking their synthesis and this occurs by a reduction in the function of cyclooxygenase (COX). Leukotriene activity can also be regulated by reducing the activity of lipoxygenase. The inhibition of prostaglandins has been shown to be associated with the suppression of oral carcinogenesis.[18,26,27,28,29]
SUMMARY
It is thought that patients with head and neck premalignant changes consist of a diverse population and should be treated differently depending on their molecular genotype. Patients with minimal genetic changes may be treated with single-agent retinoids or other agents.[30] Those with more accumulated genetic changes will require combination of chemoprevention therapies. Lesions that have advanced genetic changes with mutant p53 may benefit from targeted p53 therapy and those lesions that express EGFR and COX-2 may require inhibitors of EFGR and COX-2.[25,31,32,33]
However, the challenge today is achieving long-lasting efficacy with retinoids and/or new agents and determining the optimal dose and duration of therapy while maintaining acceptable toxicities.[34,35,36]
ACKNOWLEDGEMENT
I take this opportunity with much pleasure to thank all the people who have helped me through the course of my journey towards producing this paper.
My sincere gratitude also goes to all those who instructed and taught me through the years.
Finally, this paper would not have been possible without the confidence, endurance and support of my family, my children in particular and the questfulness of my PG students so far. I wish to thank my parents for their constant support and blessings.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Washington: National Academy of Sciences, National Academy Press; 1981. Committee on Nitrite and Alternative Curing Agents in Food. The health effects of nitrate, nitrite, and N-nitroso compounds. Available from: http://www.archive.org/stream/healtheffectsofn004248mbp/healtheffectsofn004248mbp_djvu.txt . [Google Scholar]
- 2.Gonzalez FJ. Genetic polymorphism and cancer susceptibility: Fourteenth Sapporo Cancer Seminar. Cancer Res. 1995;55:710–5. [PubMed] [Google Scholar]
- 3.Meyskens FL., Jr Biomarker intermediate endpoints and cancer prevention. J Natl Cancer Inst Mongr. 1992;13:177–81. [PubMed] [Google Scholar]
- 4.Kerr JF, Winterford CM, Harmon BV. Apoptosis. Its significance in cancer and cancer therapy. Cancer. 1994;73:2013–26. doi: 10.1002/1097-0142(19940415)73:8<2013::aid-cncr2820730802>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz JL. The inhibition of oral carcinogenesis through the induction of programmed cell death. Cancer Res. 1996;35:631. [Google Scholar]
- 6.Cheng KC, Loeb LA. Genomic instability and tumor progression: Mechanistic considerations. Adv Cancer Res. 1993;60:121–56. doi: 10.1016/s0065-230x(08)60824-6. [DOI] [PubMed] [Google Scholar]
- 7.Hockenberry DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 function in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–51. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 8.Shin DM, Hittelman WN, Hong WK. Biomarkers in upper aerodigestive tract tumorigenesis: A review. Cancer Epidemiol Biomark Prev. 1994;3:697–709. [PubMed] [Google Scholar]
- 9.Wali RK, Kunte DP, De La Cruz M, Tiwari AK, Brasky J, Weber CR, et al. Topical polyethylene glycol as a novel chemopreventive agent for oral cancer via targeting of epidermal growth factor response. PloS One. 2012;7:e38047. doi: 10.1371/journal.pone.0038047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bodhade AS, Dive AM. Chemoprevention of premalignant and malignant lesions of oral cavity. Recent Trends. Eur J Dent. 2013;7:246–50. doi: 10.4103/1305-7456.110198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bisen PS, Bundela SS, Sharma A. Ellagic acid- chemopreventive role in oral cancer. J Cancer Sci Ther. 2012;4:23–30. [Google Scholar]
- 12.Lotan R, Xu X-C, Lippman SM, Ro JY, Lee JS, Lee JJ, et al. Suppression of retinoic acid receptor-β in premalignant oral lesions and its upregulation by isotretinoin. N Engl J Med. 1995;332:1405–1410. doi: 10.1056/NEJM199505253322103. [DOI] [PubMed] [Google Scholar]
- 13.Sharma S, Stutzman JD, Kelloff GJ, Steele VE. Screening of potential chemopreventive agents using biochemical markers of carcinogenesis. Cancer Res. 1994;54:5848–55. [PubMed] [Google Scholar]
- 14.Wattenberg LW. Chemoprevention of cancer by naturally occurring and synthetic compounds. In: Wattenberg LW, Lipkin M, Boone CW, Kelloff GJ, editors. Cancer Chemoprevention. Boca Raton: CRC Press Inc; 1992. pp. 19–39. [Google Scholar]
- 15.Daly MB. The chemopreventive of cancer: Directions for the future. Cancer Epidemiol Biomark Prev. 1993;2:509–12. [PubMed] [Google Scholar]
- 16.Palan PR, Mikhail MS, Goldberg GL, Basu J, Runowicz CD, Romney SL. Plasma levels of beta-carotene, lycopene, canthaxanthin, retinol, and alpha- and tau-tocopherol in cervical intraepithelial neoplasm and cancer. Clin Cancer Res. 1996;2:181–5. [PubMed] [Google Scholar]
- 17.Garewal HS, Schantz S. Emerging role of beta-carotene and other antioxidant nutrients in prevention of oral cancer. Arch Otolaryngol Head Neck Surg. 1995;121:141–4. doi: 10.1001/archotol.1995.01890020005002. [DOI] [PubMed] [Google Scholar]
- 18.Burton GW, Ingold KU. r-carotene: An unusual type of lipid antioxidant. Science. 1989;224:509–13. doi: 10.1126/science.6710156. [DOI] [PubMed] [Google Scholar]
- 19.Gridley G, McLaughlin JK, Block G, Blot WJ, Gluch M, Fraumeni JF., Jr Vitamin supplement use and reduced risk of oral and pharyngeal cancer. Am J Epidemiol. 1992;135:1083–92. doi: 10.1093/oxfordjournals.aje.a116208. [DOI] [PubMed] [Google Scholar]
- 20.Lippman SM, Heyman RA, Kurie JM, Brenner SE, Hong WK. Retinoids and chemoprevention: Clinical and basic studies. J Cell Biochem Suppl. 1995;22:1–10. doi: 10.1002/jcb.240590802. [DOI] [PubMed] [Google Scholar]
- 21.Manoharan S, Singh RB, Balakrishnan S. Chemopreventive mechanisms of natural products in oral, mammary and skin carcinogenesis: An overview. Open Nutraceuticals J. 2009;2:52–63. [Google Scholar]
- 22.Patel JB, Shukla SN, Patel HR, Kothari KK, Shah PM, Patel PS. Utility of urinary biomarkers in oral cancer. Asian Pac J Cancer Prev. 2007;8:229–35. [PubMed] [Google Scholar]
- 23.Giovanini AF, Zielak JC, Matsubara F, Urban CA, Pizzatto E. Immunoexpression of p53 and p16 proteins as biomarkers in oral carcinogenesis. Appl Cancer Res. 2009;29:83–8. [Google Scholar]
- 24.Wu JY, Yi C, Chung HR, Wang DJ, Chang WC, Lee SY, et al. Potential biomarkers in saliva for oral squamous cell carcinoma. Oral Oncol. 2010;46:226–31. doi: 10.1016/j.oraloncology.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 25.Shpitzer T, Hamzany Y, Bahar G, Feinmesser R, Savulescu D, Borovoi I, et al. Salivary analysis of oral cancer biomarkers. Br J Cancer. 2009;101:1194–8. doi: 10.1038/sj.bjc.6605290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rudin CM, Murur S. Update: Wolters Kluwer; 2013. Oct 23, Chemoprevention and screening in oral dysplasia and head and neck cancer. Available from: http://www.uptodate.com/contents/chemoprevention-and-screening-in-oral-dysplasia- and-head-and-neck-cancer . [Google Scholar]
- 27.Tsao AS, Kim ES, Hong WK. Chemoprevention of cancer. CA Cancer J Clin. 2004;54:150–80. doi: 10.3322/canjclin.54.3.150. [DOI] [PubMed] [Google Scholar]
- 28.Holpuch AS, Desai KG, Schwendeman SP, Mallery SR. Optimizing therapeutic efficacy of chemopreventive agents: A critical review of delivery strategies in oral cancer chemoprevention clinical trials. J Carcinog. 2011;10:23. doi: 10.4103/1477-3163.85185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsuda M, Ohba Y. Functional biomarkers of oral cancer. In: Dr. Kalu UE, Ogbureke, editors. Oral Cancer. Croatia (Europe): Intech Europe; 2012. ISBN: 978-953-51-0228-1. Available from: http://www.intechopen.com/books/oral-cancer/functional-biomarkers-of-oral-cancer . [Google Scholar]
- 30.Goodin S, Shiff SJ. NSAIDs for the chemoprevention of oral cancer: Promise or pessimism?: Commentary re J. L. Mulshine et al., randomized, double-blind, placebo-controlled, phase IIB trial of the cyclooxygenase inhibitor ketorolac as an oral rinse in oropharyngeal leukoplakia. Clin Cancer Res. 2004;10:1561–4. doi: 10.1158/1078-0432.ccr-0003-4. [DOI] [PubMed] [Google Scholar]
- 31.Tanaka T, Tanaka M, Tanaka T. Oral carcinogenesis and oral cancer chemoprevention: A review. Patholog Res Int. 2011 doi: 10.4061/2011/431246. 431246. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Chaparwal Y, Keerthilatha P, Vineetha R. Chemoprevention of oral cancer. J Indian Acad Oral Med Radiol. 2012;24:39–44. [Google Scholar]
- 33.Schwartz JL. Biomarkers and molecular epidemiology and chemoprevention of oral carcinogenesis. Crit Rev Oral Biol Med. 2000;11:92–122. doi: 10.1177/10454411000110010501. [DOI] [PubMed] [Google Scholar]
- 34.Thomas G, Ramdoss K. Chemoprevention of Oral Cancer. In: Manoj Pandey, Krishnan Nair M, Paul Sebastain., editors. Advances in Oncology. 1st edition. Vol. 1. New Delhi: Jaypee brothers medical publishers; 2000. p. 51. [Google Scholar]
- 35.Casto BC, Kresty LA, Kraly CL, Pearl DK, Knobloch TJ, Schut HA, et al. Chemoprevention of oral cancer by black raspberries. Anticancer Res. 2002;22:4005–15. [PubMed] [Google Scholar]
- 36.Rhee JC, Khuri FR, Shin DM. Advances in chemoprevention of head and neck cancer. Oncologist. 2004;9:302–11. doi: 10.1634/theoncologist.9-3-302. [DOI] [PubMed] [Google Scholar]