

Abstract
Background
The blood brain barrier tightly regulates the passage of molecules into the brain and becomes leaky following obstructive cholestasis. The aim of this study was to determine if increased serum bile acids observed during cholestasis permeabilize the blood brain barrier.
Methods
Rats underwent bile duct ligation or deoxycholic or chenodeoxycholic acid injections and blood brain barrier permeability assessed. In vitro, the permeability of rat brain microvessel endothelial cell monolayers, the expression and phosphorylation of occludin, ZO-1 and ZO-2 as well as the activity of Rac1 was assessed after treatment with plasma from cholestatic rats, or bile acid treatment, in the presence of a Rac1 inhibitor.
Results
Blood brain barrier permeability was increased in vivo and in vitro following bile duct ligation or treatment with bile acids. Associated with the bile acid-stimulated increase in endothelial cell monolayer permeability was elevated Rac1 activity and increased phosphorylation of occludin. Pretreatment of endothelial cell monolayers with a Rac1 inhibitor prevented the effects of bile acid treatment on occludin phosphorylation and monolayer permeability.
Conclusions
These data suggest that increased circulating serum bile acids may contribute to the increased permeability of the blood brain barrier seen during obstructive cholestasis via disruption of tight junctions.
Keywords: Occludin, bile duct ligation, Rac1, tight junction proteins
The blood brain barrier (BBB) is a protective barrier that protects the brain from the passage of molecules from the circulation [1]. Cerebral endothelial cells form tight junctions with adjacent cells via interactions of the tight junction proteins occludin and claudin-5 and intracellular docking proteins ZO-1 and ZO-2 [2]. When these tight junctions are compromised, molecules are able to passively diffuse into the brain. Opening of these tight junctions can be mediated by a number of signal transduction pathways including Rac1 [3], Protein kinase C (PKC) [4], or Jun-N-terminal kinase (JNK) [5], that may result in either a rearrangement of the cytoskeleton [6, 7] or the phosphorylation of the tight junction proteins occludin [8] or claudin-5 [9].
The BBB becomes compromised during the course of various liver diseases [10–12], which may contribute to the various neurological changes associated with these diseases [13–15]. During liver damage, serum bile acid concentrations are increased. Under normal physiological conditions, ~95% of bile is recycled and stored in the gallbladder with the remaining 5% being excreted [16]. These bile acids exert their effects through the nuclear receptor farnesoid X receptor (FXR) [17] or the membrane bound receptor, TGR5 [18]. In addition to signaling through FXR, bile acids have been shown to activate an array of cell signaling pathways such as ERK1/2 and JNK [19]. However, when hepatocytes are damaged and cannot reabsorb bile acids, the serum levels are increased.
Bile acids have been shown previously to induce permeability in epithelial cells [20, 21], however, the role of bile acids in the permeabilization of the BBB during cholestasis remains unknown. Thus the aims of our study were to assess BBB integrity during cholestasis and determine if bile acids can permeabilize the barrier formed by the endothelial cells that make up the BBB in vitro as well as in vivo.
Materials & Methods
Materials
Unless otherwise indicated, all chemicals were purchased from Sigma-Aldrich (St Louis, MO) and were of the highest grade available. All primers in this study were purchased from Qiagen (SABiosciences; Frederick, MD).
Animal Treatment
Male Sprague Dawley rats (150–175g) were maintained in a temperature-controlled environment (20–22°C) with a 12:12-hour light-dark cycle with free access to drinking water and rat chow. Rats underwent sham or bile duct ligation (BDL) surgery as described previously [22]. In parallel, rats were injected with chenodeoxycholic acid or deoxycholic acid (iv; 3μg/rat/day for 1 or 5 days). All animal experiments were performed with the approval of and in accordance with the guidelines of the Scott & White IACUC Committee.
In vivo permeabilization measurements
To assess in vivo permeabilization of the BBB a modified Evan’s blue extrusion assay [23] was performed in sham and BDL rats or after tail vein injection of bile acids. Briefly, rats were anesthetized with isoflurane inhalation and Evan’s blue dye was infused (5mg/ml; 500μl) through the carotid artery and allowed to circulate for 20 minutes. Rats were then transcardially perfused with 80ml ice-cold phosphate-buffered saline, the meninges removed and the brain homogenized with 3ml ice-cold trichloroacetic acid (50% v/v) in a glass homogenizer. The resulting homogenates were centrifuged for 10 minutes at 10,000 g and absorbance of the supernatant was read at 620nm.
In vivo microvessel staining
To visualize the BBB in the experimental treatment groups, whole brains were fixed in 4% PFA, followed by cryoprotection in 30% sucrose solution (w/v in 1X phosphate buffered saline). Free-floating immunofluorescence staining of brain sections (20μm) was performed as described previously [24, 25]. Slices were stained with an anti-rat SMI-71 antibody (1:500; Covance, Princeton, NJ). SMI-71 stains for an endothelial cell-specific protein expressed in BBB but not in peripheral endothelial cells [26]. In addition, the presence of serum albumin in the brain was assessed by immunohistochemical staining [25] using a horseradish peroxidase-conjugated albumin antibody (Bethyl Laboratories, Montgomery TX).
In vitro permeabilization measurement
Monolayers of rat brain microvascular endothelial cells (RBMECs; Cell Applications, San Diego, CA) were seeded onto fibronectin-coated Transwell™ inserts. This is a primary cell line and was not used after passage 10. After cells grew into a confluent monolayer (3–4 days) cells were treated with sham or BDL rat serum (10% to 75%) or vehicle, chenodeoxycholic acid, deoxycholic acid, glycochenodeoxycholic acid, taurochenodeoxycholic acid, or ursodeoxycholic acid (10 μM or 100μM) for 24 hours. In parallel, RBMEC monolayers were pretreated with inhibitors for Rac1 (NSC23766; 50 μM [27]) Rho kinase (HA-1077; 5μM [28]), JNK (SP600125; 50μM, [29]), Ca2+ signaling (BAPTA; 1μM [30]), MAPK p42/44 (PD98059; 10 μM, [31]), and PKC (Gö6976; 1μM [32]) or EGFR (BIBX; 10μM [33]), before treating with 100μM chenodeoxycholic acid or deoxycholic acid. Additionally, RBMEC monolayers were treated with various doses of the FXR agonist Fexaramine (1–100nM). After 24 hours, media was replaced with phenol-red free RPMI media (Invitrogen, Carlsbad, CA) and 10kDa FITC-dextran (25mg/ml; 10μl) was added to the upper well. After 1 hr, fluorescence (excitation 494nm; emission 520nm) was read in the lower well. Each experiment was performed on 6 transwells per treatment and repeated at least twice and data was expressed as the average relative fluorescence units compared to control.
Quantitative in vivo bile acid measurement
At 1, 3 and 5 days post-surgery, or after tail vein injection of chenodeoxycholic acid or deoxycholic acid, circulating bile acids were assessed in the serum of sham and BDL rats via a colorimetric assay according to the manufacturer’s protocol (Diagnostic Chemicals Limited, Charlottetown, Canada).
Cell viability assay
RBMECs were seeded into each well (10,000 cells/well) of a 96 well plate and allowed to adhere overnight, after which vehicle, chenodeoxycholic acid or deoxycholic acid was added (100μM) for 24 hours. Cell viability was assessed using a colorimetric MTS assay (CellTiter 96 AQueous; Promega Corp, Madison, Wisconsin, USA) and absorbance was read at 490nm [24].
Immunofluorescence
RBMECs were seeded onto fibronectin-coated coverslips, were allowed to grow to confluence and were treated with vehicle, chenodeoxycholic acid or deoxycholic acid (100μM). After 24 hours, cells were fixed with 4% PFA for 5 minutes at room temperature, permeabilized by washing with PBS containing 0.2% Triton X-100 (PBST) and blocked using LiCor blocking buffer (Lincoln, NE). Cells were stained overnight with antibodies specific for ZO-1, ZO-2, claudin 5 and occludin (Invitrogen-Zymed, Irvine, CA), at 1:100. Fluorescent detection using Alexa 488 secondary antibody (Jackson Immuno Research, West Grove, PA) was performed and coverslips were mounted onto a microscope slide with antifade gold containing 4′,6-diamidino-2-phenylindole (DAPI) as a counter stain (Molecular Probes, Eugene, OR).
Subcellular fractionation
Analysis of tight junction protein localization was determined by utilizing the ProteoExtract Subcellular Proteome Extraction kit (Calbiochem; La Jolla, CA). Membrane fractions were separated via SDS-PAGE as previously described [24] and occludin, ZO-1, and ZO-2 localization was determined using anti-occludin, anti-ZO-1, and anti-ZO-2 antibodies. Specificity of fractions was determined by using anti-pan-cadherin antibody.
Real-time PCR
The expression occludin, ZO-1 and ZO-2 was assessed by real time polymerase chain reaction (qPCR) as described previously [34] in RBMECs treated with saline, chenodeoxycholic acid or deoxycholic acid for 1, 4, and 24 hours and using specific commercially available primers. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression was used for normalization. A ΔΔCT analysis was performed [35] using sham serum or vehicle as controls. Data are expressed as relative mRNA±SEM (an n of at least 4 was used).
Immunoblotting
RBMECs were treated with chenodeoxycholic acid or deoxycholic acid (100 μM) for various time points and the expression of Occludin, ZO-1 and ZO-2 was assessed by immunoblotting [24] using the specific antibodies described above. The expression of GAPDH was assessed as an internal loading control.
Immunoprecipitation
To assess the effects of bile acid treatment on the phosphorylation of occludin, RBMEC’s were treated with chenodeoxycholic acid or deoxycholic acid (100 μM) for 4 hr in the presence or absence of the Rac1 inhibitor (NSC23766; 50 μM [27], EMD Millipore, Billerica, MA). Cells were then lysed in RIPA buffer containing a protease inhibitor cocktail (Sigma-Aldrich, Saint Louis, MI). Immunoprecipitation was then performed on 50 μg of protein from each treatment using 2 μg of anti-phosphotyrosine antibody (Abcam, Cambridge, MA) and 20 μL of protein A/G agarose. The subsequent amounts of occludin pulled down were assessed by immunoblotting as described above. In parallel, the relative amounts of target proteins was assessed in 10 μg of total protein from each treatment by immunoblotting as an indication of the input into the immunoprecipitation assay.
Rac1 activity assay
RBMEC’s were treated with chenodeoxycholic acid or deoxycholic acid (100 μM) for 1 hr in the presence or absence of the Rac1 inhibitor (NSC23766; 50 μM [27], EMD Millipore, Billerica, MA). Rac1 activity was assessed using the Rac1 activity kit (Cytoskeleton Inc, Denver, CO) following the protocol described by the vendor.
Statistical analysis
All statistical analyses were performed using Graphpad Prism software (Graphpad Software, La Jolla, CA). Results are expressed as mean ± SEM. For data that passed normality tests, significance was established using the Student t test when differences between two groups were analyzed, and analysis of variance when differences between three or more groups were compared followed by the appropriate post hoc test. If tests for normality failed, two groups were compared with a Mann-Whitney U test or a Kruskal-Wallis ranked analysis when more than two groups were analyzed. Differences were considered significant when the p value was less than 0.05.
RESULTS
Cholestasis induces permeabilization of the BBB in vivo
In order to assess the effects of cholestasis on BBB integrity, experimental cholestasis was induced using the BDL model which exhibited a significant increase in BBB permeability as shown by increased Evan’s blue content in brain homogenates compared to sham-operated rats (Figure 1A). Immunofluorescent staining with SMI-71 showed continuous cerebral microvessels with several branch points (indicated by arrows) in sham-operated rats, whereas BDL rats have discontinuous sporadic staining, suggesting a loss of BBB integrity in BDL rats (Figure 1B). Furthermore, cholestatic rats had a higher degree of albumin immunoreactivity in the brain (Figure 1C). Treatment of RBMEC monolayers with serum from BDL rats increased permeability of RBMEC monolayers compared to sham serum-treated monolayers (Figure 1D), suggesting that a circulating factor is leading to permeabilization of the BBB observed in our cholestatic model in vivo.
Figure 1.

Cholestasis leads to blood brain barrier permeability via circulating factors. (A) Permeability of the blood brain barrier was assessed via an Evan’s blue assay in sham and bile duct ligated rats 5 days post-surgery. BDL rats have a significant increase in blood brain barrier permeability compared to sham rats. (* denotes p<0.05, n=4) (B) Brain slices of sham and bile duct ligated rats were stained with a SMI-71 antibody (red) allowing for microvessel visualization and counterstained with DAPI (blue). Sham rats have long continuous staining of microvessels with several branch points (white arrows). Bile duct ligated rats show dispersed staining, indicating a loss of microvessel integrity. Scale bar represents 100μm. (C) Albumin immunoreactivity was assessed in sham and Bile duct ligated rats via immunohistochemistry. Bile duct ligated rats have a higher degree of albumin immunoreactivity in brain slices, particularly the hippocampus and the cortex compared to sham operated rats, indicating a loss of blood brain barrier integrity. (D) Confluent monolayers of brain endothelial cells were treated for 24 hours with serum extracted from either sham or bile duct ligated rats 5 days post-surgery and permeability was measured via the passive diffusion of 10kDa fluorescein isothiocyanate-dextran. Monolayers treated with various concentrations of bile duct ligated serum have increased permeability compared to 75% sham serum. (* denotes p<0.05, n=6) Data are expressed as average permeability coefficient ± SEM. (BDL; bile duct ligation, HP; hippocampus, CTX, cortex)
Circulating bile acids increase during cholestasis and can permeabilize the BBB
Circulating bile acid levels increased approximately 20 fold, 1 day after BDL compared to sham-operated rats, and remained elevated up to 5 days after surgery (Figure 2A). In order to assess whether bile acids alone have the capacity to induce BBB permeability, we treated RBMEC monolayers with various bile acids. Treatment with chenodeoxycholic acid or deoxycholic acid, but not glycochenodeoxycholic acid, taurochenodeoxycholic acid, or ursodeoxycholic acid for 24 hours led to significantly increased permeability of RBMEC monolayers (Figure 2B and data not shown). To parallel the bile acid effect seen after BDL, rats were injected daily with chenodeoxycholic acid or deoxycholic acid for 5 days, a strategy that resulted in serum total bile acid concentrations of 47.18 ± 2.35 μM (compared to 22.58 ± 1.42 μM in vehicle-treated animals). This treatment increased the permeability of the BBB as demonstrated using the Evan’s blue extravasation assay (Figure 2C). Furthermore, increased Evan’s blue could be observed 4 hr after a single tail vein injection of these bile acids (data not shown). Fluorescent imaging of cerebral microvessels using a SMI-71 antibody demonstrated discontinuous sporadic staining in deoxycholic acid-treated rats compared to vehicle-treated rats, which have continuous staining with several branch points (Figure 2D). Lastly, there was an increase in albumin immunoreactivity in brains from deoxycholic acid-treated rats compared to controls (Figure 2E).
Figure 2.

Bile acids are increased in the circulation of bile duct ligated rats and have the capacity to permeabilize the blood brain barrier both in vitro as well as in vivo. (A) Circulating bile acids were measured with a colorimetric assay in sham and bile duct ligated rats 5 days post-surgery. BDL rats have ~7 fold higher circulating bile acids than sham rats. (* denotes p<0.05, n=3) (B) Confluent monolayers were treated with vehicle or chenodeoxycholic acid, or deoxycholic acid (10μM or 100μM) for 24 hours and permeability was measured via fluorescein isothiocyanate-dextran flux. (* denotes p<0.001, n=6) (C) Permeability was measured in vivo via Evan’s blue assay in rats tail vein-injected with either vehicle, chenodeoxycholic acid or deoxycholic acid for 5 days. chenodeoxycholic acid- or deoxycholic acid-treated rats have a significant increase in blood brain barrier permeability as measured by Evan’s blue concentration compared to vehicle treated rats. (* denotes p<0.05, an n of at least 3 was used) (D) Brain slices were stained with a SMI-71 antibody (red) and counterstained with DAPI (blue) in vehicle- and deoxycholic acid-treated rats to visualize cerebral microvessels. Similar to sham rats, vehicle-treated rats have long continuous staining with several bifurcations (white arrows) compared to deoxycholic acid-treated rats that have discontinuous staining. Scale bar represents 100μm. (E) Albumin immunohistochemistry was performed on rats injected with vehicle or deoxycholic acid for 5 days. Rats treated with deoxycholic acid show a higher degree of albumin immunoreactivity in brain slices, in particular the hippocampus and cortex regions, indicating a disruption of the BBB. (BDL; bile duct ligation, DCA; deoxycholic acid, CDCA; chenodeoxycholic acid, UDCA; ursodeoxycholic acid HP; hippocampus, CTX, cortex)
Chenodeoxycholic acid or deoxycholic acid disrupts endothelial cell tight junctions
Since there was increased permeability of the RBMEC monolayers to chenodeoxycholic acid or deoxycholic acid treatment, we first examined if this was due to increased endothelial cell death. Stimulation of cells with chenodeoxycholic acid or deoxycholic acid hours did not result in a significant decrease in cell viability as demonstrated by MTS assays (Supplement Figure S1). We then assessed the cellular distribution of the tight junction proteins occludin and claudin-5 along with the intracellular docking proteins ZO-1 and ZO-2 in RBMECs treated with vehicle, chenodeoxycholic acid or deoxycholic acid. In vehicle-treated cells, these tight junction proteins show continuous staining along the cell borders between adjacent cells (Figure 3A), whereas after chenodeoxycholic acid or deoxycholic acid treatment occludin, ZO-1 and ZO-2 immunoreactivity was disrupted with gaps between cell borders (Figure 3A). This redistribution of tight junction proteins was further confirmed in deoxycholic acid-treated cells by subcellular fractionation. Deoxycholic acid-treated cells had reduced occludin, ZO-1, and ZO-2 localization in the membrane than in vehicle-treated cells (Figure 3B). No effect on the cellular distribution of claudin-5 immunoreactivity or subcellular distribution after any treatment was demonstrated (data not shown).
Figure 3.
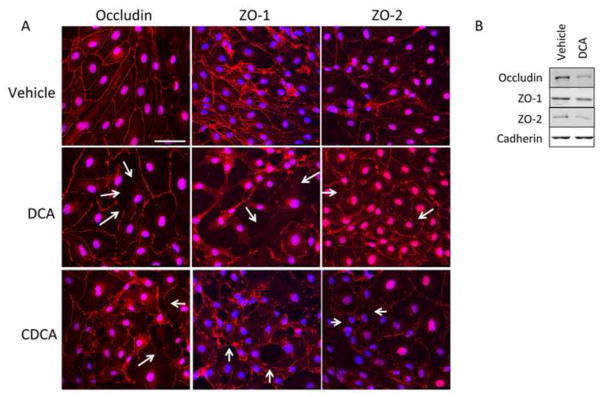
Chenodeoxycholic acid and deoxycholic acid disrupts endothelial tight junctions. (A) Confluent monolayers of brain microvessel endothelial cells were treated with either vehicle, chenodeoxycholic acid or deoxycholic acid and the tight junction protein occludin and tight junction-associated proteins ZO-1 and ZO-2 (red) were stained and cells were counterstained with DAPI (blue). Vehicle-treated cells show continuous staining along cell margins indicating intact tight junctions. Treatment with 100μM chenodeoxycholic acid or deoxycholic acid for 24 hours disrupts tight junctions leaving gaps at the cell margins. Scale bar represents 50 μm. Arrows indicate sites of disruption (B) Brain microvessel endothelial cells were treated with vehicle or deoxycholic acid and membrane localization of occludin, ZO-1 and ZO-2 were determined via immunoblot analyses. Vehicle-treated cells show a higher degree of occludin, ZO-1 and ZO-2 expression in the membrane compared to deoxycholic acid-treated cells. Pan-cadherin was used as a membrane loading control. (DCA; deoxycholic acid, CDCA; chenodeoxycholic acid)
We then determined if the redistribution of occludin, ZO-1 and ZO-2 were due to changes in their expression. Indeed, there was no change in the mRNA or protein expression of any of the tight junction or tight junction-associated proteins studied up to 24 hr after treatment (Supplemental Figure S2A–D). Intracellular redistribution of tight junction proteins is often a downstream consequence of various signal transduction pathways. In particular, phosphorylation of tight junction proteins, such as occludin, is known to increase the permeability of endothelial cell layers and disrupts their ability to interact with ZO-1 and ZO-2 [28, 36]. Therefore we assessed the phosphorylation of occludin after chenodeoxycholic acid or deoxycholic acid treatment. Four hours after bile acid treatment, there was an increased amount of occludin protein pulled down with a phospho-tyrosine antibody, indicating increased phosphorylation of occludin after bile acid treatment (Figure 4A). No change in phosphorylation of tyrosine residues on claudin-5, ZO-1 or ZO-2 could be detected after bile acid treatment (data not shown).
Figure 4.
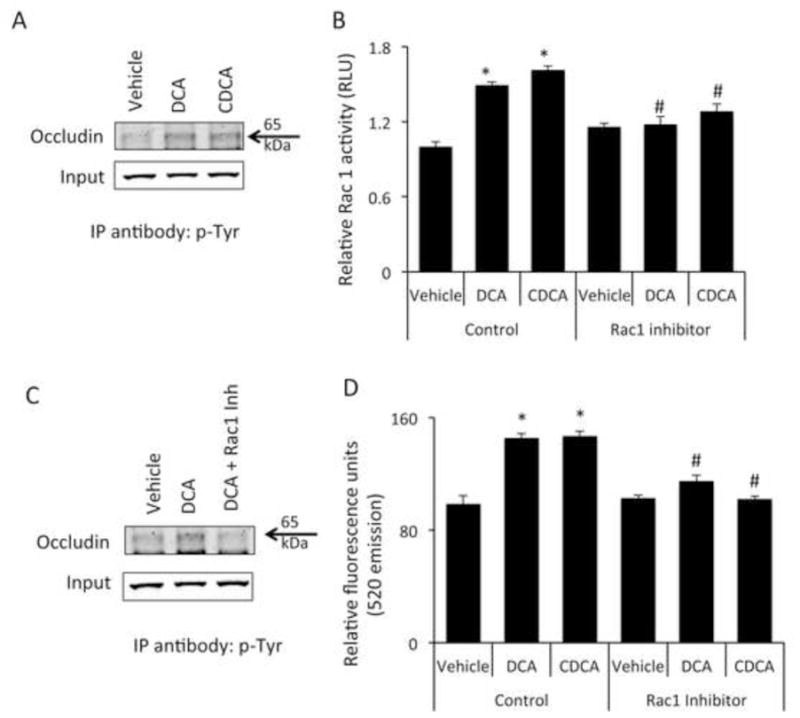
Bile acids increase the phosphorylation of occludin via a Rac1-dependent mechanism (A) Brain microvessel endothelial cells were treated with vehicle, chenodeoxycholic acid or deoxycholic acid for 1 hour and phosphorylated occludin levels were assessed by immunoprecipitating 50μg of total protein with a phospho-tyrosine specific antibody and immunoblotting using and anti-occludin antibody. 10μg total protein lysate was used as input. Cells treated with bile acids show a higher degree of tyrosine phosphorylation of occludin as shown by increased immunoprecipitation with anti-phospho-tyrosine antibody. (B) Confluent monolayers of brain microvessel endothelial cells were treated for 1 hour with vehicle, chenodeoxycholic acid or deoxycholic acid and Rac1 activity was measured. chenodeoxycholic acid or deoxycholic acid induce a significant increase in Rac1 activity compared to vehicle-treated cells, which could be blocked by the pre-treatment with the Rac1 inhibitor NSC23766. (* denotes p<0.05 compared to vehicle; # denotes p<0.05 compared to chenodeoxycholic acid or deoxycholic acid in the absence of NSC23766; n=5). (C) Brain microvessel endothelial cells were treated with vehicle, deoxycholic acid or deoxycholic acid + Rac1 inhibitor (NSC23766) for one hour and phosphorylation of occludin was determined by immunoprecipitating protein lysates with anti-phospho-tyrosine antibody and immunoblotting for occludin. 10μg total protein lysates were loaded for input. Pretreatment with the Rac1 inhibitor NSC23766 inhibited the deoxycholic acid-mediated phosphorylation of occludin. (D) Confluent monolayers of Brain microvessel endothelial cells were treated for 24 hours with vehicle, chenodeoxycholic acid or deoxycholic acid in either the presence or absence of Rac1 inhibitor NSC23766 and fluorescein isothiocyanate-dextran permeability was measured. Pretreatment with NSC23766 inhibited the bile acid-mediated permeabilization of brain endothelial cell monolayers. (* denotes p<0.05 compared to vehicle, # denotes p<0.05 compared to chenodeoxycholic acid or deoxycholic acid in the absence of NSC23766; n=6). (DCA; deoxycholic acid, CDCA; chenodeoxycholic acid; IP immunoprecipitation)
Chenodeoxycholic acid or deoxycholic acid induce occludin phosphorylation and permeability via a Rac1-dependent mechanism
Recent studies have demonstrated that occludin can be phosphorylated by a Rac1-dependent mechanism [37] therefore we assessed the relative Rac1 activity in RBMEC monolayers. Rac1 activity was increased 1 hr after treatment of RBMECs with chenodeoxycholic acid or deoxycholic acid, an effect that could be eliminated with pre-treatment with NSC23766, an inhibitor of Rac1 activity (Figure 4B). Furthermore, the phosphorylation of occludin by deoxycholic acid could be inhibited by pretreatment with the Rac1 inhibitor (Figure 4C). Lastly, the increased permeability of the RBMEC monolayers after chenodeoxycholic acid or deoxycholic acid could be prevented by pre-treatment with a Rac1 inhibitor (Figure 4D).
Previous studies have demonstrated that associated with bile acid-induced activation of Rac1 is an increased activation of the pathways mediated by Rho, PKC, MAPK p42/44 and the bile acid receptor FXR [38]. Furthermore, bile acids have previously been shown to cause permeability in epithelial monolayers via the activation of EGFR [21]. However, pretreatment of RBMECs with specific inhibitors of Rho kinase, PKC, MAPK p42/44 or EGFR had no effect on the bile acid-induced increase in RBMEC monolayer permeability, nor did the inhibition of Ca2+-mediated pathways by the Ca2+ chelator BAPTA A/M (Figure 5). Moreover, activation of the FXR receptor with the specific agonist fexaramine had no effect on RBMEC monolayer permeability (data not shown). In addition, the effects of Rac1 on the phosphorylation of occludin has previously been shown to depend on the activation of the JNK pathway, however pretreatment with the JNK inhibitor failed to prevent the bile acid-mediated increase in RBMEC monolayer permeability (Figure 5).
Figure 5.
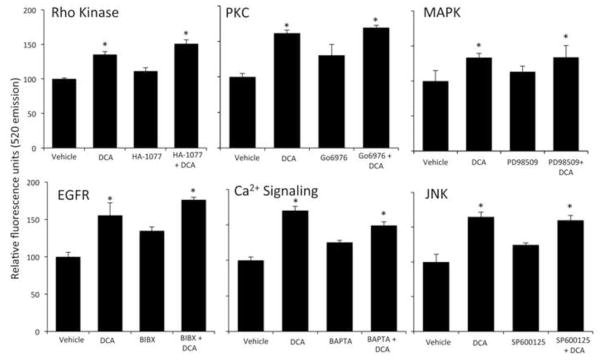
Deoxycholic acid does not disrupt endothelial tight junctions via known signal transduction pathways. Confluent monolayers of brain microvessel endothelial cells were treated for 24 hours with vehicle or deoxycholic acid in either the presence or absence of specific inhibitors for Rho kinase (HA-1077), PKC (Gö6976), ERK1/2 (PD98059), EGFR (BIBX), Ca2+ signaling (BAPTA) or JNK (SP600125), and permeability to fluorescein isothiocyanaate-dextran was measured. Pretreatment with these inhibitors failed to prevent the deoxycholic acid-induced permeability of RBMEC monolayers. (* denotes p<0.05 compared to vehicle, n=6).
Discussion
The major findings of this study pertain to the alterations that occur to the BBB during the course of cholestatic liver disease and the pathogenic role of bile acids. We present data showing that bile acids are key in the pathogenic breakdown of the BBB during cholestatic liver disease, and that these permeabilizing effects were due to the activation of Rac1 and downstream phosphorylation of the tight junction protein occludin, leading to disruption of tight junctions. This study identifies bile acids as a potential culprit in the BBB permeability observed during cholestatic liver disease.
Previous studies have identified pathogenic changes that occur to the BBB during the course of liver disease [11]. Rats with acute galactosamine-induced liver failure exhibit regional cerebral edema, indicating that that the BBB had lost, at least in part, its barrier function [10]. Evidence that the BBB is altered during the course of liver disease is further confirmed in portal hypertensive rats where there was a region-specific breakdown of the BBB at the hippocampus [12]. The current study examines the pathogenic role of bile acids in the permeabilization of the BBB. We employed the BDL model of cholestasis to examine BBB permeability and found that: i) there was a significant increase in circulating bile acids, ii) there was a compromise in the integrity of the BBB and iii) bile acids alter tight junction structure at the BBB. Increases in serum bile acids are not just a feature of biliary disorders. Spill over of bile acids into the circulation are also observed during acute liver failure [39], acute on chronic liver disease [39] as well as in non-alcoholic steatohepatitis [40]. Therefore our data may be relevant to not only biliary disorders, but also other nonbiliary liver diseases as well.
The alteration of tight junction structure by bile acids has been shown previously [20, 21]. Hydrophobic bile acids cholic acid, chenodeoxycholic acid and deoxycholic acid, but not the hydrophilic bile acid ursodeoxycholic acid, induces permeability in Caco-2 monolayers [21] via the activation of the EGFR. In contrast, we demonstrate that the bile acid-induced permeability of the RBMEC monolayers is via a mechanism involving the Rac1-dependent phosphorylation of occludin. The activation of Rac1 by bile acids has previously been shown in colorectal cancer cells and is associated with FXR expression [38]. However, the data presented here suggests that the effects of bile acids on Rac1 activation is not via an FXR-mediated event, but involves the phosphorylation of occludin.
Since Rac1 does not possess kinase activity itself, we assessed the role of some of the kinases known to be associated with Rac1 activity. We blocked rho kinase, JNK, MAPK p42/44, PKC and Ca2+ signaling and were not able to dampen the permeabilizing effects of deoxycholic acid. We did not block the cAMP/PKA pathway because it has been shown to be required for healthy barrier function [41], therefore specific inhibitors of these pathways would be expected to increase permeability independently of the bile acid effects. Other signaling pathways, such as myosin light chain kinase, that have been shown to alter BBB function [42] cannot be ruled out, however, the depletion of Ca2+ via pretreatment with BAPTA would presumably inhibit this Ca2+ dependent kinase [43], indicating that these pathways are more than likely not involved. Therefore the specific kinase involved in the bile acid-induced phosphorylation of occludin in our model is unknown and warrants further investigation.
Collectively, we present data indicating that the BBB is compromised in our model of cholestasis and that chenodeoxycholic acid or deoxycholic acid can permeabilize endothelial monolayers in vitro. We also determined if chenodeoxycholic acid or deoxycholic acid has the capacity to permeabilize the BBB in vivo. Injection of chenodeoxycholic acid or deoxycholic acid led to a modest but significant permeabilization of the BBB in normal rats compared to rats injected with saline. While this permeabilization effect is not nearly as robust as the permeabilization observed during cholestasis, this could possibly be explained by the fact that other factors are released from the liver during the course of cholestasis such as pro-inflammatory cytokines [44] that could have a synergistic effect with bile acids in permeabilizing the BBB. It should be mentioned that our tail vein injection experiments were performed in rats that were absent of liver injury. It is plausible that in these animals the healthy liver was able to take up the increased circulating bile acids and was then able to recycle it into the enterohepatic circulation, thus, clearing the majority of it from the circulation. Indeed, the concentration of serum bile acids reached after tail vein injection was elevated compared to controls but was significantly less than those observed after BDL.
Our results are consistent with previous findings that bile acids disrupt tight junctions. Indeed bile acids have been shown to transiently open the BBB when acutely administered [45]. The authors found that at high concentrations, bile acids permeabilize the BBB via lytic actions on cell membranes, but at lower concentrations, comparable to the concentrations used in this study, exert their actions by inhibiting the two junctions from fusing [45]. Bile acids could be the major contributor to the pathogenic breakdown of the BBB observed during cholestatic liver disease, as they have been shown to open the BBB in hepatic failure induced by D-galactosamine injection [46].
Since the neurological decline associated with liver damage is a major burden on quality of life and lowers priority level for transplantation [47] it is of utmost importance to determine its pathogenesis, which could be initiated by the breakdown of the BBB. The current study provides evidence that lowering circulating bile acids could provide an attractive treatment strategy to possibly ameliorate or delay the onset of neurological decline in patients with liver disease.
Supplementary Material
Acknowledgments
Source of support: This work was supported by an NIH R01 award (DK082435) and an NIH K01 award (DK078532) to Dr. DeMorrow. This material is the result of work supported with resources and the use of facilities at the Central Texas Veterans Health Care System, Temple, Texas.
We would also like to acknowledge Dr. Binu Tharakan for his help with the monolayer experiments and Dr. Dinorah Leyva-Illades for technical assistance.
Footnotes
Conflicts of interest: The authors of this manuscript have no financial arrangements to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bernacki J, Dobrowolska A, Nierwinska K, et al. Physiology and pharmacological role of the blood-brain barrier. Pharmacol Rep. 2008;60:600–22. [PubMed] [Google Scholar]
- 2.Ueno M. Molecular anatomy of the brain endothelial barrier: an overview of the distributional features. Curr Med Chem. 2007;14:1199–206. doi: 10.2174/092986707780597943. [DOI] [PubMed] [Google Scholar]
- 3.Kahles T, Luedike P, Endres M, et al. NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke. 2007;38:3000–6. doi: 10.1161/STROKEAHA.107.489765. [DOI] [PubMed] [Google Scholar]
- 4.He F, Yin F, Omran A, et al. PKC and RhoA signals cross-talk in Escherichia coli endotoxin induced alterations in brain endothelial permeability. Biochem Biophys Res Commun. 2012;425:182–8. doi: 10.1016/j.bbrc.2012.07.063. [DOI] [PubMed] [Google Scholar]
- 5.Urrutia A, Rubio-Araiz A, Gutierrez-Lopez MD, et al. A study on the effect of JNK inhibitor, SP600125, on the disruption of blood-brain barrier induced by methamphetamine. Neurobiol Dis. 2013;50:49–58. doi: 10.1016/j.nbd.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 6.Hicks RR, Baldwin SA, Scheff SW. Serum extravasation and cytoskeletal alterations following traumatic brain injury in rats. Comparison of lateral fluid percussion and cortical impact models. Mol Chem Neuropathol. 1997;32:1–16. doi: 10.1007/BF02815164. [DOI] [PubMed] [Google Scholar]
- 7.Rist RJ, Romero IA, Chan MW, et al. F-actin cytoskeleton and sucrose permeability of immortalised rat brain microvascular endothelial cell monolayers: effects of cyclic AMP and astrocytic factors. Brain Res. 1997;768:10–8. doi: 10.1016/s0006-8993(97)00586-6. [DOI] [PubMed] [Google Scholar]
- 8.Rao R. Occludin phosphorylation in regulation of epithelial tight junctions. Ann N Y Acad Sci. 2009;1165:62–8. doi: 10.1111/j.1749-6632.2009.04054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shen W, Li S, Chung SH, et al. Tyrosine phosphorylation of VE-cadherin and claudin-5 is associated with TGF-beta1-induced permeability of centrally derived vascular endothelium. Eur J Cell Biol. 2011;90:323–32. doi: 10.1016/j.ejcb.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 10.Gove CD, Hughes RD, Ede RJ, et al. Regional cerebral edema and chloride space in galactosamine-induced liver failure in rats. Hepatology. 1997;25:295–301. doi: 10.1002/hep.510250207. [DOI] [PubMed] [Google Scholar]
- 11.Livingstone AS, Potvin M, Goresky CA, et al. Changes in the blood-brain barrier in hepatic coma after hepatectomy in the rat. Gastroenterology. 1977;73:697–704. [PubMed] [Google Scholar]
- 12.Scorticati C, Prestifilippo JP, Eizayaga FX, et al. Hyperammonemia, brain edema and blood-brain barrier alterations in prehepatic portal hypertensive rats and paracetamol intoxication. World J Gastroenterol. 2004;10:1321–4. doi: 10.3748/wjg.v10.i9.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swain MG, Maric M, Carter L. Defective interleukin-1-induced ACTH release in cholestatic rats: impaired hypothalamic PGE2 release. Am J Physiol. 1995;268:G404–9. doi: 10.1152/ajpgi.1995.268.3.G404. [DOI] [PubMed] [Google Scholar]
- 14.Swain MG, Patchev V, Vergalla J, et al. Suppression of hypothalamic-pituitary-adrenal axis responsiveness to stress in a rat model of acute cholestasis. J Clin Invest. 1993;91:1903–8. doi: 10.1172/JCI116408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quinn M, Ueno Y, Pae HY, et al. Suppression of the HPA axis during extrahepatic biliary obstruction induces cholangiocyte proliferation in the rat. Am J Physiol Gastrointest Liver Physiol. 2012;302:G182–93. doi: 10.1152/ajpgi.00205.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirschfield GM, Heathcote EJ, Gershwin ME. Pathogenesis of cholestatic liver disease and therapeutic approaches. Gastroenterology. 2010;139:1481–96. doi: 10.1053/j.gastro.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 17.Stanimirov B, Stankov K, Mikov M. Pleiotropic functions of bile acids mediated by the farnesoid X receptor. Acta gastro-enterologica Belgica. 2012;75:389–98. [PubMed] [Google Scholar]
- 18.Kawamata Y, Fujii R, Hosoya M, et al. A G protein-coupled receptor responsive to bile acids. The Journal of biological chemistry. 2003;278:9435–40. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 19.Hylemon PB, Zhou H, Pandak WM, et al. Bile acids as regulatory molecules. J Lipid Res. 2009;50:1509–20. doi: 10.1194/jlr.R900007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen X, Oshima T, Tomita T, et al. Acidic bile salts modulate the squamous epithelial barrier function by modulating tight junction proteins. Am J Physiol Gastrointest Liver Physiol. 2011;301:G203–9. doi: 10.1152/ajpgi.00096.2011. [DOI] [PubMed] [Google Scholar]
- 21.Raimondi F, Santoro P, Barone MV, et al. Bile acids modulate tight junction structure and barrier function of Caco-2 monolayers via EGFR activation. Am J Physiol Gastrointest Liver Physiol. 2008;294:G906–13. doi: 10.1152/ajpgi.00043.2007. [DOI] [PubMed] [Google Scholar]
- 22.Alpini G, Lenzi R, Sarkozi L, et al. Biliary physiology in rats with bile ductular cell hyperplasia. Evidence for a secretory function of proliferated bile ductules. J Clin Invest. 1988;81:569–78. doi: 10.1172/JCI113355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hawkins BT, Egleton RD. Fluorescence imaging of blood-brain barrier disruption. J Neurosci Methods. 2006;151:262–7. doi: 10.1016/j.jneumeth.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 24.DeMorrow S, Glaser S, Francis H, et al. Opposing actions of endocannabinoids on cholangiocarcinoma growth: recruitment of Fas and Fas ligand to lipid rafts. J Biol Chem. 2007;282:13098–113. doi: 10.1074/jbc.M608238200. [DOI] [PubMed] [Google Scholar]
- 25.Goodenough S, Davidson M, Kidd G, et al. Cell death and immunohistochemistry of p53, c-Fos and c-Jun after spermine injection into the rat striatum. Exp Brain Res. 2000;131:126–34. doi: 10.1007/s002219900254. [DOI] [PubMed] [Google Scholar]
- 26.Ghabriel MN, Lu JJ, Hermanis G, et al. Expression of a blood-brain barrier-specific antigen in the reproductive tract of the male rat. Reproduction. 2002;123:389–97. doi: 10.1530/rep.0.1230389. [DOI] [PubMed] [Google Scholar]
- 27.Gao Y, Dickerson JB, Guo F, et al. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A. 2004;101:7618–23. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagumo H, Sasaki Y, Ono Y, et al. Rho kinase inhibitor HA-1077 prevents Rho-mediated myosin phosphatase inhibition in smooth muscle cells. Am J Physiol Cell Physiol. 2000;278:C57–65. doi: 10.1152/ajpcell.2000.278.1.C57. [DOI] [PubMed] [Google Scholar]
- 29.Thiel A, Heinonen M, Rintahaka J, et al. Expression of cyclooxygenase-2 is regulated by glycogen synthase kinase-3beta in gastric cancer cells. J Biol Chem. 2006;281:4564–9. doi: 10.1074/jbc.M512722200. [DOI] [PubMed] [Google Scholar]
- 30.Francis H, Glaser S, DeMorrow S, et al. Small mouse cholangiocytes proliferate in response to H1 histamine receptor stimulation by activation of the IP3/CaMK I/CREB pathway. Am J Physiol Cell Physiol. 2008;295:C499–513. doi: 10.1152/ajpcell.00369.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alessi DR, Cuenda A, Cohen P, et al. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–94. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 32.LeSage GD, Alvaro D, Glaser S, et al. Alpha-1 adrenergic receptor agonists modulate ductal secretion of BDL rats via Ca(2+)- and PKC-dependent stimulation of cAMP. Hepatology. 2004;40:1116–27. doi: 10.1002/hep.20424. [DOI] [PubMed] [Google Scholar]
- 33.Solca FF, Baum A, Langkopf E, et al. Inhibition of epidermal growth factor receptor activity by two pyrimidopyrimidine derivatives. J Pharmacol Exp Ther. 2004;311:502–9. doi: 10.1124/jpet.104.069138. [DOI] [PubMed] [Google Scholar]
- 34.DeMorrow S, Francis H, Gaudio E, et al. The endocannabinoid anandamide inhibits cholangiocarcinoma growth via activation of the noncanonical Wnt signaling pathway. Am J Physiol Gastrointest Liver Physiol. 2008;295:G1150–8. doi: 10.1152/ajpgi.90455.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 36.Kale G, Naren AP, Sheth P, et al. Tyrosine phosphorylation of occludin attenuates its interactions with ZO-1, ZO-2, and ZO-3. Biochem Biophys Res Commun. 2003;302:324–9. doi: 10.1016/s0006-291x(03)00167-0. [DOI] [PubMed] [Google Scholar]
- 37.Walsh TG, Murphy RP, Fitzpatrick P, et al. Stabilization of brain microvascular endothelial barrier function by shear stress involves VE-cadherin signaling leading to modulation of pTyr-occludin levels. J Cell Physiol. 2011;226:3053–63. doi: 10.1002/jcp.22655. [DOI] [PubMed] [Google Scholar]
- 38.Debruyne PR, Bruyneel EA, Karaguni IM, et al. Bile acids stimulate invasion and haptotaxis in human colorectal cancer cells through activation of multiple oncogenic signaling pathways. Oncogene. 2002;21:6740–50. doi: 10.1038/sj.onc.1205729. [DOI] [PubMed] [Google Scholar]
- 39.Benyoub K, Muller M, Bonnet A, et al. Amounts of bile acids and bilirubin removed during single-pass albumin dialysis in patients with liver failure. Therapeutic apheresis and dialysis: official peer-reviewed journal of the International Society for Apheresis, the Japanese Society for Apheresis, the Japanese Society for Dialysis Therapy. 2011;15:504–6. doi: 10.1111/j.1744-9987.2011.00980.x. [DOI] [PubMed] [Google Scholar]
- 40.Tanaka N, Matsubara T, Krausz KW, et al. Disruption of phospholipid and bile acid homeostasis in mice with nonalcoholic steatohepatitis. Hepatology. 2012;56:118–29. doi: 10.1002/hep.25630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ishizaki T, Chiba H, Kojima T, et al. Cyclic AMP induces phosphorylation of claudin-5 immunoprecipitates and expression of claudin-5 gene in blood-brain-barrier endothelial cells via protein kinase A-dependent and -independent pathways. Exp Cell Res. 2003;290:275–88. doi: 10.1016/s0014-4827(03)00354-9. [DOI] [PubMed] [Google Scholar]
- 42.Haorah J, Heilman D, Knipe B, et al. Ethanol-induced activation of myosin light chain kinase leads to dysfunction of tight junctions and blood-brain barrier compromise. Alcohol Clin Exp Res. 2005;29:999–1009. doi: 10.1097/01.alc.0000166944.79914.0a. [DOI] [PubMed] [Google Scholar]
- 43.Mackay HJ, Twelves CJ. Targeting the protein kinase C family: are we there yet? Nat Rev Cancer. 2007;7:554–62. doi: 10.1038/nrc2168. [DOI] [PubMed] [Google Scholar]
- 44.Lechner AJ, Velasquez A, Knudsen KR, et al. Cholestatic liver injury increases circulating TNF-alpha and IL-6 and mortality after Escherichia coli endotoxemia. Am J Respir Crit Care Med. 1998;157:1550–8. doi: 10.1164/ajrccm.157.5.9709067. [DOI] [PubMed] [Google Scholar]
- 45.Greenwood J, Adu J, Davey AJ, et al. The effect of bile salts on the permeability and ultrastructure of the perfused, energy-depleted, rat blood-brain barrier. J Cereb Blood Flow Metab. 1991;11:644–54. doi: 10.1038/jcbfm.1991.116. [DOI] [PubMed] [Google Scholar]
- 46.Tominaga S, Watanabe A, Tsuji T. Synergistic effect of bile acid, endotoxin, and ammonia on brain edema. Metab Brain Dis. 1991;6:93–105. doi: 10.1007/BF00999907. [DOI] [PubMed] [Google Scholar]
- 47.Butterworth RF. Hepatic encephalopathy: a central neuroinflammatory disorder? Hepatology. 2011;53:1372–6. doi: 10.1002/hep.24228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.