

Abstract
Site-specific incorporation of nonstandard amino acids (NSAAs) into proteins enables the creation of biopolymers, proteins, and enzymes with new chemical properties, new structures, and new functions. To achieve this, amber (TAG codon) suppression has been widely applied. However, the suppression efficiency is limited due to the competition with translation termination by release factor 1 (RF1), which leads to truncated products. Recently, we constructed a genomically recoded Escherichia coli strain lacking RF1 where 13 occurrences of the amber stop codon have been reassigned to the synonymous TAA codon (rEc.E13.ΔprfA). Here, we assessed and characterized cell-free protein synthesis (CFPS) in crude S30 cell lysates derived from this strain. We observed the synthesis of 190 ± 20 μg/mL of modified soluble superfolder green fluorescent protein (sfGFP) containing a single p-propargyloxy-l-phenylalanine (pPaF) or p-acetyl-l-phenylalanine. As compared to the parent rEc.E13 strain with RF1, this results in a modified sfGFP synthesis improvement of more than 250%. Beyond introducing a single NSAA, we further demonstrated benefits of CFPS from the RF1-deficient strains for incorporating pPaF at two- and five-sites per sfGFP protein. Finally, we compared our crude S30 extract system to the PURE translation system lacking RF1. We observed that our S30 extract based approach is more cost-effective and high yielding than the PURE translation system lacking RF1, ∼1000 times on a milligram protein produced/$ basis. Looking forward, using RF1-deficient strains for extract-based CFPS will aid in the synthesis of proteins and biopolymers with site-specifically incorporated NSAAs.
Keywords: cell-free protein synthesis, PURE translation, nonstandard amino acid, release factor 1, genomically recoded organisms
Expanding the chemistry of life is an essential component in synthetic biology.1−4 Dominant among frontier applications is the site-specific incorporation of nonstandard amino acids (NSAAs) into proteins. Introducing NSAAs engenders proteins with new functional and structural features that are not possible with the canonical 20 amino acid building blocks.5 For example, proteins with unique chemical handles for precise conjugation can be generated to enable next generation antibody-drug conjugate therapeutics.6 In another exemplary illustration, incorporating a uniquely reactive NSAA for site-specific conjugation of polyethylene glycol resulted in a modified human growth hormone with increased potency and reduced injection frequency.7 Furthermore, recent breakthroughs showing the direct synthesis of phosphoproteins8 and selenoproteins9 showcase the utility of NSAA incorporation. To date, more than 70 NSAAs have been site-specifically incorporated into proteins in Escherichia coli, yeast, and mammalian cells.5 To achieve this, orthogonal suppressor tRNA (o-tRNA)/aminoacyl-tRNA synthetase (aaRS) pairs as well as ribosomes have been evolved to reinterpret the genetic code in response to nonsense or quadruplet codons.10,11 Importantly, these o-tRNA/aaRS pairs are designed to operate parallel to and independent of the cell’s endogenous machinery.
To expand the genetic lexicon, the amber (TAG) stop codon, one of three nonsense codons, is typically reprogrammed as a sense codon for the incorporation of NSAAs. This is called amber suppression.4 An engineered tRNACUATyr/TyrRS pair derived from Methanocaldococcus jannaschii has been used most extensively to incorporate NSAAs in response to the amber codon in E. coli.4 More recent demonstrations of the technology have used variants of the pyrrolysine system, tRNACUAPyl/PylRS from Methanosarcinaceae species,12 which naturally recognizes the nonsense amber codon.4 A challenge for amber suppression is the fact that evolved o-tRNAs must outcompete the endogenous release factor 1 (RF1). It is known that the presence of RF1 can result in the production of truncated proteins, particularly when multiple site-specific incorporations are desired, resulting in low yields of the target protein product.8 To address this limitation, recent studies have sought to create RF1 deletion strains. In one approach, RF1 deletion was enabled by creating gain of function mutations in release factor 2 that allow it to recognize the TAG stop signal.13 In another approach, efforts to reassign the TAG codon to the TAA synonym can enable RF1 deletion. This was initially achieved for seven essential genes using a bacterial artificial chromosome.14 More recently, precise chromosome manipulation in vivo is enabling genomically recoded organisms (GROs) with TAG nonsense codons reassigned. These efforts have shown utility for improving suppression efficiency for site-specific NSAA incorporation in vivo.
In vitro (or cell-free) protein synthesis (CFPS) is emerging as a powerful technology platform for site-specific incorporation of NSAAs.1,15,16 The driving force behind this recent growth is 3-fold. First, a technical renaissance has enabled high-yielding (>1 g/L) and long-lasting (>10 h in batch operation) protein production.15 Second, cost-effective energy generation systems by mimicking the E. coli cytoplasmic environment17,18 and optimization of extract activity, codon usage, and redox folding conditions have enabled microscale to manufacturing scale (100 L), achieving linear scalability over a 6 orders of magnitude range in volume.19 These developments are covered in detail in two recent reviews.15,20 Third, the open nature of CFPS platform brings an unprecedented level of control and freedom of design compared to in vivo systems.21 For example, new components (natural and non-natural) can be added or synthesized and can be maintained at precise ratios. In contrast to in vivo systems, there are no transport limitations for getting NSAAs into the cell and there is flexibility for reprogramming the genetic code because cellular viability need not be maintained, noting that this reprogramming must not significantly affect the ability of the organism to grow and provide the necessary components for CFPS using bacterial extract. To this point, a benefit is that not all components necessary for genetic reprogramming need to be produced in the same strain. For example, the open nature of CFPS could address growth problems by adding purified orthogonal synthetase into the reaction and enabling in situ expression of orthogonal tRNA.22
Over the past decade, several groups have demonstrated the power of using crude extract CFPS systems for site-specific incorporation of NSAAs. In one exemplary illustration, Albayrak and Swartz demonstrated a modular and efficient cell-free platform that yields up to 0.9–1.7 mg/mL of a modified (i.e., containing a NSAA) soluble superfolder green fluorescent protein (sfGFP) in which the o-tRNA and the modified protein are produced simultaneously.22 Notably, CFPS yields were higher than comparative studies in vivo.22 Other examples include efforts to (i) leverage cell-free systems to avoid solubility and transport limitations typically encountered in vivo in the incorporation of the tyrosine analog p-propargyloxy-l-phenylalanine (pPaF) as well as p-azido-l-phenylalanine for demonstrating a one-step, site-specific direct protein–protein conjugation using copper(I)-catalyzed azide–alkyne [3 + 2] cycloaddition,23,24 (ii) explore drug discovery by site-specific tagging with NSAA,25 and (iii) enhance enzyme activity through NSAAs otherwise difficult with natural amino acids.26
Despite the fact that CFPS expression systems offer advantages and open new opportunities for NSAA incorporation,27 competition between RF1 in the extract and o-tRNAs remains a limitation.22 RF1-deficient CFPS systems have been constructed by removing RF1 tagged with chitin-binding domains28 and by using an RF1-deleted strain enabled by reassigning seven amber codons to their synonymous TAA.29 However, extracts from GROs lacking RF1 have not been attempted.
In this article, we describe the development of a CFPS platform from a genomically recoded E. coli strain lacking RF1 (encoded by prfA), called rEc.E13.ΔprfA.30 The goal was to characterize suppression efficiency in the presence and absence of RF1 and assess the ability to introduce NSAAs, pPaF or p-acetyl-l-phenylalanine (pAcF), at single and multiple sites (Figure 1). For characterization purposes, we further compared our S30 crude extract based CFPS platform to the PURE (Protein synthesis Using Recombinant Elements) translation system31 lacking RF1. In both the PURE system and extract based CFPS approach, we demonstrate that removal of RF1 increases the amount of full-length modified NSAA-containing protein more than 250%, providing benefits for incorporating NSAA at multiple in-frame positions.
Figure 1.

Scheme of cell-free protein synthesis reaction incorporating nonstandard amino acid to investigate the effect of RF1. Cell extracts containing transcription and translation machinery are prepared from rEc.E13 or rEc.E13.ΔprfA strains. Plasmid DNA template of sfGFP containing single or multiple amber codon sites, orthogonal tRNA/aaRS, NSAA, T7 RNA polymerase, and other cofactors are added as necessary to activate the cell-free protein synthesis (CFPS) reaction.
Results and Discussion
rEc.E13.ΔprfA as a Chassis Strain for CFPS
In anticipation of completion of fully recoded GRO wherein all occurrences of the stop TAG codon are reassigned to the synonymous TAA codon, we created pilot strains having either seven essential genes (coaD, had, hemA, mreC, murF, lolA, and lpxK)32 or these genes plus an additional six genes (yafF, pgpA, sucB, fabH, fliN, and atpE) recoded to create the strains rEc.E7 or rEc.E13,30 respectively. Next, we introduced the gene that confers resistance to spectinomycin (specR) into the prfA locus to create RF1 knockout strains (rEc.E7.ΔprfA and rEc.E13.ΔprfA). While the rEc.E7.ΔprfA had a severe growth phenotype, the rEc.E13.ΔprfA grew nearly twice as fast, providing a suitable RF1 deletion chassis strain for this work. The TAG codon reassignment to TAA of the six additional genes in rEc.E13.ΔprfA strain might allow better fitness in the cell growth by alleviating natural suppression pressure.30 The doubling time for rEc.E13.ΔprfA and the parent rEc.E13 was 64.6 ± 0.9 min and 47 ± 2 min, respectively, in 2xYTPG media (Figure 2), and in LB, 74.8 ± 1.2 min for rEc.E13.ΔprfA and 50.3 ± 0.7 min for rEc.E13 (Supporting Information Figure S1). The rEc.E13.ΔprfA chassis strain grows slower than the rEc.E13 because there is only partial recoding of the genome.30
Figure 2.

Growth rate comparison of rEc.E13 and rEc.E1.ΔprfA strains. Growth of rEc.E13 and ΔprfA in 2xYTPG medium at 34 °C in 96 well plates. Each data point is the average of ten replicate wells from two independent cultures.
RF1 Deletion Enhances pPaF Incorporation in CFPS
The impact of RF1 deletion on NSAA incorporation in CFPS reactions was assessed in S30 crude cell extract CFPS reactions. The o-tRNA was expressed constitutively under the control of lpp promoter in extract source strain during cell growth as described previously.24 Following extract preparation, we used the PANOx-SP CFPS system developed by Jewett and Swartz17 to quantitatively test the incorporation of pPaF into sfGFP with an in-frame amber codon at position 132 (sfGFP132pPaF). Initially, we carried out a series of optimization experiments in extracts from rEc.E13 to examine the effects of temperature, incubation time, and pPaFRS concentrations on CFPS (Figure 3). Combined transcription and translation reactions were carried out in 15 μL batch reactions. Figure 3a shows wild-type sfGFP and sfGFP132pPaF synthesis at 30 and 37 °C throughout the batch reaction duration. For sfGFP132pPaF, we additionally added 0.5 mg/mL pPaFRS (see Figure 3b for optimization) and 2 mM pPaF.24 Active protein synthesis yields, as assessed by fluorescence, were approximately 2.5-fold higher at 30 °C for both wild-type and modified sfGFP. Our data are consistent with previous results, which have shown advantages for sfGFP folding33 and NSAA incorporation24 at 30 °C as compared to 37 °C. Because sfGFP synthesis was most productive in 20 h batch reactions at 30 °C, all further CFPS reactions were carried out using these conditions.
Figure 3.
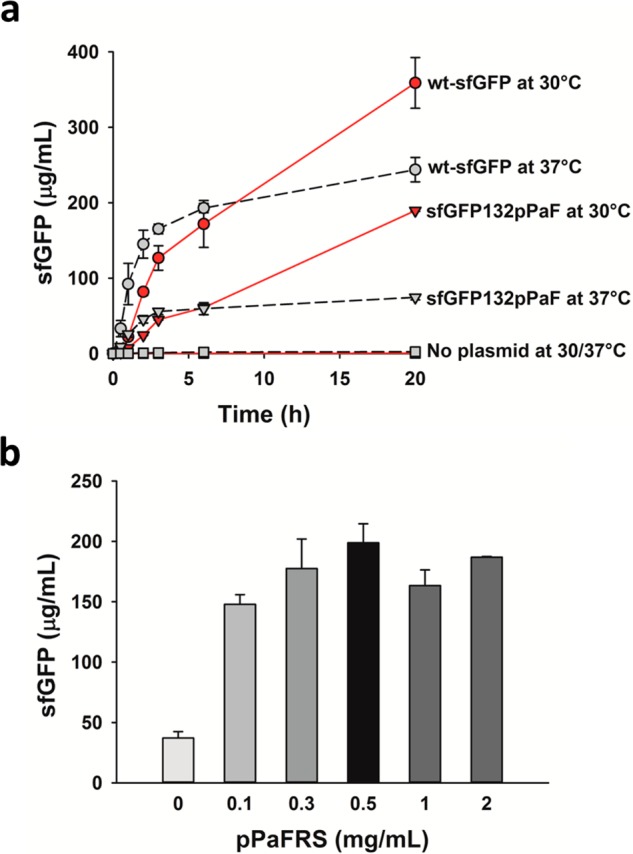
Optimization of pPaF incorporation. (a) Time course sfGFP synthesis in extracts derived from rEc.E13.ΔprfA illustrates the effect of temperature on CFPS (pPaF = 2 mM, pPaFRS = 0.5 mg/mL, temperature = 30 and 37 °C for 20 h). Wild-type sfGFP (wt-sfGFP) was produced from pY71-sfGFP plasmid, modified sfGFP with pPaF at single amber site corresponding at E132 (sfGFP132pPaF) was produced from pY71-sfGFP-E132amb, and no plasmid was added as a control (no plasmid). (b) pPaF incorporation using pY71-sfGFP-E132amb plasmid with 0.1 to 2 mg/mL of pPaFRS at 2 mM pPaF. Each data point is the average of three independent reactions, and one standard deviation is shown.
We next compared sfGFP132pPaF synthesis in extracts from rEc.E13 with and without RF1 (ΔprfA) (Figure 4, Table 1). As a control, active wild-type sfGFP as assessed by fluorescence was synthesized at equivalent levels, indicating similar protein synthesis activity (Figure 4a). Strikingly, extracts from rEc.E13.ΔprfA synthesized active sfGFP132pPaF yields more than 2.5-fold from 71 ± 6 μg/mL to 190 ± 20 μg/mL when compared to RF1-present rEc.E13 extracts (suppression efficiency: 53% in the RF1-deficient vs 21% in the RF1-present) (Figure 4a, Table 1). This result highlights significant benefits for using RF1-deficient strains. Upon closer examination, we observed that much of this benefit is due to an increase in full-length modified sfGFP synthesis. Specifically, introduction of 14C-leucine into the CFPS reaction allowed us to use autoradiograms to show that the ratio of full-length to truncated form of sfGFP132pPaF in the absence of RF1 was ∼8:2 (Figure 4b, Table 1), whereas the ratio in the presence of RF1 was ∼2:8 (Figure 4b, Table 1). As a control, only full-length wild-type sfGFP was synthesized regardless of the presence or absence of RF1 (Figure 4b). Total protein and soluble sfGFP synthesis yields quantified using 14C-leucine incorporation into trichloroacetic acid-precipitable radioactivity were consistent with the above analysis. When RF1 is present during protein biosynthesis, the o-tRNA competes with RF14 and in turn reduces the incorporation efficiency of NSAA. This results in the major portion of synthesized proteins being truncated (Figure 4b, Table 1). However, when RF1 is absent, the o-tRNA must only compete against natural suppression mechanisms,34 and thereby the majority of protein produced with pPaF is full-length (Figure 4b, Table 1). Hence, our RF1-deficient CFPS system enhances incorporation of NSAA into a protein.
Figure 4.

Effect of RF1 deletion of production of sfGFP with single site pPaF incorporation. (a) sfGFP synthesis in the presence or absence of RF1 in cell-extract based CFPS system at 30 °C for 20 h. Wild-type sfGFP (wt-sfGFP) was produced from pY71-sfGFP plasmid, modified sfGFP with pPaF (sfGFP132pPaF) was produced from pY71-sfGFP-E132amb with 2 mM pPaF and 0.5 mg/mL pPaFRS, and no pPaFRS (sfGFP132amb/no pPaFRS) or pPaF (sfGFP132amb/no pPaF) was added. At least three independent reactions were performed for each sample, and one standard deviation is shown. (b) Autoradiogram of 14C-leucine labeled proteins. FL indicates full-length sfGFP (27.1 kDa), and TR indicates truncated sfGFP at E132 (14.8 kDa).
Table 1. Yield of Native and Modified Proteinsa.
| protein | total protein (μg/mL)b | soluble protein (μg/mL)b | active protein (μg/mL)c | suppression efficiency (%)d | full-length protein (%)e | truncated protein (%)e |
|---|---|---|---|---|---|---|
| RF1+ | ||||||
| wt-sfGFP | 403 ± 2 | 349 ± 9 | 360 ± 40 | 100 | 0 | |
| sfGFP132pPaF | 330 ± 20 | 236 ± 8 | 71 ± 6 | 21 | 20 | 80 |
| sfGFP132amb/no pPaFRS | 330 ± 10 | 220 ± 20 | 1.8 ± 0.4 | 1 | 1 | 99 |
| RF1– | ||||||
| wt-sfGFP | 348 ± 5 | 320 ± 10 | 360 ± 30 | 100 | 0 | |
| sfGFP132pPaF | 282 ± 3 | 270 ± 30 | 190 ± 20 | 53 | 78 | 22 |
| sfGFP132amb/no pPaFRS | 252 ± 6 | 220 ± 4 | 37 ± 5 | 11 | 32 | 68 |
The extract-based CFPS reaction was performed at 30 °C for 20 h.
Total and soluble protein yields were calculated from liquid scintillation counting of TCA precipitated radioactive 14C-labeled protein. Soluble proteins were obtained from the supernatant of the CFPS samples after centrifugation.
Active protein yield was calculated by measuring fluorescence.
Suppression efficiency is the ratio of the active modified protein yields to the active native protein.
Percentages of full-length and truncated proteins were determined by the densitometry of protein bands in autoradiogram.
RF1-Deficient CFPS System Increases pPaF Incorporation at Multiple Sites
We next assessed the impact of RF1-deficient extracts on the incorporation of pPaF at multiple different sites in sfGFP. For these experiments, we built sfGFP constructs with 1, 2, 5, and 10 amber sites in loop regions (Figure 5a), so as to avoid deleteriously impacting fluorescence.13 Then, we compared CFPS yields in the presence or absence of RF1 as determined by fluorescence. Consistent with in vivo NSAA incorporation data,13 we observed that increasing the number of amber incorporation sites decreased the sfGFP yield, regardless of the presence or absence of RF1 (Figure 5b). Yields of sfGFP with E132, N212, or T216 replaced by pPaF (single site NSAA incorporation) were two to 6-fold higher in the absence of RF1 (Figure 5b). In addition, multiple site incorporations were much more efficient in the absence of RF1; we observed a 10-fold increase in the modified protein yield with two incorporation sites and a 12-fold increase with five incorporation sites (Figure 5b). For constructs with 10 TAG replacements, fluorescent protein was not detected (Figure 5b). Taken together, our results highlight the critical importance of RF1-deficient extracts for synthesizing proteins bearing NSAAs at single as well as multiple different sites.
Figure 5.

Multiple site pPaF incorporation is enabled by RF1 deletion extracts and improved by in situ o-tRNA synthesis. (a) pPaF incorporation sites are shown in sfGFP using Pymol. Single site incorporation was performed with amber codons at positions E132, N212, and T216, two site incorporation at N212 and T216, five site incorporation at D36, K101, E132, D190, and E213, and ten site incorporation at D36, K101, D102, E132, D133, K140, D190, V193, E213, and D218. (b) sfGFP synthesis with single and multiple site pPaF incorporation in the presence or absence of RF1 with or without optimized o-tRNA (o-tRNAopt) at 30 °C for 20 h with 2 mM pPaF and 0.5 mg/mL pPaFRS. (c) Linear DNA templates for producing different types of o-tRNAs under the control of T7-promoter. In order to create transzyme, hammerhead ribozyme sequence was added between T7 promoter and o-tRNA sequence. Sequence differences between o-tRNAopt and o-tRNA are shown in red nucleotides. (d) pPaF incorporation with different types of o-tRNAs (10 ng/μL) to synthesize sfGFP132pPaF using BL21(DE3) extract with 2 mM pPaF and 0.5 mg/mL pPaFRS. (e) Optimization of DNA concentration of transzyme o-tRNAopt in pPa incorporation using BL21(DE3) extract. At least three independent reactions were performed for each sample, and one standard deviation is shown for parts b, d, and e.
In situ Synthesis of Optimized o-tRNA Enhances pPaF Incorporation
Previously, it has been shown that o-tRNA concentration in CFPS is limiting for NSAA incorporation.24 To address this issue, Albayrak and Swartz recently developed a novel approach for in situ synthesis of the o-tRNA in the reaction, which significantly improves suppression efficiency.22 Their key advance was the direct addition of a transzyme construct to the reaction mixture. In this construct, the hammerhead ribozyme was fused to o-tRNA. Once synthesized, the transzyme cleaves itself liberating active o-tRNA (i.e., 5′–OH tRNA transcript starting with the proper nucleotide).35 To test the impact of the transzyme approach in our system, we constructed linear o-tRNA templates by PCR amplification with or without hammerhead ribozyme for the original o-tRNA24 and an optimized o-tRNA (o-tRNAopt) that was evolved to enhance the efficiency of NSAA incorporation in vivo(35,36) (Figure 5c). To protect possible degradation of linear DNA template for the o-tRNA, 500 bp of sequence upstream of the T7 promoter was added during amplification (Figure 5c). After generation of the PCR amplicons, we examined modified protein synthesis with pPaF by cotranscribing o-tRNA or o-tRNAopt and sfGFP from the linear DNA template and the plasmid, respectively.
To prove that the transzyme construct was functionally active, we first used BL21(DE3) cell extract that does not contain any o-tRNA. Transzymes of o-tRNA and o-tRNAopt enabled the synthesis of the modified protein, while nontranszymes did not make the modified protein (Figure 5d) due to a lack of active tRNA formation without correct cleavage after transcription.35 As a control, the modified protein was not produced when no DNA template for o-tRNA or o-tRNAopt was added (Figure 5d). In addition, o-tRNAopt resulted in more protein yield than the original o-tRNA (Figure 5d), which is consistent with a previous report36 and the fact that the o-tRNAopt is a better suppressor tRNA than the original o-tRNA when in competition with translation termination by RF1. We further showed that modified sfGFP yield could be increased by increasing the amount of the linear DNA encoding the transzyme o-tRNAopt up to 10 ng/μL (Figure 5e).
We then used transzyme o-tRNAopt in single and multiple site pPaF incorporation in the rEc.E13 extracts with or without RF1 to measure if extra o-tRNAopt increases the modified sfGFP yield. Indeed, the addition of 10 ng/μL o-tRNAopt DNA template to CFPS reactions increased pPaF incorporation into sfGFP in the presence of RF1, having a more pronounced effect on multiple site incorporation (Figure 5b). In RF1-deficient extracts, in situ o-tRNAopt synthesis resulted in a 2-fold higher yield in modified protein synthesis with two and five incorporation sites, although it did not show increased protein yield at single site incorporations in the absence of RF1 (Figure 5b). Our results suggest that o-tRNA concentrations are limiting in RF1-deficient extracts only for multisite incorporations. More broadly, our results highlight efficiency advantages with RF1-deficient extracts for incorporating multiple NSAAs into the same polypeptide. Another possibility is that there is competition with natural suppressors, which we discuss in detail below.
Quantitative Mass Spectrometry Data Discovers High Suppression Efficiencies for RF1-Deficient Extracts
While synthesis of sfGFP132pPaF is increased more than 2.5-fold in RF1-deficient extracts, we curiously observed a significant increase in the amount of active sfGFP synthesized in the absence of pPaFRS or pPaF in RF1-deficient extracts (Figure 4a). These data imply mis-incorporation in the absence of RF1 when components of the orthogonal translation system are not added, which has also been observed in vivo in an RF1-deletion strain.37 It is known that the ribosome can stall due to incomplete mRNA,38 rare codons,38 and at specific peptide sequences.39 To bypass this issue, natural suppressor mechanisms exist to rescue ribosomes34 (e.g., SsrA-mediated peptide tagging40) or noncognate tRNA interactions can install an incorrect amino acid, the latter of which we hypothesized was occurring in our system. This is because in the absence of both RF1 and a component of the orthogonal translation system (e.g., pPaF), the ribosome is expected to stall at in-frame amber codons. If this occurs, a canonical amino acid may be mis-incorporated to rescue the stalled ribosome. Previous studies reported that tyrosine,36,37 phenylalanine,36 tryptophan,37 and glutamine8,37,41 are misincorporated at amber codon. In addition, our previous in vivo work has shown direct genetic, Western blot, and mass spectrometry evidence for different types of natural mechanisms in the rEc.E13.ΔprfA strain.30
We therefore used top-down (TD) mass spectrometry (i.e., MS analysis of intact proteins) to detect and provide semiquantitative information on the incorporation of pPaF into sfGFP. In typical proteomic workflows involving enzymatic digestion, truncations and mis-incorporated amino acid residues are difficult to detect, since information on intact protein length and sequence is lost. The TD methodology used here not only provides confirmation of single amino acid incorporation but is also capable of identifying truncations and mis-incorporation events at the same time. Specifically, intact sfGFP generated by CFPS was analyzed by nanocapillary LC-MS without prior sample purification. We observed that, for the samples without pPaFRS, glutamic acid (E) was incorporated at amber codon corresponding to position at E132 (Supporting Information Figure S2a) and N212 of sfGFP (Supporting Information Figure S2b). The CFPS reaction contains more than 150 mM glutamate as it is used as the dominant anion in the system. Thus, it appears that this high concentration relative to the other amino acids (2 mM) as well as the fact that the near cognate GAG codon assigned to glutamic acid tRNAGlu is only a single bp different from the TAG amber codon allows for the natural suppression of the amber codon.
The above results could suggest that we additionally observe mis-incorporation when the entire orthogonal translation system is present when RF1 is absent. However, this is not the case. In the presence of all units of the orthogonal translation, NSAA incorporation is the major driving force of translation for amber codon suppression. Indeed, pPaF was correctly incorporated approximately 99% in sfGFP216pPaF (Figure 6a) and more than 90% of the time in sfGFP132pPaF (Supporting Information Figure S2a). Hence, the presence of all orthogonal translation components promotes site-specific NSAA incorporation by outcompeting natural suppression mechanisms. We also analyzed site-specific pPaF incorporation at two amber sites in sfGFP (sfGFP212pPaF216pPaF). We did not observe any mis-incorporation (Figure 6b).
Figure 6.

Mass spectrometry analysis of pPaF incorporation at single and multiple amber sites in sfGFP shows high NSAA incorporation in RF1 deletion extracts. (a) pPaF incorporation was examined to the single amber site corresponding to the position of T216, (b) the two amber site corresponding to the positions of N212 and T216, and (c) the five amber site corresponding to the positions of D36, K101, E132, D190, and E213. Theoretical modified protein peaks of multiple site pPaF incorporation are shown.
We further analyzed site-specific pPaF incorporation at five different amber sites per sfGFP (Figure 6c). Increasing the number of pPaF incorporation sites decreased active protein synthesis yields (Figure 5b). Likewise, correct pPaF incorporation efficiency to the multiple amber sites was decreased. There are 32 possible combinations with five incorporation sites in pPaF incorporation. We observed that less than 10% of the protein contained pPaF-incorporated at all five positions (D36, K101, E132, D190, and E213). The remaining 90% included various modified proteins with different numbers of pPaF incorporated (Figure 6c).
We next asked if we observed a positional dependence of nonsense suppression efficiency in extracts from the RF1-deletion strain. Previously it has been shown that the specific location of an amber codon in the gene sequence has an impact in total protein yield and suppression efficiency.22 Given the complexity of our data when trying to incorporate five NSAAs per sfGFP, we chose to focus on only single and double pPaF incorporation. When we altered the amber position, we observed a significant positional effect. For example, only half of the modified protein sfGFP212pPaF contained the correct pPaF incorporation, whereas the other half of the protein was mis-incorporation of E (Supporting Information Figure S2b). In the case of sfGFP132pPaF190pPaF, approximately 20% of the modified sfGFP comprised pPaF-incorporated at both sites, while 60% contained pPaF-incorporated at only one of two amber sites (data not shown). Taken together, our mass spectrometry data highlight that extracts from RF1 deletion extracts provide advantages for site-specific NSAA incorporation. However, further strategies will be necessary to avoid natural suppression efficiencies, with one obvious step being the completion of a completed recoded organism.30
pPaF Incorporation in PURE System
We next studied the effect of RF1 on pPaF incorporation in PURE translation system31 by adding 2 mM pPaF, 0.5 mg/mL pPaFRS, and 4.5 ng/μL linear DNA for o-tRNAopt. Corroborating the results in our extract-based system, the yield of the modified sfGFP with pPaF in the absence of RF1 was increased as compared to the system in the presence of RF1 in PURE CFPS (41 ± 3 μg/mL vs 19 ± 4 μg/mL, respectively). We subsequently confirmed by autoradiography that this was due to fewer truncated products in the absence of RF1 (Figure 7a). Furthermore, we tested the effect of RF1 by adding different amount of RF1 in PURE reaction. As the amount of RF1 is increased, production of full-length modified sfGFP132pPaF was decreased, and the truncated protein was increased (Figure 7b). Thus, the presence of RF1 inhibits NSAA incorporation in response to amber codon, and RF1 may be removed to improve protein synthesis yield containing NSAA. We directly compared our crude S30 extract system to the PURE translation system lacking RF1 in terms of the modified protein synthesis yield and the production cost. We observed that our S30 extract based approach produces about 5 times more protein (190 ± 20 μg/mL in extract based CFPS vs 41 ± 3 μg/mL in PURE) and is more cost-effective (less than $0.05/reaction in extract based CFPS vs more than $10.00/reaction in PURE) than the PURE translation system lacking RF1.
Figure 7.

pPaF incorporation in the PURE translation system. (a) Autoradiogram of radioactive labeled proteins. Wild-type sfGFP was produced from pY71-sfGFP plasmid, modified sfGFP was produced from pY71-sfGFP-E132amb with or without pPaFRS in the presence of pPaF. (b) Effect of RF1 concentration in pPaF incorporation. RF1 was diluted to 1/8, 1/4, and 1/2 of the amount that manufacturer suggested. FL indicates full-length sfGFP (27.1 kDa), and TR indicates truncated sfGFP at E132 (14.8 kDa).
pAcF Incorporation
To demonstrate the benefit of RF1-deficient extracts for CFPS more broadly, we extended our approach to another NSAA, namely pAcF. pAcF serves as a bio-orthogonal handle and has recently been incorporated into proteins and functionalized to engender more potent and stable human growth hormone7 and to improve the activity of antibody-drug conjugates.6 The suppressor o-tRNA used for pPaF incorporation is compatible to pAcF.36 Thus, we used same cell-extract with the addition of pAcF and pAcFRS to examine pAcF incorporation. Based on the final yield of modified sfGFP after a 20 h CFPS reaction, pAcF incorporation at a single site (sfGFP132pAcF) was about 20% higher in RF1-deficient extracts (Figure 8). While the overall % increase was lower than pPaF, our data consistently show that removing RF1 is beneficial for CFPS of proteins containing NSAAs.
Figure 8.

pAcF incorporation. sfGFP synthesis with pAcF (2 mM) and pAcFRS (0.5 mg/mL) in the presence or absence of RF1 in CFPS system at 30 °C for 20 h. Wild-type sfGFP (wt-sfGFP) was produced from pY71-sfGFP plasmid; modified sfGFP with pAcF (sfGFP132pAcF) was produced from pY71-sfGFP-E132amb; and no pAcFRS was added (sfGFP132amb/no pAcFRS). Three independent reactions were performed for each sample, and one standard deviation is shown.
Genetic code expansion is a promising means in protein engineering to develop new functional and structural biomolecules.5 In this study, we utilized CFPS to systematically examine NSAA incorporation in response to the amber codon in cell-free translation systems lacking RF1. We showed that omitting RF1 enhances NSAA incorporation at single site, as well as multiple different sites, in a single protein. Moreover, RF1 deletion enables a significant shift from truncated to full-length product, which provides advantages over previously reported systems. Furthermore, we were able to easily modulate components of the orthogonal translation system to levels that might otherwise be toxic to cells in order to maximize NSAA incorporation. The production of orthogonal components often causes impaired cellular fitness in vivo. For example, we observed impairment of cell growth upon maximizing pPaFRS or pAcFRS overproduction (Supporting Information Figure S3a and b). This fitness impairment can be avoided using CFPS technologies.23
Although we show that deleting RF1 is critical to enhance NSAA incorporation, the overall protein synthesis yields presented here are lower than those published recently by Albayrak and Swartz.22 We hypothesize that this is because the strain used in that study had been subjected to more than a decade of strain engineering efforts to stabilize reaction substrates. Indeed it is well-known that strain modification is a powerful tool for increasing CFPS yields.15 For example, stabilization of amino acid substrates by deleting genes related to substrate depletion,42 stabilization of DNA by deleting genes encoding endonuclease I,43 and reduction of protein degradation by disrupting proteases44 enabled high-yield protein synthesis in CFPS systems. In addition to eliminating deleterious pathways, activating beneficial pathways may improve protein production, as shown that adding elongation factors to CFPS reactions increased protein synthesis rates and yields.45 Because the parent rEc.E13.ΔprfA strain has not been previously optimized for CFPS, we expect that future efforts to design and construct synthetic genomes that upon cell lysis lead to improved extract performance will increase synthesis yields of proteins bearing NSAAs. For example, multiplex automated genome engineering (MAGE)46 could be used to make numerous genomic changes upregulating positive effectors and downregulating negative effectors both individually and in combinations insofar as these genomic changes do not have a deleterious effect on the growth of the organism.
Looking forward, additional strain engineering efforts could also be pursued to reduce and remove natural suppression mechanisms. As described above, in the absence of some orthogonal translation components, we observe glutamate incorporation at the amber codon in RF1 deficient extracts from a partially recoded strain (13 of 321 amber sites in the genome). We hypothesize this phenotype is caused by the fact that the strain lacking RF1 induces natural suppression mechanisms during cell growth prior to extract generation to help deal with ribosomal pausing at amber sites present in the genome. Replacing the remaining amber codons in the genomes (i.e., the fully recoded E. coli strain) should reduce natural suppression mechanisms, as was observed in vivo.30
In summary, we described a novel CFPS platform derived from the RF1-deficient strain rEc.E13.ΔprfA with advantages for improved amber suppression. While it is still early in the development of crude extract based CFPS system derived from RF1 deficient strains, our advances, along with the potential for future strain engineering, suggest promise for future efforts to generate sequence-defined proteins or polymers with many NSAAs at high yields and purity. Indeed, we anticipate that RF1 deficient chassis strains will achieve greater than 1g/L yields in the upcoming years, becoming a significant player on the stage with other CFPS technologies for synthesis of unnatural biopolymers with high efficiency and high fidelity.
Methods
Bacterial Strains and Plasmids
The bacterial strains and plasmids used in this study are listed in Table 2. We used rEc.E13 (a modified EcNR2 strain),30rEc.E13.ΔprfA,30 and BL21(DE3) strains for making S30 cell extract and BL21(DE3) for producing the orthogonal amino acyl tRNA synthetases. Chloramphenicol (25 μg/mL) was used for preculturing rEc.E13, spectinomycin (20 μg/mL) for preculturing prfA knockout mutant, and kanamycin (50 μg/mL), chloramphenicol (25 μg/mL), and tetracycline (20 μg/mL) for maintaining plasmids as necessary.
Table 2. Strains and Plasmids Used in This Studya.
| strains and plasmids | genotype/relevant characteristics | source |
|---|---|---|
| Strains | ||
| rEc.E13 | EcNR246 (CmR, ApR) with 13 TAG termination reassignments to TAA at coaD, hda, hemA, mreC, murF, lolA, lpxK, yafF, pfpA, sucB, fabH, fliN, and atpE | (30) |
| rEc.E13.ΔprfA | rEc.E13 ΔprfA Ω SpR | (30) |
| BL21(DE3) | fhuA2 [lon] ompT gal (λ DE3) [dcm] ΔhsdS λ DE3 = λ sBamHIo ΔEcoRI-B int::(lacI::PlacUV5::T7 gene1) i21 Δnin5 | New England Biolabs |
| Plasmids | ||
| pY71-sfGFP | KmR, PT7::sfGFP, C-terminal Strep-tag | (24) |
| pY71-sfGFP-E132amb | pY71-sfGFP with amber codon at E132 | this study |
| pY71-sfGFP-N212amb | pY71-sfGFP with amber codon at N212 | this study |
| pY71-sfGFP-T216amb | pY71-sfGFP with amber codon at T216 | (24) |
| pY71-sfGFP-2amb | pY71-sfGFP with amber codon at N212 and T216 | this study |
| pY71-sfGFP-5amb | pY71-sfGFP with amber codon at D36, K101, E132, D190, and E213 | this study |
| pY71-sfGFP-10amb | pY71-sfGFP with amber codon at D36, K101, D102, E132, D133, K140, D190, V193, E213, and D218 | this study |
| pY71-pPaFRS | KmR, PT7::pPaFRS, C-terminal 6x histidine tag | (24) |
| pEVOL-pAcF | CmR, PglnS::pAcFRS, ParaBAD::pAcFRS, PproK::o-tRNAopt | (36) |
| pY71-pAcFRS | KmR, PT7::pAcFRS, C-terminal 6x histidine tag | this study |
| pDULE-o-tRNA | TetR, Plpp::o-tRNA | (24) |
| pY71-T7-tz-o-tRNA | KmR, PT7:: transzyme o-tRNA | this study |
| pY71-T7-tz-o-tRNAopt | KmR, PT7:: transzyme o-tRNAopt | this study |
| pY71-T7-o-tRNA | KmR, PT7::o-tRNA | this study |
| pY71-T7-o-tRNAopt | KmR, PT7::o-tRNAopt | this study |
ApR, CmR, KmR, SpR, and TetR are ampicillin, chloramphenicol, kanamycin, spectinomycin, and tetracycline resistance, respectively.
Growth Measurement
Overnight cultures of rEc.E13 and rEc.E13.ΔprfA strains grown in Luria–Bertani (LB)47 at 250 rpm at 34 °C were diluted to an OD of 0.05 at 600 nm in LB and 2xYTPG media (16 g/L tryptone, 10 g/L yeast extract, 5 g/L NaCl, 7 g/L K2HPO4, 3 g/L KH2PO4, and 18 g/L glucose; adjusted pH to 7.1 with KOH). 100 μL of the diluted cultures was added in 96-well polystyrene plates (Costar 3370; Corning Incorporated, Corning, NY). OD600 was measured at 15 min intervals for 15 h at 34 °C with fast shaking mode on a Synergy2 plate reader (Biotek, Winooski, VT). Growth data of each strain was obtained from ten replicate wells with two independent cultures. BL21(DE3) strains containing pY71-pPaFRS or pY71-pAcFRS were incubated at 37 °C in LB with different concentrations (0 mM, 0.01 mM, 0.1 mM, or 1 mM) of isopropyl-β-D-thiogalactopyranoside (IPTG) (Sigma, St. Louis, MO) in 96 well plates. Growth data of each strain was obtained from six replicate wells with three independent cultures.
Cell Extract Preparation
E. coli cell extracts were prepared from rEc.E13 and rEc.E13.ΔprfA strains containing pDULE-o-tRNA. Cells were grown in a BIOSTAT C-plus fermentor (Sartorious AG, Goettingen, Germany) to OD at 600 nm of 3.0 in 10 L of 2xYTPG media at 34 °C. Cells were pelleted by centrifuging for 15 min at 6000 × g at 4 °C, washed twice with cold S30 buffer (10 mM tris-acetate pH 8.2, 14 mM magnesium acetate, 60 mM potassium acetate, 1 mM dithiothreitol),48 and stored at −80 °C. Thawed cells were suspended in 1 mL of S30 buffer per gram cells and lysed in EmulsiFlex-C3 homogenizer (Avestin, Manheim, Germany) with a single pass at pressure of 20 000 to 25 000 psi. Cell debris and insoluble components were removed by two rounds of centrifugation for 30 min at 30 000 × g at 4 °C. The clarified samples were incubated for 80 min at 120 rpm at 37 °C to optimize the extract activity and centrifuged for 15 min at 15 000 × g at 4 °C. The supernatant was flash-frozen using liquid nitrogen and stored at −80 °C until use. Total protein concentration of the extracts was approximately 50 mg/mL, as measured by Quick-Start Bradford protein assay kits (Bio-Rad, Hercules, CA).
Preparation of NSAAs
pPaF and pAcF were used to examine NSAA incorporation. pPaF was synthesized as described previously,49 and pAcF was purchased through Chem-Impex International (PN 24756; Wood Dale, IL). Stock solutions (500 mM) of pPaF and pAcF in 0.5 N NaOH were prepared. Working concentration of pPaF or pAcF was 2 mM in CFPS reaction. Adding the same volume of 0.5 N NaOH to CFPS reactions did not affect protein synthesis efficiency.
Plasmid Construction
Amber codons were introduced at the corresponding positions of E132, D190, N212 in sfGFP using inverse PCR with TAG insertion primers (Supporting Information Table S1) on pY71-sfGFP plasmid. PCR was performed using Phusion High-Fidelity DNA polymerase (New England Biolabs, Ipswich, MA) at 98 °C for 30 s, with 30 cycles of 98 °C for 10 s, 60 °C for 30 s, and 72 °C for 3 min, and a final extension of 72 °C for 5 min followed by ligation and DpnI digestion. The constructed plasmids were electroporated in to BL21(DE3) competent cells. In order to introduce two TAGs in sfGFP, an amber codon was added at the codon corresponding to N212 on pY71-sfGFP-T216amb, resulting in pY71-sfGFP-2amb. sfGFP with five amber sites (corresponding to positions D36, K101, E132, D190, and E213) or ten amber sites (corresponding to positions D36, K101, D102, E132, D133, K140, D190, V193, E213, and D218) was synthesized through GenScript (Piscataway, NJ) and cloned into pY71 vector using NdeI and SalI restriction sites to construct pY71-sfGFP-5amb and pY71-sfGFP-10amb. pAcFRS gene was amplified from pEVOL-pAcF using pAcF-NdeI-f and pAcF-SalI-r primers (Supporting Information Table S1) and cloned into pY71 vector using NdeI and SalI sites to construct pY71-pAcFRS. All constructs were confirmed by DNA sequencing using pY71-f and pY71-r primers (Supporting Information Table S1).
o-tRNA Synthetase Purification
o-tRNA synthetases were purified as described previously23 with modifications. BL21(DE3) harboring pY71-pPaFRS or pY71-pAcFRS was grown in 1 L LB to OD at 600 nm of 1.0 at 250 rpm and 37 °C. pPaFRS and pAcFRS were produced by adding 0.2 mM IPTG for 3 h. Cells were harvested at 5000 × g for 30 min at 4 °C, washed with S30 buffer, and stored at −80 °C. The frozen cell pellet was thawed in loading buffer (300 mM NaCl, 10 mM imidazole, 50 mM NaH2PO4, 5 mM Tris–HCl, pH 8.0) and lysed using a homogenizer at 20 000–25 000 psi. After clarification by centrifuging at 16 000 × g at 4 °C for 30 min, the supernatant was loaded into a 1 mL Ni-NTA column using a BioLogic DuoFlow FPLC system (Bio-Rad, Hercules, CA). The column was washed with wash buffer (300 mM NaCl, 50 mM imidazole, 50 mM NaH2PO4, 5 mM Tris–HCl, pH 8.0), and His-tagged proteins were eluted with 250 mM imidazole in wash buffer. The eluent was dialyzed three times for 2 h each using 6000–8000 MWCO dialysis tubing (Spectrum, New Brunswick, NJ) with 10 volumes of phosphate buffer (pH 7.4) for pPaFRS23 and the aaRS-buffer (20 mM Tris-HCl pH 8.0, 150 mM KCl, 15 mM MgCl2, and 5 mM β-mercaptoethanol) for pAcFRS.50 The proteins were concentrated using Amicon Ultracel YM-3 centrifugal filter (Millipore, Billerica, MA). Protein purity was confirmed by 4–12% NuPAGE SDS-PAGE (Life Technologies, Grand Island, NY). Concentrations were determined by Quick-Start Bradford protein assay kit (Bio-Rad, Hercules, CA).
CFPS Reaction
CFPS reactions were performed testing for incorporation of pPaF and pAcF using a modified PANOx-SP system.17 Briefly, 15 uL of CFPS reaction in a 1.5 mL microcentrifuge tube was prepared by mixing the following components: 1.2 mM ATP; 0.85 mM each of GTP, UTP, and CTP; 34.0 μg/mL folinic acid; 170.0 μg/mL of E. coli tRNA mixture; 13.3 μg/mL plasmid; 100 μg/mL T7 RNA polymerase; 2 mM each of 20 standard amino acids; 0.33 mM nicotinamide adenine dinucleotide (NAD); 0.27 mM coenzyme-A (CoA); 1.5 mM spermidine; 1 mM putrescine; 4 mM sodium oxalate; 130 mM potassium glutamate; 10 mM ammonium glutamate; 12 mM magnesium glutamate; 33 mM phosphoenolpyruvate (PEP); 2 mM pPaF or pAcF; 0.1 to 2 mg/mL pPaFRS or pAcFRS and 27% v/v of cell extract. The sample was incubated for 20 h at 30 °C unless noted otherwise.
Quantification of the Synthesized sfGFP
Total and soluble protein yields were quantified by determining radioactive 14C-Leu incorporation using trichloroacetic acid (TCA).48 Radioactivity of TCA-precipitated samples was measured using liquid scintillation counting (MicroBeta2, PerkinElmer, Waltham, MA). Active sfGFP protein yields were quantified by measuring fluorescence. Two microliters of CFPS reaction was added in the middle of the flat bottom of 96-well half area black plates (Costar 3694; Corning Incorporated, Corning, NY). sfGFP was excited at 485 nm while measuring emission at 528 nm with a 510 nm cutoff filter. The fluorescence of sfGFP was converted to concentration (μg/mL) according to a standard curve (Supporting Information Figure S4).
Autoradiography Analysis
Radioactive 35S-Met was added in CFPS reactions. 2.5 μL of each reaction was loaded on 4–12% NuPAGE SDS-PAGE gel after denaturing the sample. The gel was soaked in Gel Drying Solution (Bio-Rad, Hercules, CA) for 30 min, fixed with cellophane films, dried applying heat for 1 h in GelAir Dryer (Bio-Rad, Hercules, CA), and exposed overnight on Storage Phosphor Screen (GE Healthcare Biosciences, Pittsburgh, PA). Autoradiogram was scanned using Storm Imager (GE Healthcare Biosciences, Pittsburgh, PA) and analyzed using Quantity One software (Bio-Rad, Hercules, CA).
Construction of Linear DNA Templates for Expressing o-tRNA
DNA oligomers of o-tRNA and optimized o-tRNA bearing a T7 promoter with or without hammerhead ribozyme (Figure 5c), were synthesized via gBlocks (Integrated DNA technologies, Coralville, IA). DNA sequences of o-tRNA and optimized o-tRNA (Figure 5c) were obtained from plasmid pDULE-o-tRNA24 and pEVOL-pAcF,35 respectively. The hammerhead ribozyme sequence (Figure 5c) was added between the T7-promoter and o-tRNA sequence.22 The gBlock fragments were amplified using T7-PCRamp-f and T7-PCRamp-r primers (Supporting Information Table S1), cloned into pY71 plasmid using BglII and SalI restriction sites, and confirmed by DNA sequencing using tRNA-seq-f and tRNA-seq-r primers (Supporting Information Table S1). Linear DNA templates of original or optimized o-tRNA were generated by PCR amplification using T7tRNA500-f and T7tRNA-r or T7tRNAopt-r primers (Supporting Information Table S1).
CFPS Using PURE System
NSAA incorporation was examined in the PURE system using PURExpress ΔRF123 Kit (New England Biolabs, Ipswich, MA). The reaction components of PURExpress with and without RF1 were mixed with 5 μg/mL sfGFP plasmids, 2 mM pPaF, 0.5 mg/mL pPaFRS, and 4.5 ng/μL linear DNA template of optimized o-tRNA in a 10 μL reaction. The reaction was incubated for 20 h at 30 °C.
Full-length sfGFP Purification
To confirm pPaF incorporation at corresponding amber codon positions, mass spectrometry analysis was performed with purified sfGFP with NSAA incorporated. Full-length sfGFP was purified using C-terminal strep-tags and 0.2 mL gravity-flow Strep-Tactin Sepharose mini-columns (IBA GmbH, Gottingen, Germany). Eluted protein samples were concentrated using Microcon centrifugal filter columns YM-10 (Millipore, Billerica, MA).
Mass Spectrometry
The semipurified protein was analyzed by nanocapillary LC-MS using a 100 mm × 75 μm ID PLRP-S column in-line with an Orbitrap Elite (ThermoFisher, Waltham, MA). All MS methods included the following events: (1) FT scan, m/z 400–2000, 120 000 resolving power and (2) data-dependent MS/MS on the top 2 peaks in each spectrum from scan event 1 using higher-energy collisional dissociation (HCD) with normalized collision energy of 25, isolation width 50 m/z, and detection of ions with resolving power of 60 000. All data were analyzed using QualBrowser, part of the Xcalibur software packaged with the ThermoFisher Orbitrap Elite.
Acknowledgments
This work was supported by the Office of Naval Research (N00014-11-1-0363), the Defense Advanced Research Projects Agency (N66001-12-C-4211), the David and Lucile Packard Foundation (M.C.J.), the National Institutes of Health (GM 067193 and NIH-MSTP-TG-T32GM07205), the Chicago Biomedical Consortium with support from the Searle Funds at the Chicago Community Trust, and the Arnold and Mabel Beckman Foundation (F.J.I.). We thank Prof. Bradley Bundy and Prof. James R. Swartz for providing the pY71 plasmid, Prof. Peter G. Schultz for providing the pEVOL plasmid, Corinna Tuckey from New England Biolabs for providing PURE system kits, Dr. Xinhao Ye for the help with synthetase purification, and Dr. D. Calvin Harris for synthesizing pPaF.
Supporting Information Available
This information is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
S.H.H. and M.C.J. designed the experiments, S.H.H. performed the experiments, A.D.H. and F.J.I. constructed the strains, and I.N. and N.L.K. performed mass spectrometry and analyzed MS data. S.H.H. and M.C.J. wrote the manuscript. All authors discussed the results and commented on the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Harris D. C.; Jewett M. C. (2012) Cell-free biology: Exploiting the interface between synthetic biology and synthetic chemistry. Curr. Opin. Biotechnol. 23, 672–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sismour A. M.; Benner S. A. (2005) Synthetic biology. Expert Opin. Biol. Ther. 5, 1409–1414. [DOI] [PubMed] [Google Scholar]
- Kim C. H.; Axup J. Y.; Schultz P. G. (2013) Protein conjugation with genetically encoded unnatural amino acids. Curr. Opin. Chem. Biol. 17, 412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoesl M. G.; Budisa N. (2012) Recent advances in genetic code engineering in Escherichia coli. Curr. Opin. Biotechnol. 751–757. [DOI] [PubMed] [Google Scholar]
- Liu C. C.; Schultz P. G. (2010) Adding new chemistries to the genetic code. Annu. Rev. Biochem. 79, 413–444. [DOI] [PubMed] [Google Scholar]
- Axup J. Y.; Bajjuri K. M.; Ritland M.; Hutchins B. M.; Kim C. H.; Kazane S. A.; Halder R.; Forsyth J. S.; Santidrian A. F.; Stafin K.; Lu Y.; Tran H.; Seller A. J.; Biroc S. L.; Szydlik A.; Pinkstaff J. K.; Tian F.; Sinha S. C.; Felding-Habermann B.; Smider V. V.; Schultz P. G. (2012) Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. U.S.A. 109, 16101–16106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H.; Daniel T.; Buechler Y. J.; Litzinger D. C.; Maio Z.; Putnam A.-M. H.; Kraynov V. S.; Sim B.-C.; Bussell S.; Javahishvili T.; Kaphle S.; Viramontes G.; Ong M.; Chu S.; GC B.; Lieu R.; Knudsen N.; Castiglioni P.; Norman T. C.; Axelrod D. W.; Hoffman A. R.; Schultz P. G.; DiMarchi R. D.; Kimmel B. E. (2011) Optimized clinical performance of growth hormone with an expanded genetic code. Proc. Natl. Acad. Sci. U.S.A. 108, 9060–9065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H.-S.; Hohn M. J.; Umehara T.; Guo L.-T.; Osborne E. M.; Benner J.; Noren C. J.; Rinehart J.; Söll D. (2011) Expanding the genetic code of Escherichia coli with phosphoserine. Science 333, 1151–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y.; Bröcker M. J.; Sekine S.-i.; Hammond G.; Suetsugu S.; Söll D.; Yokoyama S. (2013) Decameric SelA•tRNASec ring structure reveals mechanism of bacterial selenocysteine formation. Science 340, 75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K.; Neumann H.; Peak-Chew S. Y.; Chin J. W. (2007) Evolved orthogonal ribosomes enhance the efficiency of synthetic genetic code expansion. Nat. Biotechnol. 25, 770–777. [DOI] [PubMed] [Google Scholar]
- Neumann H.; Wang K.; Davis L.; Garcia-Alai M.; Chin J. W. (2010) Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature 464, 441–444. [DOI] [PubMed] [Google Scholar]
- Polycarpo C. R.; Herring S.; Bérubé A.; Wood J. L.; Söll D.; Ambrogelly A. (2006) Pyrrolysine analogues as substrates for pyrrolysyl-tRNA synthetase. FEBS Lett. 580, 6695–6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D. B. F.; Xu J.; Shen Z.; Takimoto J. K.; Schultz M. D.; Schmitz R. J.; Xiang Z.; Ecker J. R.; Briggs S. P.; Wang L. (2011) RF1 knockout allows ribosomal incorporation of unnatural amino acids at multiple sites. Nat. Chem. Biol. 7, 779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai T.; Hayashi A.; Iraha F.; Sato A.; Ohtake K.; Yokoyama S.; Sakamoto K. (2010) Codon reassignment in the Escherichia coli genetic code. Nucleic Acids Res. 38, 8188–8195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson E. D.; Gan R.; Hodgman C. E.; Jewett M. C. (2012) Cell-free protein synthesis: Applications come of age. Biotechnol. Adv. 30, 1185–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz J. R. (2012) Transforming biochemical engineering with cell-free biology. AIChE J. 58, 5–13. [Google Scholar]
- Jewett M. C.; Swartz J. R. (2004) Mimicking the Escherichia coli cytoplasmic environment activates long-lived and efficient cell-free protein synthesis. Biotechnol. Bioeng. 86, 19–26. [DOI] [PubMed] [Google Scholar]
- Jewett M. C.; Calhoun K. A.; Voloshin A.; Wuu J. J.; Swartz J. R. (2008) An integrated cell-free metabolic platform for protein production and synthetic biology. Mol. Syst. Biol. 4, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawada J. F.; Yin G.; Steiner A. R.; Yang J.; Naresh A.; Roy S. M.; Gold D. S.; Heinsohn H. G.; Murray C. J. (2011) Microscale to manufacturing scale-up of cell-free cytokine production—A new approach for shortening protein production development timelines. Biotechnol. Bioeng. 108, 1570–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz J. (2006) Developing cell-free biology for industrial applications. J. Ind. Microbiol. Biotechnol. 33, 476–485. [DOI] [PubMed] [Google Scholar]
- Hodgman C. E.; Jewett M. C. (2012) Cell-free synthetic biology: Thinking outside the cell. Metab. Eng. 14, 261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albayrak C.; Swartz J. R. (2013) Cell-free co-production of an orthogonal transfer RNA activates efficient site-specific non-natural amino acid incorporation. Nucleic Acids Res. 41, 5949–5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goerke A. R.; Swartz J. R. (2009) High-level cell-free synthesis yields of proteins containing site-specific non-natural amino acids. Biotechnol. Bioeng. 102, 400–416. [DOI] [PubMed] [Google Scholar]
- Bundy B. C.; Swartz J. R. (2010) Site-specific incorporation of p-propargyloxyphenylalanine in a cell-free environment for direct protein–protein click conjugation. Bioconjug. Chem. 21, 255–263. [DOI] [PubMed] [Google Scholar]
- Ugwumba I. N.; Ozawa K.; Cruz L. d. l.; Xu Z.-Q.; Herlt A. J.; Hadler K. S.; Coppin C.; Brown S. E.; Schenk G.; Oakeshott J. G.; Otting G. (2011) Using a genetically encoded fluorescent amino acid as a site-specific probe to detect binding of low-molecular-weight compounds. Assay Drug Dev. Technol. 9, 50–57. [DOI] [PubMed] [Google Scholar]
- Ugwumba I. N.; Ozawa K.; Xu Z.-Q.; Ely F.; Foo J.-L.; Herlt A. J.; Coppin C.; Brown S.; Taylor M. C.; Ollis D. L.; Mander L. N.; Schenk G.; Dixon N. E.; Otting G.; Oakeshott J. G.; Jackson C. J. (2010) Improving a natural enzyme activity through incorporation of unnatural amino acids. J. Am. Chem. Soc. 133, 326–333. [DOI] [PubMed] [Google Scholar]
- Whittaker J. W. (2013) Cell-free protein synthesis: the state of the art. Biotechnol. Lett. 35, 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscha K. V.; Herlt A. J.; Qi R.; Huber T.; Ozawa K.; Otting G. (2012) Multiple-site labeling of proteins with unnatural amino acids. Angew. Chem., Int. Ed. Engl. 51, 2243–2246. [DOI] [PubMed] [Google Scholar]
- Mukai T.; Yanagisawa T.; Ohtake K.; Wakamori M.; Adachi J.; Hino N.; Sato A.; Kobayashi T.; Hayashi A.; Shirouzu M.; Umehara T.; Yokoyama S.; Sakamoto K. (2011) Genetic-code evolution for protein synthesis with non-natural amino acids. Biochem. Biophys. Res. Commun. 411, 757–761. [DOI] [PubMed] [Google Scholar]
- Lajoie M. J.; Rovner A. J.; Goodman D. B.; Aerni H.-R.; Haimovich A. D.; Kuznetsov G.; Mercer J. A.; Wang H. H.; Carr P. A.; Mosberg J. A.; Rohland N.; Schultz P. G.; Jacobson J. M.; Rinehart J.; Church G. M.; Isaacs F. J. (2013) Genomically recoded organisms expand biological functions. Science 342, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y.; Inoue A.; Tomari Y.; Suzuki T.; Yokogawa T.; Nishikawa K.; Ueda T. (2001) Cell-free translation reconstituted with purified components. Nat. Biotechnol. 19, 751–755. [DOI] [PubMed] [Google Scholar]
- Heinemann I. U.; Rovner A. J.; Aerni H. R.; Rogulina S.; Cheng L.; Olds W.; Fischer J. T.; Söll D.; Isaacs F. J.; Rinehart J. (2012) Enhanced phosphoserine insertion during Escherichia coli protein synthesis via partial UAG codon reassignment and release factor 1 deletion. FEBS Lett. 586 3716–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett M. C.; Fritz B. R.; Timmerman L. E.; Church G. M. (2013) In vitro integration of ribosomal RNA synthesis, ribosome assembly, and translation. Mol. Syst. Biol. 9, 678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiler K. C. (2008) Biology of trans-translation. Annu. Rev. Microbiol. 62, 133–151. [DOI] [PubMed] [Google Scholar]
- Fechter P.; Rudinger J.; Giegé R.; Théobald-Dietrich A. (1998) Ribozyme processed tRNA transcripts with unfriendly internal promoter for T7 RNA polymerase: Production and activity. FEBS Lett. 436, 99–103. [DOI] [PubMed] [Google Scholar]
- Young T. S.; Ahmad I.; Yin J. A.; Schultz P. G. (2010) An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol. 395, 361–374. [DOI] [PubMed] [Google Scholar]
- Johnson D. B. F.; Wang C.; Xu J.; Schultz M. D.; Schmitz R. J.; Ecker J. R.; Wang L. (2012) Release factor one is nonessential in Escherichia coli. ACS Chem. Biol. 7, 1337–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes C. S.; Bose B.; Sauer R. T. (2002) Proline residues at the C terminus of nascent chains induce SsrA tagging during translation termination. J. Biol. Chem. 277, 33825–33832. [DOI] [PubMed] [Google Scholar]
- Nakatogawa H.; Ito K. (2002) The ribosomal exit tunnel functions as a discriminating gate. Cell 108, 629–636. [DOI] [PubMed] [Google Scholar]
- Roche E. D.; Sauer R. T. (1999) SsrA-mediated peptide tagging caused by rare codons and tRNA scarcity. EMBO J. 18, 4579–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa A.; Hayami M.; Sando S.; Aoyama Y. (2012) A concept for selection of codon-suppressor tRNAs based on read-through ribosome display in an in vitro compartmentalized cell-free translation system. J. Nucleic Acids 2012, 538129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel-Reydellet N.; Calhoun K.; Swartz J. (2004) Amino acid stabilization for cell-free protein synthesis by modification of the Escherichia coli genome. Metab. Eng. 6, 197–203. [DOI] [PubMed] [Google Scholar]
- Michel-Reydllet N.; Woodrow K.; Swartz J. (2005) Increasing PCR fragment stability and protein yields in a cell-free system with genetically modified Escherichia coli extracts. J. Mol. Microbiol. Biotechnol. 9, 26–34. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Oohira K.; Iwasaki Y.; Nakano H.; Ichihara S.; Yamane T. (2002) Reduction of protein degradation by use of protease-deficient mutants in cell-free protein synthesis system of Escherichia coli. J. Biosci. Bioeng. 93, 151–156. [DOI] [PubMed] [Google Scholar]
- Underwood K. A.; Swartz J. R.; Puglisi J. D. (2005) Quantitative polysome analysis identifies limitations in bacterial cell-free protein synthesis. Biotechnol. Bioeng. 91, 425–435. [DOI] [PubMed] [Google Scholar]
- Wang H. H.; Isaacs F. J.; Carr P. A.; Sun Z. Z.; Xu G.; Forest C. R.; Church G. M. (2009) Programming cells by multiplex genome engineering and accelerated evolution. Nature 460, 894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J. and Russell D. W. (2001) Molecular Cloning, A Laboratory Manual, 3rd ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Swartz J. R., Jewett M. C., and Woodrow K. A. (2004) Cell-free protein synthesis with prokaryotic combined transcription-translation In Recombinant Gene Expression (Balbás P., and Lorence A., Eds.), pp 169–182, Humana Press. [DOI] [PubMed] [Google Scholar]
- Deiters A.; Cropp T. A.; Mukherji M.; Chin J. W.; Anderson J. C.; Schultz P. G. (2003) Adding amino acids with novel reactivity to the genetic code of Saccharomyces cerevisiae. J. Am. Chem. Soc. 125, 11782–11783. [DOI] [PubMed] [Google Scholar]
- Nehring S.; Budisa N.; Wiltschi B. (2012) Performance analysis of orthogonal pairs designed for an expanded eukaryotic genetic code. PLoS ONE 7, e31992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.