

Abstract
Wound healing, angiogenesis and hair follicle maintenance are often impaired in the skin of diabetic patients, but the pathogenesis has not been well understood. Here, we report that circulation levels of kallistatin, a member of the serine proteinase inhibitor (SERPIN) superfamily with anti-angiogenic activities, were elevated in Type 2 diabetic patients with diabetic vascular complications. To test the hypothesis that elevated kallistatin levels could contribute to a wound healing deficiency via inhibition of Wnt/β-catenin signaling, we generated kallistatin-transgenic (KS-TG) mice. KS-TG mice had reduced cutaneous hair follicle density, microvascular density, and panniculus adiposus layer thickness as well as altered skin microvascular hemodynamics and delayed cutaneous wound healing. Using Wnt reporter mice, our results showed that Wnt/β-catenin signaling is suppressed in dermal endothelium and hair follicles in KS-TG mice. Lithium, a known activator of β-catenin via inhibition of glycogen synthase kinase-3β, reversed the inhibition of Wnt/β-catenin signaling by kallistatin and rescued the wound healing deficiency in KS-TG mice. These observations suggest that elevated circulating anti-angiogenic serpins in diabetic patients may contribute to impaired wound healing through inhibition of Wnt/β-catenin signaling. Activation of Wnt/β-catenin signaling, at a level downstream of Wnt receptors, may ameliorate the wound healing deficiency in diabetic patients.
INTRODUCTION
Globally, every 30 seconds, a limb is amputated due to pathologic complications associated with diabetes mellitus (Margolis et al., 2011; Rajamani et al., 2009; Tseng, 2006). There is a strong clinical need to identify biomarkers or therapeutic targets in the circulation and skin that modulate skin maintenance and repair in diabetes.
In humans, 20 extracellular serine proteinase inhibitors (serpins) comprise approximately 10 percent of proteins by mass in the human circulation (Goettig et al., 2010; Irving et al., 2000). Serpins α1-antitrypsin (SERPINA1), pigment epithelium-derived factor (PEDF, SERPINF1) and kallistatin (SERPINA4) have displayed anti-angiogenic activities (Dawson et al., 1999; McMahon et al., 2001; Miao et al., 2002). Recently, we have shown that kallistatin binds with low-density lipoprotein receptor-related protein 6 (LRP6), an essential co-receptor of the canonical Wnt pathway, and suppresses the activation of Wnt signaling by Wnt ligands (Liu et al., 2013). Here, we explore the concept that kallistatin regulates skin hair follicle development and wound healing through interactions with the canonical Wnt signaling pathway.
Canonical Wnt signaling in adult tissues up-regulates expression of direct T-cell factor/Lymphoid enhancer factor-1 (TCF/LEF-1) target genes that modulate hair follicle growth (DasGupta and Fuchs, 1999), cell proliferation (He et al., 1998) and angiogenesis (Zhang et al., 2001). Wnt ligands, such as Wnt3a, bind to a co-receptor complex consisting of frizzled (Fz) receptors and LRP6, causing phosphorylation of LRP6 and recruitment of a degradation complex consisting of casein kinase 1 (CK-1), glycogen synthase kinase-3β (GSK-3β) and adenopolyposis coli protein (APC). In the absence of Wnt ligand, this kinase complex phosphorylates β-catenin, leading to degradation of β-catenin in the cytoplasm (MacDonald et al., 2009). Phosphorylation and degradation of β-catenin is prevented when Wnt ligands activate the pathway, and the stabilized β-catenin translocates to the nucleus and dimerizes with the T-Cell Factor (TCF)-Groucho complex, activating transcription of corresponding direct target genes. Canonical Wnt signaling is crucial for development and plays key roles in cancer progression (MacDonald et al., 2009); yet its role in wound healing has only recently been studied (Fathke et al., 2006; Whyte et al., 2012; Whyte et al., 2013; Wu et al., 2011). Although Wnt signaling has been shown to promote angiogenesis (Barcelos et al., 2009; Chen et al., 2011; Chen et al., 2009; Dejana, 2010; Parmalee and Kitajewski, 2008; Phng et al., 2009; Zerlin et al., 2008; Zhang and Ma, 2010) and be essential for the morphogenesis of hair follicles (Enshell-Seijffers et al., 2010; Ito et al., 2007), the role of anti-angiogenic serpins in modulating Wnt signaling and wound healing in adult skin has not been investigated. We undertook this study to explore the role of kallistatin in modulation of wound healing and identified a potential pharmacological rescue strategy to attenuate negative effects of anti-angiogenic serpins in wound healing.
RESULTS
Elevation of serum kallistatin levels in type 2 diabetic patients with vascular complications
We analyzed kallistatin levels in the sera of healthy individuals and type 2 diabetic patients with or without clinically evident diabetes-related vascular complications (Table S1). In diabetic patients with complications, 87.5% of patients had microvascular complications, including 46.9% with peripheral neuropathy (Table S1). Circulating kallistatin levels differed significantly across the three subject groups (Fig. 1). Kallistatin levels were significantly higher in the type 2 diabetic patients with vascular complications compared to non-diabetic control subjects and diabetic patients without complications (Fig. 1). Kallistatin levels in diabetic patients showed correlations with various clinical parameters related to vascular health, including HbA1c, albumin-to-creatinine ratio, large artery elasticity and small artery elasticity (Table S2).
Figure 1. Elevation of serum kallistatin levels in Type 2 diabetic patients with vascular complications of diabetes.
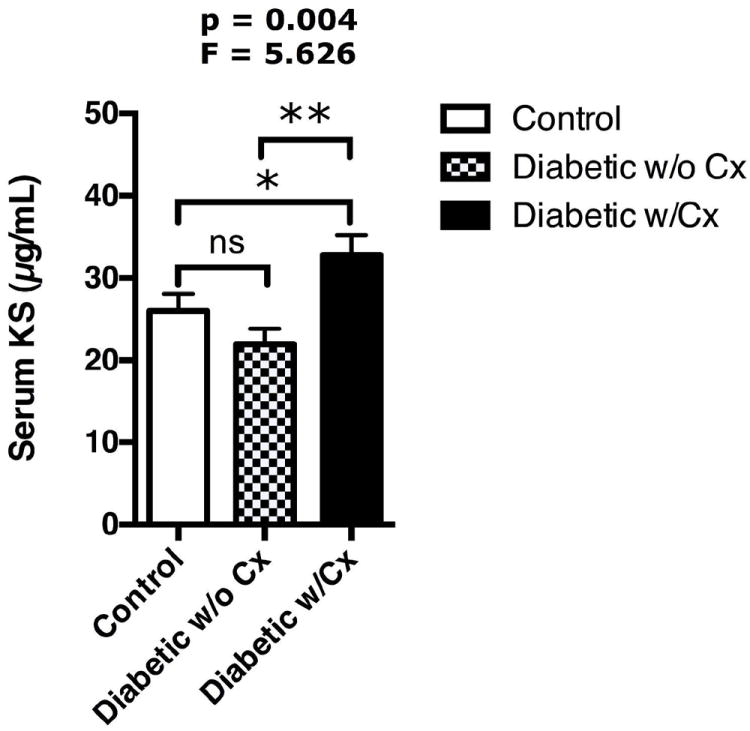
Non-diabetic subjects (N=45), diabetic patients without vascular complications (DM w/o Cx, N=36) and diabetic patients with vascular complications (DM w/Cx, N=44). Mean ± S.E.M., ANOVA: p=0.004, F= 5.626. Post-hoc analysis group vs. group comparison indicated with bars: ns= not significant, *p<0.05, **p<0.01.
Reduced hair follicle density and skin microvascular density in kallistatin transgenic (KS-TG) mice
To understand the impacts of elevated kallistatin levels, we generated KS-TG mice overexpressing and secreting human kallistatin into the circulation and tissues (Fig. S1a-b). Circulating levels of endogenous mouse kallistatin (SERPINA3C) were approximately 1 μg/ml in wild-type (WT) mice (Fig. S1c), while KS-TG mice had circulating levels of kallistatin at 5 μg/mL (Fig. S1d). The increases of serum levels of human kallistatin in KS-TG mice were comparable with the fold increase of kallistatin in diabetic patients with vascular complications. The elevated kallistatin levels were detected in the skin of KS-TG mice at sufficient amounts for studying a potential skin phenotype (Fig. S1d).
In vitro tissue kallikrein activity assays showed that KS-TG mice had no detectable change in tissue kallikrein activity in wounded skin or serum, compared to the WT mice (Fig. 3j and Fig. S1e, respectively). Comparing the amino acid sequence of human kallistatin (SERPINA4) with endogenous mouse kallistatin (SERPINA3C) revealed that the sequence of the reactive center loop of human kallistatin that interacts with and is cleaved by tissue kallikrein is not identical to the sequence in mouse kallistatin (Fig. S1f), suggesting human kallistatin probably does not significantly impact the activity of mouse tissue kallikrein.
Newborn KS-TG mice at postnatal day 0 (P0) had reduced hair follicle density compared to WT littermates (Fig. 1a-b; quantification in Fig. 2i). Skin with telogen phase follicles in 3-month-old KS-TG mice showed significantly decreased dorsal skin thickness (Fig. 2c-d; quantification in Fig. 2k) and reduced hair follicle density (Fig. 2j). Furthermore, as seen after synchronized induction of anagen by depilation, induction of anagen phase hair follicles was attenuated in KS-TG mice relative to WT littermates (Fig. S4). The decrease in 3-month-old KS-TG mouse skin thickness was largely due to a decrease in the thickness and cell population in the panniculus adiposus layer, with nuclei numbers in the dermis and panniculus carnosus layers being similar (Fig. 2e, 2f, 2l). The microvascular density in the skin of KS-TG mice was significantly reduced (Fig. 2g and 2h; quantification in Fig. 2m).
Figure 2. Kallistatin affects skin structure and function.
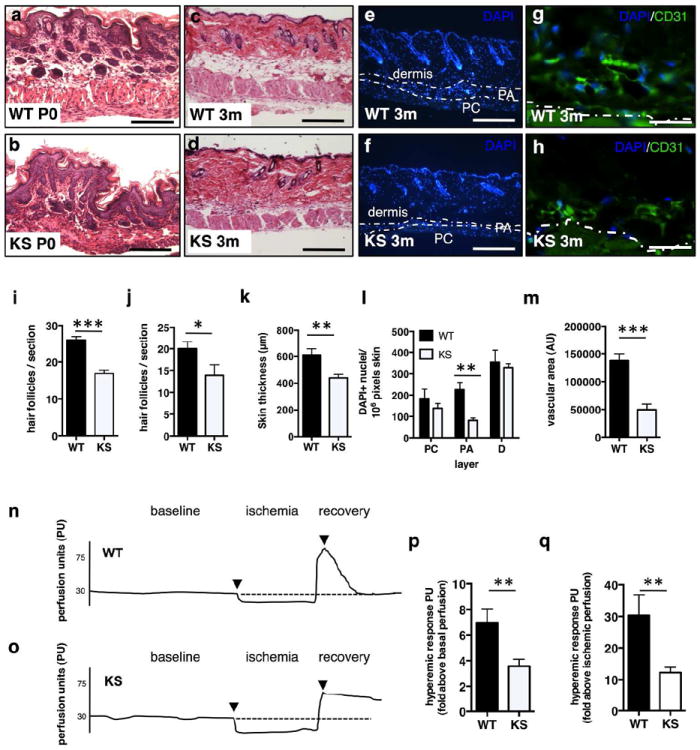
(a-d) H&E, dorsal skin, (a, b) newborn (P0) WT and KS-TG littermates, Scale =100 μm; (c, d) 3-month-old littermates, Scale =500 μm. (e, f) DAPI staining; dotted lines indicate boundaries of the skin dermis, panniculus adiposus, and panniculus carnosus layers; Scale =500 μm. (g, h) FITC-anti-CD31 antibody, Scale =50 μm. (i, j) hair follicle density at P0 (i) and 3 months (j); (k) skin thickness; (l) nuclei between dotted lines in panniculus carnosus (PC), panniculus adiposus (PA), dermis; (m) microvascular density. (n, o) Laser Doppler flowmetry in hindlimb skin, (n) WT and (o) KS-TG mice. (p, q) Hyperemic responses. N= 5 or >5 in all analyses with multiple sections/tissues per analysis, Mean ± S.E.M. * p<0.05, **p< 0.01, ***p < 0.001.
Kallistatin overexpression impairs skin hyperemic response to ischemia
Pressure to the skin causes local ischemia. Upon release of pressure, blood flow increases immediately, peaks and rapidly stabilizes due to vascular reactivity (Tur et al., 1991). We measured skin blood flow after standardized local ischemia in adult WT and KS-TG mice (Fig. 2n-o). WT skin blood flow dynamics appeared as expected, with a sharp rise and rapid return to baseline (Fig. 2n). KS-TG mice, however, had a blunted response above both baseline levels and ischemia levels (Fig. 2o; quantified in Fig. 2p-q) and had a delay in returning to baseline (Fig. 2o).
Kallistatin transgenic mice have delayed skin wound repair
Skin wound healing assay demonstrated that wound closure in KS-TG mice lagged behind WT littermates (Fig. 3a). Vascular density in the wound area was reduced in KS-TG mice than in WT mice at day 7 of wound healing, a peak phase of endothelial cell proliferation during wound healing (Nissen et al., 1998) (Fig. 3f-i). There was no difference in tissue kallikrein activity in wounds at day 7 (Fig. 3j). Vascular density in wound beds at day 7 was significantly decreased in KS-TG mice (Fig. 3k). The expression of vegf-a was significantly lower at both the mRNA and protein levels in KS-TG mice vs. WT mice during day 7 of wound healing (Fig. 3l and m).
Figure 3. Kallistatin delays wound closure and inhibits wound angiogenesis.
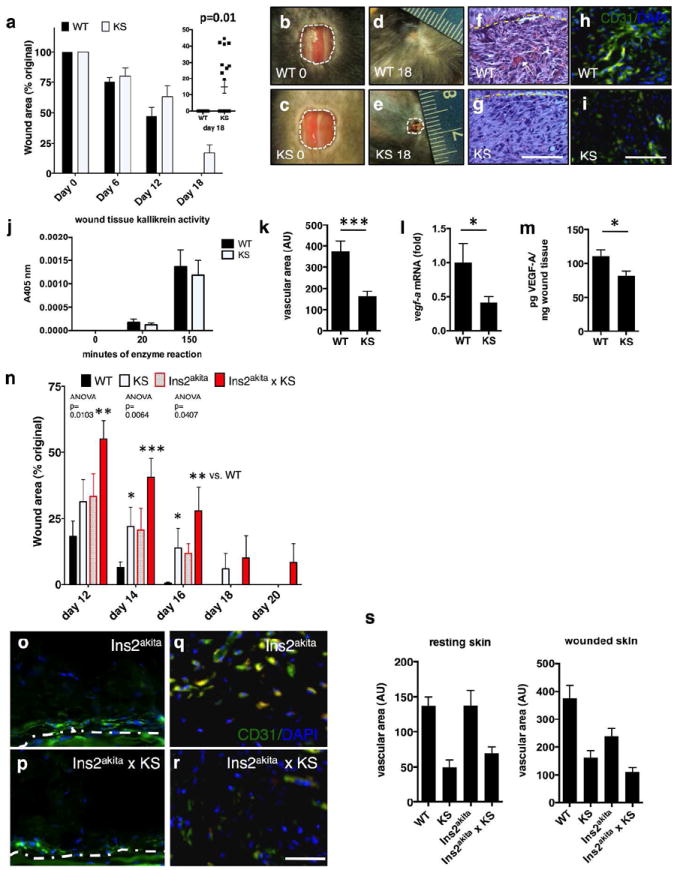
(a) Wound healing rate (3-month-old male littermates). (b-e) images of representative wounds. (f, g) H&E, wound bed at day 7 (Scale bar=50 μm); (h, i) CD31, wound beds; (j) normalized tissue kallikrein activity in wounds; (k) wound vascular area; (l) vegf-a mRNA levels in wounds; (m) VEGF-A in wound homogenates; (n) wound areas in 3-month-old male mice; (o, p) CD31+ cells in resting skin in Ins2akita and Ins2akita × KS-TG mice; (q, r) CD31+ endothelial cells, wounded skin, Ins2akita and Ins2akita × KS-TG mice. Scale bar in (o-r): 50 μm. (s) CD31+ area. Mean ± S.E.M., N= 5 or >5 in all analyses with multiple sections/tissues per analysis, * p<0.05, **p< 0.01, ***p < 0.001.
Kallistatin overexpression exacerbates wound-healing delay in diabetic mice
Ins2akita mice represent a model of diabetes caused by an insulin 2 gene mutation (Wang et al., 1999). While KS-TG mice and Ins2akita mice (3-month-old) alone showed mild delays in skin wound healing, Ins2akita × KS-TG mice had the slowest wound-healing rate of all groups (Fig. 3n). Although having thinner skin than WT, Ins2akita mice had better angiogenic responses in wounds compared to Ins2akita × KS-TG mice, at age 3 months (Fig. 3o, p are resting skin; Fig. 3q, r are wounds; quantification in Fig. 3s).
Kallistatin transgenic mice have reduced activation of Wnt/TCF/β-catenin signaling in skin and wounds
We examined if kallistatin overexpression affects Wnt/TCF/β-catenin signaling in the skin by crossing KS-TG mice with Wnt/TCF/β-catenin-reporter BAT-gal mice, which express the β-galactosidase reporter gene under the control of a promoter containing TCF/β-catenin binding sites. X-gal staining indicated that Wnt signaling was activated in the periphery of the wounded skin (Fig. 4a, b) and in cells that had an endothelial-like morphology and were co-stained with CD31 (Fig. 4c, d) in the wound beds and hair follicles of BAT-gal mice. In the wounded Wnt reporter mice, the number of hair follicles in the immediate periphery of the wound with Wnt reporter activity was significantly higher than in resting skin (Fig. 4e). The resting skin of BAT-gal mice had more than 25% of the total population of hair follicles with Wnt activation, while resting skin of BAT-gal × KS-TG mice had less than 10% of hair follicles with Wnt signaling activation (Fig. 4e). During the proliferative phase of wound healing, BAT-gal mice had over 45% of Wnt-activated hair follicles adjacent to the wound area, while BAT-gal × KS-TG mice had less than 10% activation (Fig. 4e). In wound beds in the proliferative stage, BAT-gal mice had higher densities of cells with active Wnt signaling (Fig. 4h), and these cells were associated with CD31 in wound beds, compared to BAT-gal × KS-TG mice (Fig. 4i; quantification in Fig. 4j).
Figure 4. Kallistatin is associated with reduction in Wnt signaling in hair follicles and during wound healing.
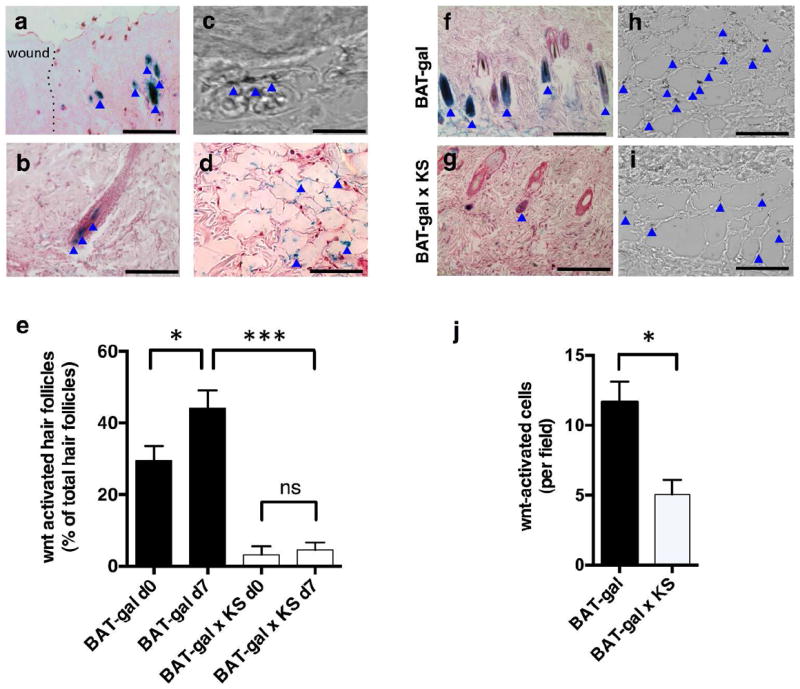
(a) X-gal-stained hair follicles surrounding wound area, Wnt-reporter BAT-gal mice; (b) Wnt activation in various positions in hair follicle adjacent to wound; (c) Differential interference contrast image, X-gal+ endothelial cell in skin; (d) X-gal+ endothelial cells, wound bed; (e) quantification, X-gal+ hair follicles; (f, g) X-gal+ hair follicles surrounding wounds; (h, i) X-gal+ cells, day 7 wound beds; (j) quantification of X-gal+ cells. In all panels, blue arrows indicate X-gal staining. Scale bars= (a) 200 μm, (b) 100 μm, (c) 50 μm, (d, f, g) 200 μm, (h, i) 50 μm. N= 5 or >5 in all analyses with multiple sections/tissues per analysis. Mean ± S.E.M. * p<0.05, **p< 0.01, ***p < 0.001.
Kallistatin inhibits Wnt/β-catenin signaling in primary human dermal microvascular endothelial cells
To dissect the effect of kallistatin on endothelial Wnt signaling, we treated primary human dermal microvascular endothelial cells (HDMVECs) with 30% Wnt3a-conditioned-media (WCM) or with L-cell-conditioned media (LCM) as control. In an in vitro angiogenesis assay, kallistatin reduced WCM-induced tube and branch formation from HDMVECs after 12- hr treatment (Fig. 5a-c). WCM stimulated HDMVEC proliferation over 72 hr, compared to LCM control (Fig. 5d). Purified kallistatin inhibited WCM-induced proliferation of the dermal microvascular endothelial cells, compared to BSA control (Fig. 5d). Kallistatin reduced Wnt3a-induced phosphorylation of LRP6, an essential co-receptor of canonical Wnt signaling and levels non-phosphorylated β-catenin (NP-β-catenin) in HDMVECs, suggesting an inhibitory effect on Wnt signaling in endothelial cells (Fig. 5e). To assess kallistatin’s effect on Wnt3a/TCF/β-catenin-dependent transcription in HDMVECs, we delivered vectors via lentivirus for TCF/β-catenin-driven luciferase and constitutively expressed renilla luciferase. Luciferase assay revealed that HDMVECs harbor the endogenous machinery for canonical Wnt signaling and respond to Wnt3a ligand in WCM vs. LCM (Fig. 5f). Furthermore, kallistatin dose-dependently reduced transcriptional activity of β-catenin in HDMVECs (Fig. 5f). Expression of a direct angiogenic Wnt/TCF/β-catenin target gene, vegf-a, was shown to be upregulated in HDMVECs by WCM and downregulated by kallistatin (Fig. 5g). Taken together, these data support that kallistatin impairs dermal angiogenesis, at least in part, by inhibition of canonical Wnt/TCF/β-catenin signaling in skin endothelial cells.
Figure 5. Kallistatin reduces Wnt3a-induced dermal endothelial cell angiogenesis and Wnt3a-induced TCF/β-catenin-dependent transcription.
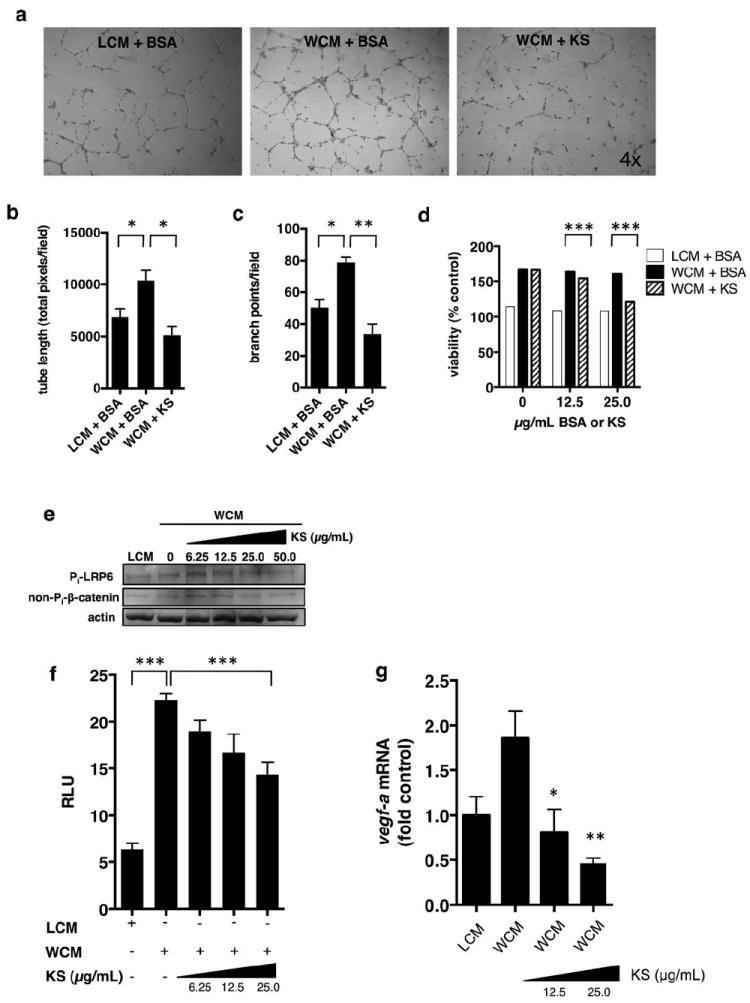
In vitro angiogenesis assay, primary HDMVECs; (a) 30% LCM + 25 μg/mL BSA; 30% WCM + 25 μg/mL BSA; 30% WCM + 25 μg/mL kallistatin (KS); (b) total tube length quantification; (c) branch points; (d) HDMVECs treated simultaneously with 30% WCM and purified KS or BSA, 48 hr. Cell viability via MTT assay; (e) Western blot analysis, phosphorylated LRP6 (Pi-LRP6); HDMVECs; (f) HDMVECs, infected with lentivirus expressing luciferase driven by TCF/β-catenin (renilla luciferase for normalization). HDMVECs were treated with 30% LCM or 30% WCM and different concentrations of KS for 16 hr. (g) vegf-a mRNA levels in HDMVECs treated as indicated for 16 hr. Mean ± S.E.M., *p<0.05; **<0.01; ***p<0.001.
Lithium attenuates the effects of kallistatin on skin angiogenesis and wound healing
To confirm that the effect of kallistatin on wound healing is through inhibition of Wnt signaling by blocking LRP6, we activated TCF/β-catenin intracellularly via pharmacological inhibition of GSK-3β and subsequent stabilization of β-catenin using LiCl. HDMVECs formed more branches and longer tubes in the presence of 5 mM LiCl versus 5 mM NaCl (Fig. 6a-c). Addition of 25 μg/mL of purified kallistatin was unable to significantly attenuate HDMVEC tube formation induced by 5 mM LiCl (Fig. 6a-c). Consistently, the same concentration of kallistatin, while able to reduce Wnt3a-induced TCF/β-catenin-driven transcription and tube formation (Fig. 5c), was unable to decrease TCF/β-catenin-driven transcription induced by LiCl (Fig. 6d).
Figure 6. Lithium attenuates the anti-angiogenic and anti-Wnt effects of kallistatin.
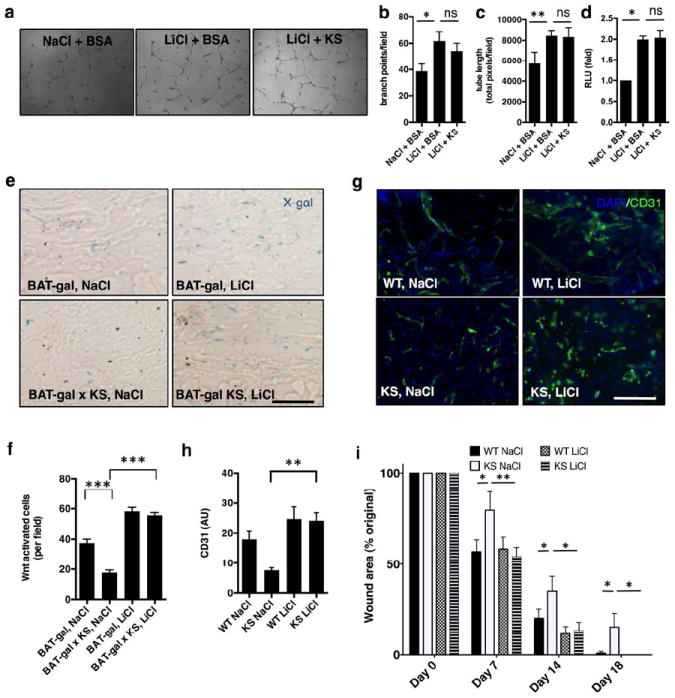
(a) Tube formation assay with HDMVECs; (b) total branch points, n=3; (c) total tube length, n=3; (d) TCF/β-catenin transcriptional activity in HDMVECs; (e) X-gal staining showing Wnt-activation in day 7 wounds of Wnt reporter mice in response to NaCl or LiCl topical treatments. (f) Quantification, Wnt-reporter X-gal activity; (g) CD31 signaling in day 7 wound beds after topical treatments; (h) quantification, CD31+ cells, day 7 wound beds (AU=arbitrary fluorescence units); (i) Overall skin wound healing rate expressed by wound area; N=7-10 age-matched male mice; n=5 or >5. Mean±S.E.M *p<0.05; **<0.01; ***p<0.001; ANOVA and Tukey’s post-hoc significance analysis performed. Scale bars (e, g): 50 μm.
To test whether or not lithium has the capacity to rescue the wound healing delay associated with kallistatin overexpression, wounded mice were treated topically with 20 mM LiCl in DMEM, applied directly to the wounds, a dose previously shown to activate Wnt signaling in BAT-gal mice in vivo (Fathke et al., 2006), twice daily for the first 7 days of wound healing, followed by once daily for days 8-10 of wound healing. BAT-gal × KS mice treated with topical LiCl showed a robust increase in the numbers of cells with activated Wnt signaling which were associated with CD31+ areas in the wound, compared with BAT-gal × KS mice treated with topical NaCl (Fig. 6e-f). The LiCl treatment of KS-TG mice increased the endothelial cell density in wound beds significantly, compared to KS-TG mice treated with 20 mM NaCl DMEM (Fig. 6g-h). As a consequence, topical LiCl treatment significantly rescued wound repair in KS-TG mice (Fig. 6i).
DISCUSSION
Our study establishes that increased circulating levels of an abundant, endogenous anti-angiogenic serpin in patients with diabetic microvascular complications contribute to impaired skin function and wound repair. Kallistatin is secreted by nearly every cell type in vivo (Chao et al., 1996); yet its roles in modulating the structure and physiology of many organs are not fully understood. Kallistatin was originally identified as a specific binding protein and inhibitor of tissue kallikrein (Chao et al., 1986). Kallistatin is a heparin-binding protein (Chen et al., 2001) and is expressed in a wide array of tissues and cell types, including endothelium, salivary glands and immune cells (Chao et al., 1996; Wolf et al., 1999). This pattern of expression and secretion, as well as its characterization as an inhibitor of angiogenesis, strongly suggests that kallistatin is involved in the regulation of vascular function and remodeling in skin.
The causes of systemic elevation of kallistatin in diabetic patients with microvascular complications are not yet known. It may be due to increased secretion and/or decreased re-uptake by the liver, as the liver has been shown to be the major recycler of the kallistatin-kallikrein complex from the circulation (Xiong et al., 1992). We demonstrated in cell culture that high glucose treatment up-regulates kallistatin expression in HepG2 cells, a cell line derived from human liver, but did not find evidence that endogenous mouse kallistatin is elevated in early diabetes in 3-month-old Ins2akita mice (Fig. S3).
Diabetic patients with retinal and renal complications are at higher risks of neuropathy and cardiovascular disease and are more likely to develop foot ulcers and require lower limb amputations (Monteiro-Soares et al., 2012). Here, we show that transgenic elevation of human kallistatin levels in mice affected the ultimate structure and histology of the skin, with resting skin being thinner in the panniculus adiposus layer, having reduced skin microvascular density and less hair follicles – features of human lower limb skin in patients with diabetes and/or peripheral vascular disease. Although thickening of some parts of skin may occur in diabetic patients, such as with acanthosis nigricans and with diabetic pseudoscleroderma (Kostler et al., 2005), high levels of kallistatin may contribute to what is also often seen in diabetic skin – thinning of the panniculus adiposus layer that harbors the subcutaneous fat and blood vessels (Petrofsky et al., 2008). As the panniculus adiposus layer loses structural integrity and becomes thinner, there may be hair loss, reduced capillary return, neuropathy, ulceration and gangrene – signs of tissue damage that precede lower limb amputation (Hoyt, 2004; Petrofsky et al., 2008). Recent studies elucidated the crosstalk between adipocyte precursor cells, epithelial stem cells and hair follicle cycling (Festa et al., 2011; Schmidt and Horsley, 2012). Through kallistatin’s inhibition of Wnt/β-catenin signaling within hair follicles and endothelial cells, KS-TG mice likely possess defective crosstalk between hair follicles and adipose tissue. At one level, the decreased hair follicle units likely result in less stimulation of adipose tissue within the panniculus adiposus layer of the mice. Furthermore, the decreased microvascular density within the panniculus adiposus layer likely results in less support for adipocyte precursors, thus disabling the crosstalk between hair follicles and adipose tissue in coordinating proper skin structure and function.
Diabetic patients are known to have impaired skin blood flow and hemodynamic changes upon pressure or injury to the skin (Petrofsky et al., 2009). We found that overexpression of kallistatin resulted in an impaired hyperemic response to local ischemia. KS-TG mice do not develop hyperglycemia, but still have impaired local skin hemodynamics, mimicking the defective hemodynamics present in diabetic skin. Furthermore, KS-TG mice displayed delayed wound healing as well as attenuated wound vegf-a expression and wound neovascularization.
Taken together, our data suggests that kallistatin is an endogenous Wnt/β-catenin inhibitor in postnatal murine skin. Wnt signaling is known to be a significant modulator of inflammation and angiogenesis (George, 2008; Masckauchan and Kitajewski, 2006). The skin/hair follicle phenotypes of KS-TG mice are similar to what was reported in transgenic mice systemically overexpressing DKK-1, a potent and specific inhibitor of the canonical Wnt pathway (Sick et al., 2006).
Our recent study showed that kallistatin inhibits Wnt signaling by blocking LRP6, an essential co-receptor in the canonical Wnt pathway (Liu et al., 2013). To confirm the impact of kallistatin on wound healing is indeed through inhibition of Wnt signaling, we activated Wnt signaling downstream of LRP6. Lithium, a drug approved by the Food and Drug Administration (FDA) to treat mood disorders and known to increase vegf-a expression (Guo et al., 2009; Kaga et al., 2006), is a potent activator of canonical Wnt signaling by inhibiting GSK-3β and stabilizing β-catenin. Because lithium activates β-catenin downstream of LRP6, and has been shown to rescue vascular defects and re-stimulate angiogenesis during development (Curtis and Griffin, 2012; Griffin et al., 2011) and in the cardiovascular (Kaga et al., 2006) and central nervous systems (Guo et al., 2009), we chose LiCl as an agent to bypass the blocking effects of kallistatin on Wnt signaling in vivo and in vitro. Our results showed that LiCl attenuated the effects of kallistatin on wound angiogenesis and wound healing in vivo and dermal endothelial tube formation and branching in vitro. This experiment provides further evidence supporting that kallistatin causes a wound healing delay through antagonizing LRP6.
We propose the following model: excessive accumulation of anti-angiogenic serpins, such as kallistatin, inhibits Wnt/β-catenin signaling, contributing to impaired skin endothelial function and wound healing defects in diabetic patients. Activation of Wnt signaling downstream of Wnt receptors in endothelium and hair follicles, in and around wounded skin, may benefit the treatment of impaired wound healing in diabetic patients with elevated levels of anti-angiogenic serpins, reducing the overall risk of amputations.
MATERIALS AND METHODS
Human subjects
The study, which adhered to the Declaration of Helsinki Guidelines, was approved by the University of Oklahoma Health Sciences Center Institutional Review Board, and written informed consent was obtained from each subject. History and examination were performed, and clinicians confirmed diabetes-associated vascular complication status prior to this study. Diabetes-associated complications were pre-defined as having at least one of the following complications of diabetes: history of leg, foot or toe amputation, retinopathy, documented myocardial infarction or angina with ECG changes and/or positive cardiac imaging study, nephropathy, history of TIA or stroke, angioplasty, or vascular bypass surgery.
Enzyme-linked immunosorbent assay (ELISA) specific for kallistatin
Kallistatin levels in sera were quantified by ELISA (R&D Systems, Inc. Minneapolis, MN) as previously described (Jenkins et al., 2010). For mouse kallistatin ELISA, wells were coated with 2.0 μg/ml anti-mouse SERPINA3C antibody (Sinobiological, China) overnight, and recombinant SERPINA3C standard (Sinobiological, China) was used for standard curve.
Kallistatin transgenic, diabetic and Wnt reporter mice
The Institutional Animal Care and Use Committee approved all of the animal experiments described. The chicken β-actin promoter was used to drive systemic expression of human kallistatin cDNA, and cloned into the pTriE×1.1 vector (Novagen, Darmstadt, Germany).
Tissue kallikrein activity assays
Enzymatic activity of endogenous tissue kallikrein was assayed using the colorimetric substrate S-2266 (Chromogenix, Orangeburg, New York), which can be specifically cleaved by both mouse and human tissue kallikrein. Upon cleavage, the colorimetric reaction produced a yellow color, which was quantified by absorbance at 405 nm wavelength.
Laser Doppler flowmetry
After anesthesia, the hind legs of the mice were fixed in place using mild-adhesive tape, and the laser Doppler probe was fixed firmly to skin to measure perfusion units (PU) using the PerimedPeriFlux System 5000 (Perimed, Stockholm, Sweden).
Skin wound healing assay
Clippers were used on dorsal surface of anesthetized mice to remove hair but retain hair follicles. Standardized circular wounds were made with biopsy punches and Image J software (NIH) was used to trace wound areas and quantify the pixels within the wound.
Visualization of transcriptional activity of β-catenin in vivo
Skin and wounds from BAT-gal transgenic mice were stained with 5-Bromo-4-chloro-3-indolyl b-D-galactopyranoside (X-gal) according to manufacturer’s instructions (Sigma, St. Louis, MO).
Dermal microvascular endothelial cell culture and tube formation assay
Primary human dermal microvascular cells were obtained from ATCC (Manassas, VA). The cells were seeded on BD matrigel extracellular matrix mix at a density of 100,000 cells per well in presence of WCM or LCM as control or 5 mM LiCl in microvascular growth media (5 mM NaCl as control) and 25 μg/mL of purified His-tagged kallistatin (or 25 μg/mL BSA as control), and conditions were incubated at 37°C. Twelve hours post-seeding, the tube lengths and branching were imaged under microscope and quantified to reflect angiogenesis in vitro.
Topical application of lithium chloride during in vivo wound healing
During wound healing, sterile 20 mM NaCl or 20 mM LiCl in serum-free DMEM was applied topically to open wounds of single-housed mice (500 μL gently ejected from sterile pipette tips under biosafety hood) twice daily to directly bathe the wound from days 0 –7; once a day from days 7 –10. Thereafter, wounds were allowed to heal spontaneously.
Statistics
One-way ANOVA for continuous variables was used with a Tukey honest significant difference (HSD) post-hoc test for differences between two groups when ANOVA P-value was <0.05. For animal studies involving two groups, 2-tailed t-test was performed with p<0.05 considered significant.
Supplementary Material
Acknowledgments
We thank Dr. DongXu Fu for assistance in the human studies, Jeffery Smith and Carol Haaksma in the Histology Core of the Diabetes COBRE, Robert Mott at the Diabetic Animal Core of the Diabetes COBRE for assistance with the wound healing assay, Dr. Yih-Kuen Jan’s lab and Blake Hopiavuori for help with the use and analysis of laser Doppler flowmetry, and Dr. Randall Moon at the University of Washington School of Medicine for the generous gift of pBARLS, pfuBARLS, and pSL9/Ren vectors.
Footnotes
CONFLICTS OF INTEREST
None.
References
- Barcelos LS, Duplaa C, Krankel N, et al. Human CD133+ progenitor cells promote the healing of diabetic ischemic ulcers by paracrine stimulation of angiogenesis and activation of Wnt signaling. Circulation research. 2009;104:1095–102. doi: 10.1161/CIRCRESAHA.108.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao J, Schmaier A, Chen LM, et al. Kallistatin, a novel human tissue kallikrein inhibitor: levels in body fluids, blood cells, and tissues in health and disease. The Journal of laboratory and clinical medicine. 1996;127:612–20. doi: 10.1016/s0022-2143(96)90152-3. [DOI] [PubMed] [Google Scholar]
- Chao J, Tillman DM, Wang MY, et al. Identification of a new tissue-kallikrein-binding protein. The Biochemical journal. 1986;239:325–31. doi: 10.1042/bj2390325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Stahl A, Krah NM, et al. Wnt signaling mediates pathological vascular growth in proliferative retinopathy. Circulation. 2011;124:1871–81. doi: 10.1161/CIRCULATIONAHA.111.040337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VC, Chao L, Pimenta DC, et al. Identification of a major heparin-binding site in kallistatin. The Journal of biological chemistry. 2001;276:1276–84. doi: 10.1074/jbc.M005791200. [DOI] [PubMed] [Google Scholar]
- Chen Y, Hu Y, Zhou T, et al. Activation of the Wnt pathway plays a pathogenic role in diabetic retinopathy in humans and animal models. The American journal of pathology. 2009;175:2676–85. doi: 10.2353/ajpath.2009.080945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis CD, Griffin CT. The chromatin-remodeling enzymes BRG1 and CHD4 antagonistically regulate vascular Wnt signaling. Molecular and cellular biology. 2012;32:1312–20. doi: 10.1128/MCB.06222-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DasGupta R, Fuchs E. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development. 1999;126:4557–68. doi: 10.1242/dev.126.20.4557. [DOI] [PubMed] [Google Scholar]
- Dawson DW, Volpert OV, Gillis P, et al. Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science. 1999;285:245–8. doi: 10.1126/science.285.5425.245. [DOI] [PubMed] [Google Scholar]
- Dejana E. The role of wnt signaling in physiological and pathological angiogenesis. Circulation research. 2010;107:943–52. doi: 10.1161/CIRCRESAHA.110.223750. [DOI] [PubMed] [Google Scholar]
- Enshell-Seijffers D, Lindon C, Kashiwagi M, et al. beta-catenin activity in the dermal papilla regulates morphogenesis and regeneration of hair. Developmental cell. 2010;18:633–42. doi: 10.1016/j.devcel.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fathke C, Wilson L, Shah K, et al. Wnt signaling induces epithelial differentiation during cutaneous wound healing. BMC cell biology. 2006;7:4. doi: 10.1186/1471-2121-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festa E, Fretz J, Berry R, et al. Adipocyte lineage cells contribute to the skin stem cell niche to drive hair cycling. Cell. 2011;146:761–71. doi: 10.1016/j.cell.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George SJ. Wnt pathway: a new role in regulation of inflammation. Arteriosclerosis, thrombosis, and vascular biology. 2008;28:400–2. doi: 10.1161/ATVBAHA.107.160952. [DOI] [PubMed] [Google Scholar]
- Goettig P, Magdolen V, Brandstetter H. Natural and synthetic inhibitors of kallikrein-related peptidases (KLKs) Biochimie. 2010;92:1546–67. doi: 10.1016/j.biochi.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin CT, Curtis CD, Davis RB, et al. The chromatin-remodeling enzyme BRG1 modulates vascular Wnt signaling at two levels. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:2282–7. doi: 10.1073/pnas.1013751108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Arai K, Stins MF, et al. Lithium upregulates vascular endothelial growth factor in brain endothelial cells and astrocytes. Stroke; a journal of cerebral circulation. 2009;40:652–5. doi: 10.1161/STROKEAHA.108.524504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–12. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- Hoyt RE. Peripheral arterial disease in people with diabetes: response to consensus statement. Diabetes care. 2004;27:2095. doi: 10.2337/diacare.27.8.2095. [DOI] [PubMed] [Google Scholar]
- Irving JA, Pike RN, Lesk AM, et al. Phylogeny of the serpin superfamily: implications of patterns of amino acid conservation for structure and function. Genome research. 2000;10:1845–64. doi: 10.1101/gr.gr-1478r. [DOI] [PubMed] [Google Scholar]
- Ito M, Yang Z, Andl T, et al. Wnt-dependent de novo hair follicle regeneration in adult mouse skin after wounding. Nature. 2007;447:316–20. doi: 10.1038/nature05766. [DOI] [PubMed] [Google Scholar]
- Jenkins AJ, McBride JD, Januszewski AS, et al. Increased serum kallistatin levels in type 1 diabetes patients with vascular complications. Journal of angiogenesis research. 2010;2:19. doi: 10.1186/2040-2384-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaga S, Zhan L, Altaf E, et al. Glycogen synthase kinase-3beta/beta-catenin promotes angiogenic and anti-apoptotic signaling through the induction of VEGF, Bcl-2 and survivin expression in rat ischemic preconditioned myocardium. Journal of molecular and cellular cardiology. 2006;40:138–47. doi: 10.1016/j.yjmcc.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Kostler E, Porst H, Wollina U. Cutaneous manifestations of metabolic diseases: uncommon presentations. Clinics in dermatology. 2005;23:457–64. doi: 10.1016/j.clindermatol.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhang B, McBride JD, et al. Antiangiogenic and antineuroinflammatory effects of kallistatin through interactions with the canonical wnt pathway. Diabetes. 2013;62:4228–38. doi: 10.2337/db12-1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Developmental cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis DJ, Malay DS, Hoffstad OJ, et al. Data Points Publication Series. Rockville (MD): 2011. Prevalence of Diabetes, Diabetic Foot Ulcer, and Lower Extremity Amputation Among Medicare Beneficiaries, 2006 to 2008: Data Points #1. [PubMed] [Google Scholar]
- Masckauchan TN, Kitajewski J. Wnt/Frizzled signaling in the vasculature: new angiogenic factors in sight. Physiology (Bethesda) 2006;21:181–8. doi: 10.1152/physiol.00058.2005. [DOI] [PubMed] [Google Scholar]
- McMahon GA, Petitclerc E, Stefansson S, et al. Plasminogen activator inhibitor-1 regulates tumor growth and angiogenesis. The Journal of biological chemistry. 2001;276:33964–8. doi: 10.1074/jbc.M105980200. [DOI] [PubMed] [Google Scholar]
- Miao RQ, Agata J, Chao L, et al. Kallistatin is a new inhibitor of angiogenesis and tumor growth. Blood. 2002;100:3245–52. doi: 10.1182/blood-2002-01-0185. [DOI] [PubMed] [Google Scholar]
- Monteiro-Soares M, Boyko EJ, Ribeiro J, et al. Predictive factors for diabetic foot ulceration: a systematic review. Diabetes/metabolism research and reviews. 2012;28:574–600. doi: 10.1002/dmrr.2319. [DOI] [PubMed] [Google Scholar]
- Nissen NN, Polverini PJ, Koch AE, et al. Vascular endothelial growth factor mediates angiogenic activity during the proliferative phase of wound healing. The American journal of pathology. 1998;152:1445–52. [PMC free article] [PubMed] [Google Scholar]
- Parmalee NL, Kitajewski J. Wnt signaling in angiogenesis. Current drug targets. 2008;9:558–64. doi: 10.2174/138945008784911822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrofsky J, Prowse M, Lohman E. The Influence of Ageing and Diabetes on Skin and Subcutaneous Fat Thickness in Different Regions of the Body. The Journal of Applied Research. 2008;8:55–61. [Google Scholar]
- Petrofsky JS, Bains GS, Prowse M, et al. The influence of age and diabetes on the skin blood flow response to local pressure. Medical science monitor : international medical journal of experimental and clinical research. 2009;15:CR325–31. [PubMed] [Google Scholar]
- Phng LK, Potente M, Leslie JD, et al. Nrarp coordinates endothelial Notch and Wnt signaling to control vessel density in angiogenesis. Developmental cell. 2009;16:70–82. doi: 10.1016/j.devcel.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajamani K, Colman PG, Li LP, et al. Effect of fenofibrate on amputation events in people with type 2 diabetes mellitus (FIELD study): a prespecified analysis of a randomised controlled trial. Lancet. 2009;373:1780–8. doi: 10.1016/S0140-6736(09)60698-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt B, Horsley V. Unravelling hair follicle-adipocyte communication. Experimental dermatology. 2012;21:827–30. doi: 10.1111/exd.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sick S, Reinker S, Timmer J, et al. WNT and DKK determine hair follicle spacing through a reaction-diffusion mechanism. Science. 2006;314:1447–50. doi: 10.1126/science.1130088. [DOI] [PubMed] [Google Scholar]
- Tseng CH. Prevalence of lower-extremity amputation among patients with diabetes mellitus: is height a factor? CMAJ : Canadian Medical Association journal = journal de l’Association medicale canadienne. 2006;174:319–23. doi: 10.1503/cmaj.050680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tur E, Yosipovitch G, Bar-On Y. Skin reactive hyperemia in diabetic patients. A study by laser Doppler flowmetry. Diabetes care. 1991;14:958–62. doi: 10.2337/diacare.14.11.958. [DOI] [PubMed] [Google Scholar]
- Wang J, Takeuchi T, Tanaka S, et al. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. The Journal of clinical investigation. 1999;103:27–37. doi: 10.1172/JCI4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte JL, Smith AA, Helms JA. Wnt signaling and injury repair. Cold Spring Harbor perspectives in biology. 2012;4:a008078. doi: 10.1101/cshperspect.a008078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte JL, Smith AA, Liu B, et al. Augmenting endogenous wnt signaling improves skin wound healing. PloS one. 2013;8:e76883. doi: 10.1371/journal.pone.0076883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf WC, Harley RA, Sluce D, et al. Localization and expression of tissue kallikrein and kallistatin in human blood vessels. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 1999;47:221–8. doi: 10.1177/002215549904700210. [DOI] [PubMed] [Google Scholar]
- Wu X, Shen QT, Oristian DS, et al. Skin stem cells orchestrate directional migration by regulating microtubule-ACF7 connections through GSK3beta. Cell. 2011;144:341–52. doi: 10.1016/j.cell.2010.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, Tang CQ, Zhou GX, et al. In vivo catabolism of human kallikrein-binding protein and its complex with tissue kallikrein. The Journal of laboratory and clinical medicine. 1992;119:514–21. [PubMed] [Google Scholar]
- Zerlin M, Julius MA, Kitajewski J. Wnt/Frizzled signaling in angiogenesis. Angiogenesis. 2008;11:63–9. doi: 10.1007/s10456-008-9095-3. [DOI] [PubMed] [Google Scholar]
- Zhang B, Ma JX. Wnt pathway antagonists and angiogenesis. Protein Cell. 2010;1:898–906. doi: 10.1007/s13238-010-0112-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Gaspard JP, Chung DC. Regulation of vascular endothelial growth factor by the Wnt and K-ras pathways in colonic neoplasia. Cancer research. 2001;61:6050–4. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.