

Abstract
The T cell receptor (TCR):CD3 complex transduces signals that are critical for optimal T cell development and adaptive immunity. In resting T cells, the CD3ε cytoplasmic tail associates with the plasma membrane via a proximal basic-rich stretch (BRS). Here we show that mice lacking a functional CD3ε-BRS exhibited substantial reductions in thymic cellularity and limited CD4−CD8− double negative-3 (DN3) to DN4 thymocyte transition, due to enhanced DN4 TCR signaling resulting in increased cell death and TCR downregulation in all subsequent populations. Furthermore, positive, but not negative, T cell selection was affected in mice lacking a functional CD3ε-BRS, which led to limited peripheral T cell function and substantially reduced responsiveness to influenza infection. Collectively, these results indicate membrane association of the CD3ε signaling domain is required for optimal thymocyte development and peripheral T cell function.
INTRODUCTION
The T cell receptor (TCR):CD3 complex mediates numerous downstream signaling events at multiple developmental stages to ensure optimal T cell development, peripheral function, and a finely tuned and effective adaptive immune system (1, 2). The TCR is composed of a heterodimer (αβ or γδ) that contains variable regions that recognize peptide-bound MHC molecules and transmits intracellular signals via its association with the CD3 complex (εγ, δε and ζζ dimers), which possess immunotyrosine activation motifs (ITAMs) (3). TCR ligand recognition has been shown to induce conformational changes in the CD3ε cytoplasmic tail that lead to the recruitment of the Src-family proteins tyrosine kinases, Lck and Fyn, and subsequent recruitment and activation of key downstream signaling molecules such as the tandem Src homology 2 (SH2) domain-containing ζ-chain-associated protein of 70 kDa (ZAP-70) (4, 5). Despite our growing understanding of the steps required to mediate TCR signaling, the physiological importance of certain conserved motifs within the CD3 chains remain poorly understood.
Regulation of early CD4−CD8− double negative (DN) αβ T cell differentiation has been dissected by targeting various TCR signaling proteins including the TCR:CD3 complex components, signaling molecules (Lck/Fyn, Zap-70 and LAT) and signaling regulators (Csk and SHP1) (6, 7). Additionally, deletion of the CD3ε intracellular chains, but not the CD3γ or CD3δ, intracellular domains results in a block in early thymocyte development (8). However, mutating the CD3ε ITAM motifs does not affect T cell development (9), suggesting that there are additional critical motifs in the CD3ε cytoplasmic domain. Indeed, the CD3ε proline rich sequence (PRS), which recruits the adapter molecule Nck, facilitates early thymocyte development and enhances signals to low-avidity TCR:pMHC interactions (8, 10-12). More recently, a CD3ε membrane-proximal basic-rich stretch (BRS) was shown to be critical for plasma membrane binding by the CD3ε cytoplasmic tail (13, 14). This feature is not unique to CD3ε as it was previously shown that a similar motif in the CD3ζ cytoplasmic domain binds to acidic phospholipids and is required for stable TCR:CD3 synapse formation (15, 16). TCR binding to pMHC results in release of the CD3ε cytoplasmic domain from the plasma membrane (17). Membrane binding by the CD3ε cytoplasmic domain requires interaction of clusters of basic CD3ε residues with negatively charged membrane lipids, in particular phosphatidylserine. The negative charge of the inner leaflet of the plasma membrane is reduced in TCR microclusters, accounting for release of the CD3ε cytoplasmic domain following TCR triggering. This in turn may increase accessibility of ITAMs to the Lck/Fyn tyrosine kinases, promoting T cell activation. Accumulating data thus propose a model in which the CD3ε and CD3ζ cytoplasmic domains associate with the lipid membrane in resting T cells and TCR ligation induces a triggering event that initiates the release of the CD3ε and CD3ζ cytoplasmic domains, facilitating signal transduction. While the contribution of the CD3ε-BRS in mediating membrane association has been demonstrated, its physiological importance is unknown and could not be simply predicted. There are four possible scenarios. First, there may be no phenotype following the disruption of the CD3ε-BRS in mice due to functional redundancy with the CD3ζ-BRS, which may be sufficient for optimal regulation of TCR signaling. Second, mutation of the CD3ε-BRS may lead to a reduction in pTCR signaling due to inappropriate and/or inefficient recruitment of signaling molecules, block DN3-DN4 progression and limited T cell development. Third, mutation of the CD3ε-BRS may lead to enhanced TCR signaling that results in enhanced DN3-DN4 progression and T cell development and/or the development of autoreactive T cells and autoimmunity due to excessive peripheral T cell signaling and function. Fourth, mutation of the CD3ε-BRS may lead to enhanced TCR signaling that disrupts pTCR signaling due to increased apoptosis, and direct and/or indirect perturbation of T cell selection and peripheral T cell function leading to reduced immune responses. In this study, we investigated which scenario is correct.
We used TCR:CD3 retrogenic technology (12, 18, 19) to generate mice expressing CD3ε-BRS mutants with reduced or abolished plasma membrane binding. Our studies revealed an important role for these motifs in facilitating two vital developmental checkpoints: DN3-DN4 transition and positive selection. TCR signaling in CD3ε-BRS mutant DN4 thymocytes was enhanced, resulting in TCR down-modulation in all subsequent developmental populations. Reduced TCR expression in turn dampened TCR signaling in DP thymocytes and mature T cells. Finally, CD3ε-BRS mutant mice exhibited a limited protective response to influenza infection and were not able to effectively clear the virus. Our data highlight the critical physiological role performed by the CD3ε-BRS in regulating TCR signaling, thereby facilitating optimal T cell development, peripheral T cell function and the generation of protective immunity.
MATERIALS AND METHODS
Mice
Rag1−/− (recombination activating gene 1) and C57BL/6J mice were obtained from Jackson Laboratories. CD3.KO mice lacking all four CD3 chains were generated by crossing Cd3eΔP/ΔP and Cd247−/− mice as described earlier (9, 20, 21). Nur77GFP mice (22) were obtained from Kristin Hogquist (University of Minnesota) and crossed with CD3.KO mice. All animal experiments were performed in American Association for the Accreditation of Laboratory Animal Care-accredited, specific pathogen- free, helicobacter-free facilities in the St. Jude Animal Resource Center following national, state, and institutional guidelines. Animal protocols were approved by the St. Jude Animal Care and Use Committee.
Generation of CD3 multicistronic vectors
CD3 multiscistronic constructs were generated as described previously (9, 18, 19). Briefly, 2A peptide-linked CD3 constructs were generated by recombinant PCR and cloned into pMIG and pMIA, an MSCV-based retroviral vector containing an IRES-GFP or IRES-Ametrine1.1 (Addgene) cassette respectively. Specific lysines and arginines on the cytoplasmic domain of the CD3ε chain were mutated to serines and alanines by recombinant PCR and subsequent CD3 mutant constructs were then generated by appropriate subcloning.
Generation of retroviral producer cells
Retroviral producer cell lines were generated as previously described (9, 18, 19). Briefly, HEK-293T cells were transiently transfected with CD3 multicistronic vectors (4 μg), together with packaging and envelope vectors using TransIT LT1 transfection reagent (Mirus). The supernatant containing virus was collected and used to transduce GP+E86 cells in the presence of polybrene (6 μg/ml) every 12h for 3-4 days until viral titer greater than 105/ml after 24 h was obtained.
Flow cytometric analysis, intracellular staining and cell sorting
Double positive thymocytes were purified by FACS using mAbs against CD4 and CD8 (eBioscience). Peripheral naïve CD4+ T cells were purified by FACS using mAbs against CD4, CD45RB and CD25. DN3 and DN4 thymocytes were stained with mAbs against Mac1, Ter119, Gr1, B220, panNK, CD11b, CD11c, CD4, CD8 and γδ TCR do determine lineage positive populations. For flow cytometric analysis, mAbs against the following molecules were used: CD4 (RM4-5), CD44 (IM7), CD5 (53-7.3), CD8 (53-6.7), CD25 (PC61), CD69 (H1.2F3), B220 (RA3-6B2), TCRβ (H57), Foxp3 (FJK-16s). Flow cytometric analysis was performed using a LSRII (Becton Dickinson) and data analysis using FloJo.
Intracellular staining of thymocytes
Freshly obtained DN3/DN4 thymocytes were collected on ice and fixed in 4% final formaldehyde (Polysciences, Inc.) and permeablized in 95% final ice cold methanol. Permeablized cells were stained in 1% BSA PBS with pERK (Cell Signaling), pZap-70 (Cell Signaling), p-S6 Alexa-647 (Cell Signaling), and Bcl2 Alexa-647 (Biolegend). pERK and pZap-70 were detected with Alexa-647 conjugated goat secondary Ab against rabbit IgG (Life Technologies).
Transient transfection of HEK-293T cells
Transient transfection of HEK-293T cells was performed as previously described (23) with some modifications. HEK-293T cells were incubated in 6 well plates at 2×105/ well overnight at 37°C. AND TCRαβ (2A linked) plasmid (1 μg) and indicated titrated CD3 plasmid (1 μg, 0.25 μg, 0.06 μg) were transfected to HEK-293T cells using 6 μl TransIT LT1 transfection reagent (Mirus). Cells were harvested 40 h after transfection and stained with TCRβ mAbs for flow cytometry analysis.
Microscopic Analyses
Synthetic planar lipid bilayers containing anti-TCRβ (H57-597; Biolegend) and ICAM-1 were constructed as described (24, 25). For FRET analyses, sorted T cells were labeled with DiL (Life Technologies) and allowed to settle onto stimulatory lipid bilayers containing anti-TCRβ and ICAM-1, or onto non-stimulatory bilayers containing anti-Thy1.2 and ICAM-1. Cells were imaged with a spinning-disc laser-scanning confocal system and TIRF microscopy, and images were captured with Slidebook acquisition and analysis software (3i Technologies). Following acquisition of CD3ε-TFP fluorescence, the DiL acceptor was depleted using a 561 laser line and CD3ε-TFP fluorescence recovery (donor dequenching) was monitored and expressed relative to the pre-TFP fluorescence level which was assigned 100%. To determine the degree of CD3ε-TFP quenching, sorted T cells were allowed to adhere on glass coverslips coated with anti-Thy1.2. Single slice confocal images were taken at the midpoint of the z axis prior to (pre-Quench) and following (post-Quench) the addition of octadecyl rhodamine B chloride (R18; Life Technologies), with TFP fluorescence expressed relative to the pre-Quench level which was assigned 100%.
Confocal imaging was performed on a Leica SP5X microscope in RC-20H flow chamber (Harvard Apparatus) with constant flow of room temperature PBS. One sequential scan of TFP (458nm Ar laser excitation, 466-526nm detection) and octadecyl rhodamine B (R18, 560nm white light laser excitation, 568-628nm detection) was performed before and after R18 labeling for several fields within each flow cell. TFP fluorescence was measured at the plasma membrane before and after R18 labeling for each cell using ImageJ software. TFP quenching was calculated as EFRET (%) = (PQ-Q)/PQ where PQ = mean TFP prequench or pre-R18 labeling and Q = mean TFP post-R18 labeling.
Flu analysis and Tetramer staining
WT and CD3 mutant mice were infected intranasally with 104 egg infectious doses50 (EID50) of the recombinant influenza virus strain A/Aichi/2/68xA/Puerto Rico/8/34 (H3N2) (abbreviated as xX31). Bronchoalveolar lavage (BAL), spleen and mediastinal lymph nodes (MLN) were harvested on day 9 post-infection. Tetramer staining was performed as previously described (26). Briefly, influenza epitope-specific CD8+ CTL and CD4+ T cells were stained with fluorochrome-conjugated tetrameric complexes of the H2Db MHC class I glycoprotein and the influenza PA224-233 (SSLENFRAYV) peptide or IAb MHC class II glycoprotein and the influenza NP311-325 (QVYSLIRPNENPAHK) (obtained by agreement with the Trudeau Institute) in the presence of Fc block at room temperature in the dark. Following the tetramer stain, the cells were stained for surface markers (anti-CD4, anti-B220, anti-CD11b, anti-CD11c, anti-F4/80, all Pacblue conjugated for negative gating; PE-Cy7 conjugated anti-CD8, APC-Cy7 conjugated anti-CD43 and PercP-cy5.5 conjugated CD27. Intracellular staining (ICS) assays were performed as previously described (26). Briefly, cells from BAL, spleen and MLN were cultured in presence of IL2 and influenza PA223-233 peptide for 5 hrs at 37°C in a 96 well U-bottom plate. Cells were stained with PE conjugated anti-IFN-γ and APC conjugated anti-TNF-α in perm/wash buffer for 30 min on ice.
TCRβ sequencing
A 60 bp sequence of the rearranged TCRβ CDR3 region was amplified and sequenced for all samples using the immunoSEQ assay (Adaptive Biotechnologies). This is a multiplex PCR and high-throughput assay for the re-arranged DNA of T and B cells. This assay uses a combination of adjusted primer concentrations and computational corrections to correct for PCR bias common to multiplex PCR. Raw sequence data was filtered based on the TCRβ V, D and J gene definitions provided by the IMGT database (www.imgt.org) and binned using a modified nearest-neighbor algorithm to merging closely related sequences and remove both PCR and sequencing errors (27-29).
T cell proliferation assay
CD4+CD25− T cells were sorted as described above then plated at 0.2×106 cell per well in a round bottom 96 well plate with titrating amounts of plate bound anti-CD3 (2C11) and constant amount of soluble anti-CD28 (0.5μg/ml). After 48 hours, 3H was added to all wells and harvested 12hrs later.
Immunoblot analysis
Immunoblot analysis of T cell blasts expressing CD3ε was done as described. Briefly, FACS purified peripheral CD4+CD25− T cells were expanded in vitro with IL-2 for 10-14 days and then allowed to rest for 24hrs in 10% FBS without IL-2. Cells were then labeled with biotinylated anti-TCRβ (H57-597: Biolegend) for 15 min on ice. CD4 T cells were washed of excess antibody and rested for 30 min at 37°C in PBS. After resting, cells were crosslinked with streptavidin (Sigma) for indicated times followed by quick centrifugation and lysis in immunoprecipitation buffer containing protease and phosphatase inhibitors (Sigma). Proteins were transferred onto nitrocellulose membranes, nonspecific binding was blocked with 3% nonfat milk (Bio-Rad) and membranes were probed with anti-pERK, pZap70, pPLCγ and total ERK, Zap70, PLCγ PTEN (Cell Signaling Technologies) in 3% BSA-TBST. Additionally, freshly isolated CD4+CD8+ DP thymocytes were FACs purified and rested for 15 minutes at 37°C in PBS. Thymocytes were then put on ice, spun briefly and lysed in NP40 lysis buffer containing as described above.
Retroviral-mediated stem cell gene transfer
Retroviral transduction of murine bone marrow cells was performed as described earlier (9, 18, 19, 30). Briefly, we harvested bone marrow from 8- to 10-week-old donor mice 72h after treatment with 150mg/kg 5-fluoruracil (Pharmacia & Up- John). Bone marrow cells were single cell suspended and cultured in complete DMEM with 20% fetal bovine serum (FBS), murine IL-3 (20ng/ml), human IL-6 (50ng/ml), and murine SCF (50ng/ml) (Biosource-Invitrogen) for 24h. Cells were subsequently spin-transduced with filtered retroviral supernatant plus polybrene (6mg/ml) and cytokines as detailed above. Transduced bone marrow cells were collected, washed, and cells resuspended in phosphate-buffered saline (PBS) containing 2% FBS with heparin (20U/ml). Bone marrow cells (4×106 per mouse) were injected via the tail vein into irradiated (500rads) Rag1−/− recipient mice. Retrogenic mice were analyzed 4-6 weeks after bone marrow transplant.
Transduction of Phogrin-4 cell line
Transduction of Phogrin-4 cell line was performed as previously described in the Retroviral-mediated stem cell gene transfer section. Briefly, Phogrin-4 cells were transduced using retroviral supernatant from GPE86 producer cell line that express chimeric OKT CD3ε or WT CD3ε. After 2 spin-transductions, the transduced Phogrin-4 cells were purified by FACS based on the same fluorescent expression.
Statistical Analysis
Unless otherwise described, all analysis was performed using Prism 5, GraphPad Software. All pair-wise comparisons were performed using paired student’s t-test and significant results checked using the non-parametric equivalent Mann-Whitney U-test or one sample t-test. Group comparisons were done using a two-factor ANOVA unless otherwise specified in figure legends
RESULTS
CD3ε-BRS is required for efficient DN3-DN4 transition
Three CD3ε-BRS mutants (CD3εm1, CD3εm2, CD3εm3) were generated to determine the role of this region in modulating early thymocyte development (Supplemental Fig. 1A). Previous studies had shown that mutation of the first or second cluster of basic residues in the CD3ε cytoplasmic domain (corresponding to CD3εm1 and CD3εm2, respectively) reduced plasma membrane binding while mutation of both clusters (corresponding to CD3εm3) resulted in loss of membrane binding (13, 17). We used several approaches to determine if these CD3ε mutations influenced association with the rest of the TCR:CD3 complex and cell surface expression. First, all CD3ε mutants were able to pair with TCR efficiently when expressed on the surface of 293T cells (Supplemental Fig. 1B). Second, in a competitive environment in which chimeric proteins containing a human CD3ε ectodomain and a murine wild-type CD3εwt or mutant CD3εm3 cytoplasmic domain had to compete with murine CD3εwt in a murine T cell clone, comparable expression was observed with only a marginal reduction in CD3εm3, that was not significant, in TCR expression (Supplemental Fig. 1C). Thus, there does not appear to be a significant affect of these CD3ε mutations on TCR cell surface expression.
We next generated CD3 retrogenic mice (18, 19, 30) to determine the affect of CD3ε-BRS mutations on the relative association of the cytoplasmic domain with the plasma membrane in normal T cells (Supplemental Fig. 1D). This approach allowed us to express modified CD3ε chains in CD3.KO mice (Cd3eΔP/ΔP:Cd247−/− mice lack expression of all four CD3 chains, herein referred to as CD3.KO (9, 20, 21)) by retrovirus-mediated stem cell gene transfer with multicistronic 2A peptide-linked retroviral vectors. For membrane association analysis, we generated retrogenic mice in which wild type (WT) and mutant CD3ε was linked to teal fluorescent protein (TFP) and fluorescence resonance energy transfer (FRET) measured in the presence or absence of a membrane dye DIL. A small but significant reduction in FRET (Quenching) was observed with T cells expressing CD3εm3.TFP compared with CD3εwt.TFP (Supplemental Fig. 1E). In addition, mice expressing a single TCR (AND) also displayed reduced plasma membrane binding with CD3εm3.TFP by measuring FRET efficiency (Supplemental Fig. 1F). A de-quenching approach was also utilized to measure proximity of the CD3ε to the membrane. In this method, a laser bleaches the lipid dye, halting FRET and allowing TFP to fluoresce. Again, a small but significant increase in fluorescence was observed after de-quenching with T cells expressing CD3εm3.TFP and CD3εm1.TFP (Supplemental Fig. 1G). It is possible that these approaches are not sensitive enough to observe reduced membrane binding with CD3εm2, which possesses a more limited set of mutations. These data confirm previous studies suggesting that the BRS is required for optimal membrane association of the CD3ε cytoplasmic domain (13).
CD3 retrogenic mice were generated to gain insight into the role of electrostatic interactions between the CD3ε-BRS with acidic phospholipids in modulating thymocyte development. Total thymic cellularity was significantly reduced in mice expressing each of the three CD3ε-BRS mutants compared with CD3εwt control mice (Fig. 1A). Successful thymocyte development through two major checkpoints is dependent on optimal TCR:CD3 expression and signaling. The first checkpoint takes place at the DN3-DN4 transition when TCRβ is expressed on the cell surface with the pre TCRα chain (pTα), while the second checkpoint occurs at the CD4+CD8+ double positive (DP) stage when positive and negative selection occurs (31). Both checkpoints require a defined level of TCR signaling to provide optimal survival and developmental signals. Flow cytometric analysis of thymocyte subpopulations from CD3ε motif mutant mice revealed a substantial impact on T cell development, particularly at the DN3-DN4 transition. There was a significant increase in the fraction of DN thymocytes and a concomitant reduction in DP thymocytes, in particular in CD3εm3 mice (Fig. 1B). Interestingly, there was a significant increase in the percentage of the DN3 (CD25+CD44−) population compared with a dramatic loss of the DN4 (CD25−CD44−) population indicating a block at this critical maturation checkpoint (Fig. 1C and Supplemental Fig. 2A). As a result, the DN3:DN4 ratio was significantly increased in all mutants with a dramatic loss in total DN4 cell numbers.
Figure 1.
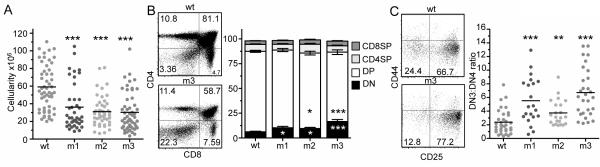
Mice with mutations in the CD3ε-BRS exhibit major deficiencies in thymic development.
(A) Rag1−/− mice reconstituted with bone marrow utilizing mutant CD3ε chains have reduced thymic cellularity (n=26-40 mice per group, at least 5 separate experiments).
(B) Abnormal frequencies of DN and DP thymocytes in mice expressing mutant CD3ε chains (n=21-34 mice per group, at least 5 separate experiments). Statistical significance was determined for all groups at the DN stage and for m2 and m3 at the DP stage when compared to the WT group.
(C) After gating on lineage negative thymocytes (B220, γδ TCR, Gr1, pan NK, CD11c, CD11b, Ter119, CD4, CD8 negative) early thymocyte stages of development were analyzed. Mice utilizing mutant CD3ε chains demonstrate a block at the DN3-DN4 transition (n=23-29 mice per group, at least 5 separate experiments). Statistical Analysis was performed using Mann Whitney t-test. *p<0.05, **p<0.01 ***p<0.001.
Mutations in the CD3ε-BRS may affect TCR signaling (13, 14), thereby altering thymocyte development. Nur77GFP reporter mice have been shown to provide a faithful readout for endogenous TCR signal strength(22). In order to determine TCR signaling activity at key thymocyte developmental checkpoints, CD3ε WT and BRS mutant retrogenic mice were generated using Nur77GFP:CD3.KO bone marrow. Interestingly, Nur77GFP expression in TCR+ DN4 thymocytes was significantly higher in the CD3ε mutant groups (Fig. 2A). Strong TCR signaling causes CD5 upregulation and TCR down-modulation. Importantly, TCR expression on the CD3ε mutant thymocytes at the DN3/4 transition was similar to or higher than CD3εwt cells, indicating normal initial TCR expression (Supplemental Fig. 2B). However, consistent with the enhanced Nur77GFP expression observed, CD5 expression was significantly higher for all CD3ε mutants and TCR expression significantly lower on DN4 thymocytes from CD3εm1 and CD3εm2 mice compared with CD3εwt retrogenic mice (Fig. 2B). Furthermore, pZAP70 and pERK expression was significantly higher in CD3ε mutant versus WT retrogenic mice (Fig. 2B, Supplemental Fig. 2B). However, Akt/TORC activity appeared to be diminished as shown by reduced pAkt.T473 and pS6 (Supplemental Fig. 2D). This was consistent with a significant decrease in Bcl2 expression and a significant increase in Annexin V staining of DN3 thymocytes for all CD3ε mutant groups, indicating pre-TCR signals may not be optimal for survival causing a developmental hiatus and cell death at prior to transition to DN4 (Fig. 2C and Supplemental Fig. 2E). However, there was no difference in Annexin V staining once the DN3 cells transitioned into DN4 thymocytes (Fig. 2C). Overall, our data indicate that mutations in the CD3ε-BRS negatively impact early thymocyte survival leading to a partial block at the first developmental checkpoint.
Figure 2.

Mice with mutations in CD3ε-BRS have enhanced pre-TCR signaling.
(A) Lineage negative (B220, γδ TCR, Gr1, pan NK, CD11c, CD11b, Ter119, CD4, CD8 negative) CD44−CD25− DN4 thymocytes were gated on TCRβ+ and analyzed for Nur77 expression (n=23-25 mice, 6 independent experiments). RFI – Relative Fluorescent Intensity.
(B) Lineage negative CD44−CD25− DN4 thymocytes, were analyzed for CD5 and TCRβ expression (n=12 mice per group, 3 independent experiments). Lineage negative CD44−CD25− DN4 thymocytes were also analyzed for basal pZap70 (n=12-14 mice per group, 3 independent experiments).
(C) Lineage negative CD44−CD25+ DN3 and CD44−CD25− DN4 thymocytes were analyzed for AnnexinV labeling (18-35 mice per group, at least 5 independent experiments). Expression is graphed as relative to the WT CD3ε chain transduced control. Statistical Analysis was performed using Mann Whitney t-test (A,B) and one sample t-test (C). *p<0.05, **p<0.01 ***p<0.001
CD3ε-BRS is required for optimal thymocyte positive selection
We next determined the influence of CD3ε-BRS mutations on positive and negative selection. Initially analysis of the ratio between DP and SP thymocytes suggested that there might not be a significant affect on T cell selection, despite substantial reductions in total thymocyte numbers (Fig. 1B). However, as multiple mechanisms have been shown to compensate for defects in positive and negative selection that can mask the affect of mutations that disrupt signaling, we examined these critical T cell developmental steps further (6).
To fully interrogate the affect of CD3ε-BRS mutations on positive and negative selection, CD3εwt and CD3εm3 retrogenic mice were generated that expressed the male antigen-specific, H-2Ab-restricted Vβ6+ Marilyn TCR, by using Rag1−/−:CD3.KO mice as bone marrow donors (Fig. 3). In this model, thymocytes expressing the Marilyn TCR are positively selected in the absence of endogenous HY antigen (female mice), but are negatively selected in the presence of HY antigen (male mice) (19). Thymic cellularity was substantially reduced and Vβ6+CD4+ SP thymocytes were completely absent in male CD3εwt and CD3εm3 retrogenic mice (Fig. 3A and 3B). In addition, there was a significant decrease in DP thymocytes in male mice compared to female, however there were equal numbers of DP thymocytes when CD3εwt and CD3εm3 male mice were compared, suggesting that negative selection was intact in the presence of the BRS mutations (Fig. 3C).
Figure 3.

CD3ε-BRS regulates positive but not negative selection.
(A) CD3εζ deficient Rag−/− bone marrow was transduced with WT or CD3εm3 chains in addition to the male antigen specific Marilyn TCR. Both male and female Rag−/− mice were then reconstituted with the transduced bone marrow to visualize selection on a negative and positive selecting background. Total cellularity for each thymus was determined (n=13-15 mice per group, 3 independent experiment).
(B) The percentage of CD4+Vβ6+ single positive thymocytes that survived negative selection is indicated. Representative and combined data are shown (n=10 mice per group, 2 independent experiments).
(C) Total double positive (CD4+CD8+Ametrine+) cellularity was determined for each male and female retrogenic mouse (n=10 mice per group, 2 independent experiments).
(D) The percentage of mature CD24loCD4+ thymocytes (gated as indicated) that survived positive selection. Representative and combined data are shown. (n=10 mice per group, 2 independent experiments).
(E) WT and mutant CD3ε CD4+Vβ6+ single positive thymocytes TCR expression was measured. Representative and combined data are shown. Expression is graphed as relative to the WT CD3ε chain transduced control (n=10 mice per group, 2 independent experiments). RFI – Relative Fluorescent Intensity. Statistical Analysis was performed using Mann Whitney t-test (A-D) and one sample t-test (E). *p<0.05, **p<0.01, ***p<0.001.
In contrast, there was a trend towards reduced thymic cellularity in female CD3εm3 compared with CD3εwt retrogenic mice, albeit not significant (Fig. 3A). However, there was a significant reduction in the percentage of Vβ6+CD4+ SP thymocytes in female CD3εm3 compared with CD3εwt retrogenic mice as well as the mature CD4+CD24lo thymocyte subpopulation (CD24 is down-regulated in mature SP thymocytes (32)) and an overall reduction in Vβ6 TCR expression (Fig. 3B,D-E). Thus, the CD3ε-BRS appears to regulate positive but not negative selection.
Given these observations, we analyzed the WT and mutant CD3ε retrogenic mice further to determine the mechanistic basis for CD3ε-BRS regulation of positive selection. Although the CD3ε-BRS mutants did not exhibit any significantly reduced capacity to form stable TCR:CD3 cell surface complexes in vitro or on DN3/4 thymocytes (Fig. 2B, Supplemental Fig. 1B and 1C), TCR expression was reduced on DP thymocytes, CD69lo DP, CD4+ and CD8+ SP thymocytes and in peripheral T cells in all CD3ε mutants groups compared with WT CD3ε retrogenic mice (Fig. 4A and Supplemental Fig. 3A). Reduced TCR expression may influence the signaling that mediates positive selection. Thymocytes undergoing positive selection express CD69 and upregulate TCR (22). There was a significant reduction in the percentage of CD69hiTCRhi DP thymocytes in all CD3ε mutant groups indicative of a reduced ability to undergo positive selection (Fig. 4B). This coincided with increased apoptosis of DP CD3 mutant thymocytes as determined by AnnexinV/7AAD staining (Fig. 4C). Lastly, there was a significant reduction in CD4+CD24lo mature thymocytes in all CD3ε mutant groups, while the reduction of CD8+CD24lo cells was less dramatic (Fig. 4D and Supplemental Fig. 3D). Collectively, these data suggest that the CD3ε-BRS facilitates optimal positive selection of polyclonal thymocytes.
Figure 4.

Thymocytes with mutations in the CD3ε-BRS display lower TCR expression and do not efficiently develop into mature thymocytes.
(A) TCR expression was measured and compared to WT CD3ε CD69lo pre-selected DP as well as CD4 and CD8 SP thymocytes. Representative and combined data are shown (n=24-30 mice per group, 5 independent experiments).
(B) Percentage of DP thymocytes undergoing positive selection (CD69hiTCR+) was measured relative to WT CD3ε expression (n=43-50 per group, 8 independent experiments). Representative and combined data are shown.
(C) Annexin V staining of DP thymocytes. Representative and combined data shown (n=24-42 mice per group, at least 5 independent experiments).
(D) Total mature CD4 thymocyte ratios (CD24loCD4+) were calculated. Representative and combined data are shown (n=28-31 mice per group).
(E) Nur77 expression (MFI) was calculated for DP thymocytes. Representative and combined data are shown (n=9-15 mice per group, 3 independent experiments).
(F) Representative and combined densitometry of pZap70 and pPLCγ by western blot analysis of sorted DP (n=3). Statistical Analysis was performed using one sample t-test (A,F) and Mann Whitney t-test (BE,G). *p<0.05, **p<0.005 ***p<0.0001
We next determined the affect of the CD3ε-BRS on downstream signaling events in DP thymocytes. In contrast to DN thymocytes, CD3ε-BRS mutant DP thymocytes expressed a lower percentage of Nur77GFP expressing cells and lower amounts than WT controls (Fig. 4E and 4F), raising the possibility that signaling was defective in CD3ε mutant DP thymocytes. Although WT DP thymocytes exhibit tonic basal Zap-70 phosphorylation (33), CD3ε-BRS mutant DP thymocytes exhibited minimal Zap-70 phosphorylation (Fig. 4G). Furthermore, there was also reduced phosphorylation of the downstream target PLCγ, in contrast to WT DP thymocytes. Lastly CD5 expression, which is known to be upregulated by TCR signaling in DP thymocytes, was significantly decreased with all three CD3ε-BRS mutants (Figure Supplemental Fig. 3E). Taken together, these data suggest that the CD3ε-BRS regulates TCR signaling in DPs thymocytes to facilitate optimal positive selection.
Finally, in order to determine if the defects observed with CD3ε mutant thymocytes are cell intrinsic, mixed bone marrow chimeras were generated. CD3.KO bone marrow cells were transduced with either CD3εwt (GFP-marked) or CD3εm3 (Ametrine-marked), mixed at a 1:1 ratio and used to reconstitute Rag1−/− mice (Fig. 5A). Equal reconstitution was verified by analysis of fluorescent protein-marked B cells (Fig. 5B). Consistent with our observations above, the CD3εm3 mutant exhibited a reduced fraction of total thymocytes, increased DN3:DN4 ratio and a reduced percentage of DP thymocytes compared with the CD3εwt controls (Fig. 5C-E). Taken together, these data suggest that CD3εm3 mutant thymocytes are at a competitive disadvantage compared with their CD3εwt counterparts, and that the defects observed in the absence of an intact CD3ε-BRS are cell intrinsic.
Figure 5.

Defects observed in CD3ε-BRS mutant thymocytes are cell intrinsic.
(A) CD3.KO bone marrow cells were transduced with either CD3εwt or CD3εm3. Rag1−/− mice were reconstituted with a 1:1 mixture using GFP to mark CD3εwt and Ametrine to mark CD3εm3. (B) The B220 transduction efficiency for each mouse was calculated. Representative and combined data are shown (n=10 mice per group, 2 independent experiments for all analysis). (C) The ratios of WT CD3ε and mutant CD3ε transduced thymocytes were calculated.
(D) The DN3:DN4 thymocyte ratio was calculated for each individual WT:mutant chimera.
(E) The ratios of WT CD3ε and mutant CD3ε transduced thymocytes were calculated for CD4+CD8+ DP thymocytes. n=10 mice per group with 2 independent experiments for all analysis. Statistical Analysis was performed using Paired two-tailed t-test. *p<0.05
CD3ε-BRS mutations limit peripheral T cell signaling and function
We next evaluated whether the defects observed in thymocyte development affected the establishment and function of peripheral T cells. Reduced TCR expression observed on DP and SP thymocytes was also observed with peripheral T cells (Supplemental Fig. 3A). However, homeostatic proliferation and the substantially lower tonic signaling events required for peripheral survival limited the contribution of the CD3ε-BRS on CD4+ and CD8+ cellularity (Supplemental Fig. 3B). Significant alterations in peripheral T cell numbers were restricted to the spleen for mice expressing CD3εm3 and to lymph nodes for mice expressing CD3εm1. All three CD3ε-BRS mutants had a somewhat greater affect on the percentage and number of CD4+Foxp3+ regulatory T cells (Tregs) in the spleen, while CD3εm1 appeared to impact Tregs in the lymph nodes (Supplemental Fig. 3C). This did not appear to be a developmental defect as the percentage of Foxp3+ thymocytes in WT and-BRS mutant CD3ε retrogenic mice was comparable (unpublished observations). However, these reductions did not appear to induce any autoimmune or inflammatory manifestations within the time frame of analysis (7 weeks post bone marrow reconstitution; unpublished observations). These data raise the possibility that Tregs may be more sensitive to aberrant TCR signaling caused by CD3ε-BRS mutations than conventional T cells in the periphery.
It is conceivable that the defective thymocyte positive selection observed in CD3ε-BRS mutant mice and subsequent homeostatic expansion to occupy the peripheral niche could have given rise to an altered TCR repertoire. However, extensive analysis by high-throughput next-generation sequencing did not reveal any clear differences in TCR Vβ usage by CD3ε-BRS mutant versus WT CD4+ SP thymocytes and peripheral CD4+ T cells despite the large spread in cellularity (Supplemental Fig. 4A-C, Supplemental Fig. 3B and Fig. 1A). Interestingly, the average CDR3 amino acid length of the TCRs is significantly shorter in peripheral CD4+ T cells (Supplemental Fig. 4D). The differences in average CD3 length was greatest for CD3εwt T cells compared with either CD3εm2 and CD3εm3 mutant T cells in the periphery versus the thymus suggesting a restricted TCR diversity, whereas there was no statistically significant difference between groups in the thymus (Supplemental Fig. 4D). Additionally, the CD3εm2 and CD3εm3 mice were more oligoclonal (higher clonality) compared with the WT peripheral CD4+ T cells again indicating a restricted diversity in the presence of CD3ε-BRS mutations (Supplemental Fig. 4E).
We next assessed TCR signaling and functional capacity of peripheral CD3ε WT and BRS mutant T cells. Phosphorylation of both proximal (PLCγ and Zap70) and distal (ERK and AKT-T308) TCR signaling proteins was substantially diminished in all CD3ε-BRS mutants (Fig. 6A). Interestingly, a known T cell activation-associated phosphatase that limits Akt activity, PTEN, was upregulated in CD3ε-BRS mutant T cells (Fig. 6B). Finally, we determined the proliferative capacity of CD3ε-BRS mutant peripheral CD4+CD25− T cells, revealing a significant reduction in responsiveness to stimulation by anti-CD3 plus anti-CD28 (Fig. 6C). Collectively, these data suggest that the CD3ε-BRS mutations limit effective TCR signaling and proliferative capacity, which may be due in part to increased PTEN expression.
Figure 6.

Diminished TCR signaling and proliferative capacity observed in CD3ε-BRS mutant T cells. (A) Expanded CD4+CD25− T cells were rested and then were stimulated at indicated time points by crosslinking H57 biotin (anti-TCRβ) with SA. Phosphorylation of downstream proteins was measured by western blot analysis (pPLC-γ, pZap-70, pERK1/2 and pAKT-T308). Representative blot shown (n=3).
(B) Total PTEN protein was measured for each group of cells at time 0 by comparing to tubulin, Representative blot shown along with relative PTEN expression at time 0 (n=3).
(C) Proliferation of freshly purified CD4+CD25− T cells with titrated plate-bound 2C11 (anti-CD3ε) and soluble CD28. Raw combined data on left, normalized to each experiment (compared to WT) on right (n=4). Statistical Analysis was performed using Mann Whitney t-test. *p<0.05
Finally, to determine the physiological importance of the CD3ε-BRS in facilitating the generation of an optimal immune response to viral infection, we assessed the capacity of CD3εwt and CD3εm3 retrogenic mice to respond to X31 influenza virus. The number and percentage of CD4+ and CD8+ CD3εwt and CD3εm3 T cells in the bronchoalveolar lavage (BAL) and spleen were unchanged 10 days post-infection (Fig. 7A and unpublished observations). In contrast, there was a significant reduction in the percentage of influenza-specific CD4+ and CD8+ T cells in the BAL of CD3εm3 compared with CD3εwt infected mice (Fig. 7B and 7C). There was also a significant reduction in the percentage of TNFα+IFNγ+ CD8+ BAL CD3εm3 T cells following in vitro restimulation with influenza PA peptide (Fig. 7D), as well as in the TNFα+IFNγ+:total IFNγ+ CD8+ T cell ratio (Fig. 7E), indicating that the quality and functional capacity of the CD3εm3 T cell population is impaired (34). Importantly, defects in the accumulation of influenza-specific T cells in the lungs of CD3εm3 infected mice led to a 10-fold increase in viral titer compared with CD3εwt mice (Fig. 7F). Taken together, these data demonstrate that the CD3ε-BRS is required for the optimal development of a protective immune response to viral infection.
Figure 7.

CD3ε-BRS is required for the optimal development of a protective immune response to viral infection.
(A) Rag1−/− mice were reconstituted with CD3.KO bone marrow transduced with either WT CD3ε or mutant CD3ε. After 5 weeks of reconstitution, mice were challenged with a low dose (1×104 EID50) of the X31 influenza virus strain. Mice were monitored for 10 days and then analyzed. The ratio and cell number of total CD8+ and CD4+ T cells in the spleen and BAL.
(B) The ratio of CD4+ tetramer (IAbNP311-325) –specific cells was measured (9-10 mice per group, 2 independent experiments).
(C) The ratio of CD8+ PA tetramer (DbPA224-233–specific T cells in the BAL (11-12 mice per group, 3 independent experiments).
(D) Cells isolated from the BAL were stimulated with PA224-233 peptide and CD8+ T cells were assessed for IFNγ and TNFα cytokine production (11-12 mice per group, 3 independent experiments).
(E) Ratio of cells that are IFNγ+TNFα+ of all IFNγ+ cytokine production (11-12 mice per group, 3 independent experiments).
(F) Total viral titer in the lung was measured (n=8-10 mice, 2 independent experiments by plaque assay. Statistical Analysis was performed using Mann Whitney t-test. *p<0.05, **p<0.01 ***p<0.001.
DISCUSSION
The induction of TCR signaling following recognition of peptide:MHC complexes is a multifaceted and tightly regulated event. Multiple layers of positive and negative regulation are required to safeguard signaling integrity. This ensures optimal T cell thymic differentiation and induction of protective immune responses to pathogens, while negating adverse inflammatory or autoimmune consequences. Recent studies have suggested that physical partitioning of the CD3ε and CD3ζ cytoplasmic signaling domains into the phospholipid membrane, thereby limiting access to the cytosolic signaling machinery, may offer a critical control mechanism for initiation of TCR signaling (13, 14). Membrane-proximal BRSs mediate the sequestration of the CD3ε and CD3ζ cytoplasmic tails into the inner leaflet of the cell membrane. However, the physiological importance of membrane sequestration and release of CD3ε and CD3ζ cytoplasmic tails remains obscure and the outcome could not be easily predicted based on what is currently known. Given that CD3ε can engage the adapter protein Nck as well as the signaling proteins ZAP-70, Lck and Fyn, membrane sequestration may offer another layer of protection from inadvertent TCR signaling (35). In addition, recent studies have shown the BRS region of CD3ε also engages the Ser/Thr kinases GRK2 and HPK1. GRK2 association with the TCR complex may allow for CXCR4 to utilize the CD3 invariant chains for it’s own signal transduction and to facilitate T cell migration (36). Therefore, it is possible CD3ε interactions with GRK2 may allow for proper migratory signals in a developmental setting such as the thymus. Here we establish a critical role for the CD3ε-BRS in T cell thymic differentiation and functional competence. It is important to note that mutating only three of the six residues in this motif significantly impacted not only DN3-DN4 differentiation but also subsequent TCR expression and signaling capacity. It was also clear that thymocytes expressing CD3ε-BRS mutations exhibited cell intrinsic defects and were at a competitive disadvantage compared with WT thymocytes, as determined in mixed bone marrow chimera experiments. Importantly, there did not appear to be a difference in the ability of WT CD3ε and the CD3ε-BRS mutants to pair and assemble with WT TCRαβ and CD3γδζ chains, even in competitive environments, although it is difficult to completely rule out an affect on assembly. Interestingly, even a reduction in CD3ε cytoplasmic tail:lipid membrane interaction (CD3εm1 and CD3εm2) had a dramatic effect on T cell thymic differentiation, emphasizing the importance of this control mechanism.
The physiological consequence of limiting CD3ε cytoplasmic tail:lipid membrane interaction resulted in limited DN3-DN4 transition, reduced thymic cellularity, increased thymocyte apoptosis and decreased Bcl-2 expression. Pre-TCR signaling at the DN3-DN4 transition induces TCRβ upregulation (37). However, thymocytes expressing the CD3ε-BRS mutations exhibited reduced TCRβ expression and increased CD5 expression, indicative of increased TCR signaling. Indeed, pZAP70, pErk and Nur77GFP, which are used as a direct readout of TCR signal strength (22), were both enhanced in mutant DN4 thymocytes. These observations are consistent with a model in which the enhanced accessibility of the CD3ε cytoplasmic tail, due to the BRS mutations, enhances ITAM phosphorylation and subsequent downstream signaling. Consistent with previous observations, increased pre-TCR signaling leads to enhanced apoptosis, reduced cellularity, increased CD5 expression and TCR downmodulation (14). However, it is unclear whether cells that survive and differentiate into DP thymocytes do so because of reduced TCR expression, attenuated TCR signaling or a combination thereof. Collectively, it is clear that the CD3ε-BRS is pivotal in optimizing pTCR signaling and early thymocyte differentiation. Although it is known that a defined minimum threshold of pTCR signaling is required to induce DN3/4 transition and TCRα rearrangement, it has been unclear whether there is a maximum, upper threshold that must not be exceeded. Future studies should address whether CD3ε association with the lipid membrane alters between thymocyte populations at the DN3/DN4 transition, at the DP stage and finally with mature single positive thymocytes. It has been shown that local changes in the lipid environment can regulate membrane binding by the CD3ε (38). Furthermore, Ca2+ regulates CD3 phosphorylation and T cell receptor activation by manipulating lipid charge (39). It would therefore be interesting to determine in future studies whether the lipid environment found around the TCR microclusters at each stage of thymocyte development is altered due to Ca2+ changes from basal TCR signaling. Our data support such a notion suggesting that pTCR signaling must occur within a defined window to ensure optimal thymocyte development. Furthermore, it is possible this window is relatively narrow as subtle changes in CD3ε-BRS membrane association and enhanced pTCR signing led to profound effects on thymocyte development and subsequent peripheral T cell immune responses to a viral pathogen. Thus, our data support the concept that the CD3ε-BRS serves as a critical quality control rheostat to ensure optimal pTCR signaling.
We also observed defective positive selection of CD3ε-BRS mutant DP thymocytes, as evidenced by reduced Marilyn TCR thymocyte differentiation in female mice, reduced differentiation of polyclonal CD69hiTCRhi DP thymocytes and CD24loCD4+ SP subpopulations. This may be due to reduced TCR signaling as CD3ε-BRS mutant DP thymocytes exhibit reduced pZap70, pPLCγ and Nur77GFP expression. It is currently unclear whether this is directly due to altered TCR signaling in DP thymocytes, reduced TCR expression resulting from an indirect consequence of disrupted pTCR signaling or combination thereof. However, given that mutant DP thymocytes express lower TCR as a consequence of disrupted pTCR signaling it seems plausible that this underlies the defective positive selection observed. While there does not appear to be an increase in the expression of negative signaling regulators, such as SHP1 or CSK (unpublished observations), we cannot rule out this possibility. Interestingly, while we observed defects in positive selection, no defects in negative selection were observed, as evidenced by comparable deletion of Marilyn TCR DP thymocytes in male CD3ε WT and BRS mutant mice. This suggests that the defects observed do not impinge on the enhanced signaling and Erk redistribution required to mediate negative selection (40). However, given that our analysis was restricted to one TCR system, we cannot rule out the possibility that negative selection of some TCRs may be affected. Moreover, we observed no alteration in TCR repertoire selection, as determined by high-throughput next-generation sequencing, even though more mice would need to by assessed to gain a full appreciation of the breadth and depth of the TCR repertoire. Taken together, it appears that the CD3ε-BRS is required for optimal positive thymocyte selection but may be dispensable for negative selection.
Finally, we also observed substantive defects in the function of peripheral T cells and the development of a protective immune response against influenza infection. TCR signaling in peripheral CD3ε-BRS mutant T cells was defective, as observed by decreased phosphorylation of the proximal and distal TCR signaling proteins PLCγ, Zap70, Erk and Akt, consistent with previous observations (14). As discussed above, it is not clear whether this is due to defective TCR signaling in peripheral T cells or an indirect consequence of disrupted pTCR signaling, reduced TCR expression and differentiation. Interestingly, CD3ε-BRS mutant T cells expressed elevated levels of the negative regulatory phosphatase PTEN, although it remains to be determined whether PTEN plays a role in the defective signaling observed or is an indirect consequence of defective signaling events in the thymus. Attenuated signaling observed with PLCγ, Zap70, Erk cannot be a direct consequence of increased PTEN, thus it will be important in future studies to determine the regulatory protein that is responsible for the results observed. Regardless, the signaling defects have detrimental physiological implications manifest in limited generation of influenza-specific T cells and poor viral clearance. These observations clearly suggest that the CD3ε-BRS is essential for the generation of a protective immune response to pathogens.
Collectively, these observations highlight the physiological importance of having TCR intrinsic molecular mechanisms, mediated in part by the CD3ε-BRS, to finely control TCR signaling and its downstream consequences. Although not directly examined, it seems plausible that the CD3ζ-BRS may also contribute critically to regulating TCR signaling and thymocyte differentiation. By enabling plasma membrane binding of the CD3ε cytoplasmic domain, the BRS ensures proper timing of signaling, thereby ensuring optimal thymocyte differentiation and protective immune response against pathogens.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Kristin Hogquist for Nur77GFP mice(22) and helpful comments, Lana McClaren and Melissa Morris for technical assistance with the X31 harvest and analysis, Karen Forbes and Amy McKenna for maintenance, breeding and genotyping of mouse colonies, Parker Ingle, Stephanie Morgan, Greg Lennon and Richard Cross of the St. Jude Immunology Flow Lab for cell sorting, the staff of the Shared Animal Resource Center at St Jude for the animal husbandry, the Hartwell Center for Biotechnology and Bioinformatics at St Jude for primers and sequencing, and the Vignali Lab for helpful discussions.
Footnotes
This work was supported by the National Institutes of Health (R01 AI52199 to D.A.A.V., R01 AI054520 to K.W.W. and R01 AI107625 to P.G.T.), Hartwell Postdoctoral Fellowship (to M.L.B.), Fellowship by the Cancer Research Institute (to E.G.), NCI Comprehensive Cancer Center Support CORE grant (CA21765, to D.A.A.V.), and ALSAC (to D.A.A.V.).
Conflict of Interests David Hamm is an employee of Adaptive Biotechnologies. The remaining authors declare no competing financial interests.
References
- 1.Morris GP, Allen PM. How the TCR balances sensitivity and specificity for the recognition of self and pathogens. Nat Immunol. 2012;13:121–128. doi: 10.1038/ni.2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moran AE, Hogquist KA. T-cell receptor affinity in thymic development. Immunology. 2012;135:261–267. doi: 10.1111/j.1365-2567.2011.03547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guy CS, Vignali DA. Organization of proximal signal initiation at the TCR:CD3 complex. Immunol Rev. 2009;232:7–21. doi: 10.1111/j.1600-065X.2009.00843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gil D, Schamel WW, Montoya M, Sanchez-Madrid F, Alarcon B. Recruitment of Nck by CD3 epsilon reveals a ligand-induced conformational change essential for T cell receptor signaling and synapse formation. Cell. 2002;109:901–912. doi: 10.1016/s0092-8674(02)00799-7. [DOI] [PubMed] [Google Scholar]
- 5.Gil D, Schrum AG, Alarcon B, Palmer E. T cell receptor engagement by peptide-MHC ligands induces a conformational change in the CD3 complex of thymocytes. The Journal of experimental medicine. 2005;201:517–522. doi: 10.1084/jem.20042036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kruisbeek AM, Haks MC, Carleton M, Michie AM, Zuniga-Pflucker JC, Wiest DL. Branching out to gain control: how the pre-TCR is linked to multiple functions. Immunol Today. 2000;21:637–644. doi: 10.1016/s0167-5699(00)01744-8. [DOI] [PubMed] [Google Scholar]
- 7.Plas DR, Williams CB, Kersh GJ, White LS, White JM, Paust S, Ulyanova T, Allen PM, Thomas ML. Cutting edge: the tyrosine phosphatase SHP-1 regulates thymocyte positive selection. J Immunol. 1999;162:5680–5684. [PubMed] [Google Scholar]
- 8.Brodeur JF, Li S, da Silva Martins M, Larose L, Dave VP. Critical and multiple roles for the CD3epsilon intracytoplasmic tail in double negative to double positive thymocyte differentiation. J Immunol. 2009;182:4844–4853. doi: 10.4049/jimmunol.0803679. [DOI] [PubMed] [Google Scholar]
- 9.Holst J, Wang H, Eder KD, Workman CJ, Boyd KL, Baquet Z, Singh H, Forbes K, Chruscinski A, Smeyne R, van Oers NS, Utz PJ, Vignali DA. Scalable signaling mediated by T cell antigen receptor-CD3 ITAMs ensures effective negative selection and prevents autoimmunity. Nat Immunol. 2008;9:658–666. doi: 10.1038/ni.1611. [DOI] [PubMed] [Google Scholar]
- 10.Tailor P, Tsai S, Shameli A, Serra P, Wang J, Robbins S, Nagata M, Szymczak-Workman AL, Vignali DA, Santamaria P. The proline-rich sequence of CD3epsilon as an amplifier of low-avidity TCR signaling. J Immunol. 2008;181:243–255. doi: 10.4049/jimmunol.181.1.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mingueneau M, Sansoni A, Gregoire C, Roncagalli R, Aguado E, Weiss A, Malissen M, Malissen B. The proline-rich sequence of CD3epsilon controls T cell antigen receptor expression on and signaling potency in preselection CD4+CD8+ thymocytes. Nat Immunol. 2008;9:522–532. doi: 10.1038/ni.1608. [DOI] [PubMed] [Google Scholar]
- 12.Szymczak AL, Vignali DA. Development of 2A peptide-based strategies in the design of multicistronic vectors. Expert Opin Biol Ther. 2005;5:627–638. doi: 10.1517/14712598.5.5.627. [DOI] [PubMed] [Google Scholar]
- 13.Xu C, Gagnon E, Call ME, Schnell JR, Schwieters CD, Carman CV, Chou JJ, Wucherpfennig KW. Regulation of T cell receptor activation by dynamic membrane binding of the CD3epsilon cytoplasmic tyrosine-based motif. Cell. 2008;135:702–713. doi: 10.1016/j.cell.2008.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deford-Watts LM, Tassin TC, Becker AM, Medeiros JJ, Albanesi JP, Love PE, Wulfing C, van Oers NS. The cytoplasmic tail of the T cell receptor CD3 epsilon subunit contains a phospholipid-binding motif that regulates T cell functions. J Immunol. 2009;183:1055–1064. doi: 10.4049/jimmunol.0900404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeFord-Watts LM, Dougall DS, Belkaya S, Johnson BA, Eitson JL, Roybal KT, Barylko B, Albanesi JP, Wulfing C, van Oers NS. The CD3 zeta subunit contains a phosphoinositide-binding motif that is required for the stable accumulation of TCR-CD3 complex at the immunological synapse. J Immunol. 2011;186:6839–6847. doi: 10.4049/jimmunol.1002721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aivazian D, Stern LJ. Phosphorylation of T cell receptor zeta is regulated by a lipid dependent folding transition. Nat Struct Biol. 2000;7:1023–1026. doi: 10.1038/80930. [DOI] [PubMed] [Google Scholar]
- 17.Gagnon E, Schubert DA, Gordo S, Chu HH, Wucherpfennig KW. Local changes in lipid environment of TCR microclusters regulate membrane binding by the CD3epsilon cytoplasmic domain. The Journal of experimental medicine. 2012;209:2423–2439. doi: 10.1084/jem.20120790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holst J, Szymczak-Workman AL, Vignali KM, Burton AR, Workman CJ, Vignali DA. Generation of T-cell receptor retrogenic mice. Nat Protoc. 2006;1:406–417. doi: 10.1038/nprot.2006.61. [DOI] [PubMed] [Google Scholar]
- 19.Holst J, Vignali KM, Burton AR, Vignali DA. Rapid analysis of T-cell selection in vivo using T cell-receptor retrogenic mice. Nat Methods. 2006;3:191–197. doi: 10.1038/nmeth858. [DOI] [PubMed] [Google Scholar]
- 20.Wang B, Wang N, Whitehurst CE, She J, Chen J, Terhorst C. T lymphocyte development in the absence of CD3 epsilon or CD3 gamma delta epsilon zeta. J Immunol. 1999;162:88–94. [PubMed] [Google Scholar]
- 21.Love PE, Shores EW, Johnson MD, Tremblay ML, Lee EJ, Grinberg A, Huang SP, Singer A, Westphal H. T cell development in mice that lack the zeta chain of the T cell antigen receptor complex. Science. 1993;261:918–921. doi: 10.1126/science.7688481. [DOI] [PubMed] [Google Scholar]
- 22.Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, Hogquist KA. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. The Journal of experimental medicine. 2011;208:1279–1289. doi: 10.1084/jem.20110308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DA. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat Biotechnol. 2004;22:589–594. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- 24.Huppa JB, Davis MM. T-cell-antigen recognition and the immunological synapse. Nat Rev Immunol. 2003;3:973–983. doi: 10.1038/nri1245. [DOI] [PubMed] [Google Scholar]
- 25.Wang H, Holst J, Woo SR, Guy C, Bettini M, Wang Y, Shafer A, Naramura M, Mingueneau M, Dragone LL, Hayes SM, Malissen B, Band H, Vignali DA. Tonic ubiquitylation controls T-cell receptor:CD3 complex expression during T-cell development. Embo J. 2010;29:1285–1298. doi: 10.1038/emboj.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas PG, Brown SA, Keating R, Yue W, Morris MY, So J, Webby RJ, Doherty PC. Hidden epitopes emerge in secondary influenza virus-specific CD8+ T cell responses. J Immunol. 2007;178:3091–3098. doi: 10.4049/jimmunol.178.5.3091. [DOI] [PubMed] [Google Scholar]
- 27.Robins H, Desmarais C, Matthis J, Livingston R, Andriesen J, Reijonen H, Carlson C, Nepom G, Yee C, Cerosaletti K. Ultra-sensitive detection of rare T cell clones. J Immunol Methods. 2012;375:14–19. doi: 10.1016/j.jim.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, Riddell SR, Warren EH, Carlson CS. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood. 2009;114:4099–4107. doi: 10.1182/blood-2009-04-217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robins HS, Srivastava SK, Campregher PV, Turtle CJ, Andriesen J, Riddell SR, Carlson CS, Warren EH. Overlap and effective size of the human CD8+ T cell receptor repertoire. Sci Transl Med. 2010;2:47ra64. doi: 10.1126/scitranslmed.3001442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bettini ML, Bettini M, Nakayama M, Guy CS, Vignali DA. Generation of T cell receptor-retrogenic mice: improved retroviral-mediated stem cell gene transfer. Nat Protoc. 2013;8:1837–1840. doi: 10.1038/nprot.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gascoigne NR, Palmer E. Signaling in thymic selection. Curr Opin Immunol. 2011;23:207–212. doi: 10.1016/j.coi.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bendelac A, Matzinger P, Seder RA, Paul WE, Schwartz RH. Activation events during thymic selection. The Journal of experimental medicine. 1992;175:731–742. doi: 10.1084/jem.175.3.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Oers NS, Killeen N, Weiss A. ZAP-70 is constitutively associated with tyrosine-phosphorylated TCR zeta in murine thymocytes and lymph node T cells. Immunity. 1994;1:675–685. doi: 10.1016/1074-7613(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 34.Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol. 2008;8:247–258. doi: 10.1038/nri2274. [DOI] [PubMed] [Google Scholar]
- 35.Kuhns MS, Davis MM. The safety on the TCR trigger. Cell. 2008;135:594–596. doi: 10.1016/j.cell.2008.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar A, Humphreys TD, Kremer KN, Bramati PS, Bradfield L, Edgar CE, Hedin KE. CXCR4 physically associates with the T cell receptor to signal in T cells. Immunity. 2006;25:213–224. doi: 10.1016/j.immuni.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 37.Hayday AC, Pennington DJ. Key factors in the organized chaos of early T cell development. Nat Immunol. 2007;8:137–144. doi: 10.1038/ni1436. [DOI] [PubMed] [Google Scholar]
- 38.Gagnon E, Schubert DA, Gordo S, Chu HH, Wucherpfennig KW. Local changes in lipid environment of TCR microclusters regulate membrane binding by the CD3epsilon cytoplasmic domain. J Exp Med. 2012;209:2423–2439. doi: 10.1084/jem.20120790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi X, Bi Y, Yang W, Guo X, Jiang Y, Wan C, Li L, Bai Y, Guo J, Wang Y, Chen X, Wu B, Sun H, Liu W, Wang J, Xu C. Ca2+ regulates T-cell receptor activation by modulating the charge property of lipids. Nature. 2013;493:111–115. doi: 10.1038/nature11699. [DOI] [PubMed] [Google Scholar]
- 40.Daniels MA, Teixeiro E, Gill J, Hausmann B, Roubaty D, Holmberg K, Werlen G, Hollander GA, Gascoigne NR, Palmer E. Thymic selection threshold defined by compartmentalization of Ras/MAPK signalling. Nature. 2006;444:724–729. doi: 10.1038/nature05269. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.